Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the
fourth leading cause of cancer-related death in the USA (1). Significant advances in the therapy for
PDAC have been achieved in the past few decades; however, the
overall 5-year survival is still less than 5% (2). The dismal prognosis is particularly
due to the occult early symptoms and the difficulty in early
diagnosis. Thus, less than 20% of tumors are resectable at the time
of diagnosis (3). Recurrence is
common for patients who undergo surgical intervention. Chemotherapy
is the most important adjuvant treatment for pancreatic cancer
patients who are recurrent and not indicated for resection.
However, PDAC is also notoriously resistant to gemcitabine (GEM),
which is the first-line chemotherapeutic agent. Therefore, it is of
utmost importance to identify new molecular targets for the
effective treatment of pancreatic cancer.
Epigenetics is a rapidly progressing field of
oncology. Modulation of epigenetic regulators has emerged as an
alternative therapeutic approach in cancer treatment. BRD4 belongs
to the bromodomain and extraterminal domain (BET) family that
contains two bromodomains in tandem and an extra terminal domain.
As a conserved epigenome regulator, BRD4 regulates the expression
of numerous genes involved in the cell cycle, cell growth and
inflammation (4,5). Recent studies have demonstrated that
BRD4 occupies super-enhancers of key oncogenic genes in certain
biological contexts (6,7). Oncogenic genes regulated by
super-enhancers are greatly sensitive to levels of BRD4. Several
studies have found that inhibition of BRD4 resulted in significant
downregulation of c-Myc in hematopoietic malignancies and
glioblastoma (8,9). In lung adenocarcinoma, FOSL1 has been
demonstrated as the main effector of BRD4 (10). Whether there is a different role for
BRD4 in different cell lines is unclear. A recent study found that
silencing of BRD4 in PDAC cells decreased the cell growth and
abolished epithelial-to-mesenchymal transition (11). However, the widespread and
heterogeneous genetic aberrations that occur in PDAC remain a major
challenge, and cause a decrease in the response of PDAC to
treatment targeting these highly altered signaling pathways. In
addition, the mechanism by which BRD4 takes part in the regulation
of PDAC cell proliferation remains elusive. Therefore, there is a
vital necessity to illustrate the function and mechanism of BRD4 in
regards to PDAC.
In the present study, we demonstrated that BRD4 is
involved in PDAC tumorigenesis. Specifically, BRD4 knockdown
suppressed tumor growth both in vitro and in vivo.
Our findings revealed the role of BRD4 in PDAC drug resistance. The
genetic inhibition of BRD4 was found to increase GEM sensitivity.
Previous studies have demonstrated that the Sonic hedgehog (Shh)
pathway induces PDAC tumorigenesis by promoting cell proliferation
and increasing drug resistance (12–14).
Therefore, we aimed to elucidate whether BRD4 is associated with
the Shh pathway in PDAC and to explore the mechanisms by which BRD4
enhances the proliferation and viability of PDAC cells. The present
study demonstrated that BRD4 promoted the Shh signaling pathway in
PDAC cells in a ligand-independent manner.
Materials and methods
Cell culture
Human PDAC cell lines (PANC-1, SW1990, COLO357,
L3.6PL, Capan-2 and MIAPaCa-2) from Shanghai Cell Bank (Shanghai,
China) were grown in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin
and 0.1 mg/ml streptomycin. Additional 2.5% horse serum was used
for the MIAPaCa-2 cells. Human pancreatic duct epithelial (HPDE)
cells were maintained in keratinocyte serum-free medium (Gibco,
Grand Island, NY, USA) supplemented with epidermal growth factor
and bovine pituitary extract. The cells were cultured in a
humidified incubator at 37°C with 5% CO2. In some
experiments, recombinant human Shh N-terminal peptide (rhShh)
(R&D Systems, Minneapolis, MN, USA) was added to the culture
medium.
Establishment of cell lines stably
expressing BRD4 shRNA
shRNA plasmids for human BRD4 were designed against
the BRD4 gene and constructed in Phblv-u6-puro vectors. A
non-target scrambled oligonucleotide served as the negative
control. To generate stable BRD4 knockdown cells, PANC-1 and
MIAPaCa-2 cells were grown in 6-well plates until they reached 50%
confluency. The medium was replaced with 1 ml of fresh culture
medium supplemented with 100 μl viral supernatant (1×108
UT/ml) and 8 μg/ml Polybrene for 24 h. The PANC-1 and MIAPaCa-2
cells were further cultured in medium containing puromycin at 3 and
2 μg/ml, respectively. Individual puromycin-resistant colonies were
isolated during drug screening. Knockdown efficiency was assessed
by immunoblotting. The shRNA sequences used in the present study
were as follows: shBRD4, 5′-GatccGCCTGGAGATGACATAG
TCTTATTCAAGAGATAAGACTATGTCATCTCCAGGTT TTTTc-3′ and shcontrol,
5′-GatccTTCTCCGAACGTGTCAC GTAATTCAAGAGATTACGTGACACGTTCGGAGAATT
TTTTg-3′.
Cell viability assay
Cell viability was measured using the Cell Counting
Kit-8 (Beyotime, Haimen, Jiangsu, China). Briefly, cells
(3,000/well) in 200 μl medium were plated into 96-well plates.
After culturing for various times, 10 μl of CCK-8 solution was
added into each well at 37°C. After 2 h, the optical density values
of each well were measured using a microplate reader at 450 nm.
Colony formation assay
PANC-1 and MIAPaCa-2 cells stably transfected with
shBRD4 or shcontrol were seeded at 200 cells/well in 6-well plates.
After 10 days of culture, the colonies were stained with crystal
violet, photographed and then scored.
Cell apoptosis analysis
Cell apoptosis was assessed by flow cytometry. For
the cell apoptosis assay, PANC-1 and MIAPaCa-2 cells stably
transfected with shBRD4 or shcontrol were incubated in the absence
or presence of 2 μmol/l GEM for 24 or 48 h. The percentage of
apoptotic cells was analyzed by staining with fluorescein
isothiocyanate-conjugated Annexin V and propidium iodide (KeyGen
Biotech, Nanjing, Jiangsu, China), immediately followed by flow
cytometry.
Real-time RT-PCR
Total RNA from the cells was isolated using TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) and then
reverse-transcribed using a reverse transcription kit (Takara,
Dalian, China) following the manufacturer’s protocol. Amplification
specificity was verified by melting curve analysis and agarose gel
electrophoresis. The relative mRNA expression of the target genes
was calculated as the inverse log of ΔΔCT and normalized to the
reference gene β-actin. The primers used for amplification were:
BRD4 forward, 5′-CATG GACATGAGCACAATCA-3′ and reverse,
5′-TCATGGTCAG GAGGGTTGTA-3′; SHH forward, 5′-CCCAATTACAACCC
CGACATC-3′ and reverse, 5′-TCACCCGCAGTTTCACTC CT-3′; PTCH1 forward,
5′-TGAGACTGACCACGGCCTG-3′ and reverse, 5′-ACCCTCAGTTGGAGCTGCTTC-3′;
GLI1 forward, 5′-AGGGCTGCAGTAAAGCCTTCA-3′ and reverse,
5′-CTTGACATGTTTTCGCAGCG-3′; β-actin forward, 5′-GA
TCATTGCTCCTCCTGAGC-3′ and reverse, 5′-ACTCCTGCT TGCTGATCCAC-3′.
Western blotting
Equal amounts of the protein from lysates of the
cultured PDAC cells were subjected to 10% SDS-PAGE and then
transferred to PVDF membranes. After blocking in 5% skim milk, the
membranes were incubated overnight at 4°C with the primary
antibodies against BRD4 (Abcam, Cambridge, MA, USA), SHH, PTCH1,
GLI1 (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA) or
β-actin (Beyotime, Haimen, Jiangsu, China). After washing thrice
with washing buffer [phosphate-buffered saline (PBS) containing
0.1% Tween-20] for 5 min, the membranes were incubated for 1 h with
the secondary antibodies at room temperature. Specific bands were
visualized with enhanced chemiluminescence reagent (KeyGen Biotech)
on an autoradiographic film.
Immunohistochemistry
Paraffin-embedded tissue slides (4-μm thick) were
deparaffinized in xylene, rehydrated through graded alcohol
solutions, blocked in methanol containing 3% hydrogen peroxide, and
then incubated with 1:200 rabbit anti-human anti-Ki67 antibody
(Santa Cruz Biotechnology) at 4°C overnight. After rinsing with PBS
solution, secondary antibodies and streptavidin peroxidase complex
reagent were applied for 1 h at room temperature. Finally, the
sections were incubated in a 3,3′-diaminobenzidine solution at room
temperature for 10 min and then counterstained with
hematoxylin.
Animal studies
All animal studies were approved by the Animal Care
and Welfare Committee of Southeast University and were conducted in
strict accordance with the guidelines of the National Animal
Welfare Law of China. Five-week old male BALB/c nude mice were
procured from the Laboratory Animal Center of Yangzhou University
(Yangzhou, China) and maintained in a specific pathogen-free
environment. Stable PANC-1 shBRD4 and PANC-1 shcontrol cells
(2×106) were subcutaneously injected into the right
flanks of athymic nude mice. Tumor volume (mm3) was
calculated every 3 days for 3 weeks using the formula V = 0.5 ×
length × width2. After 3 weeks, the mice were
euthanized. The tumors were isolated, weighed, photographed and
then processed for immunohistochemistry.
Statistical analysis
All experiments were repeated in triplicate. Unless
otherwise indicated, experimental values are expressed as mean ±
SEM. Statistical significance was determined by the unpaired
Student’s t-test using SPSS 13.0. p<0.05 was considered to
indicate a statistically significant result.
Results
BRD4 is overexpressed in PDAC
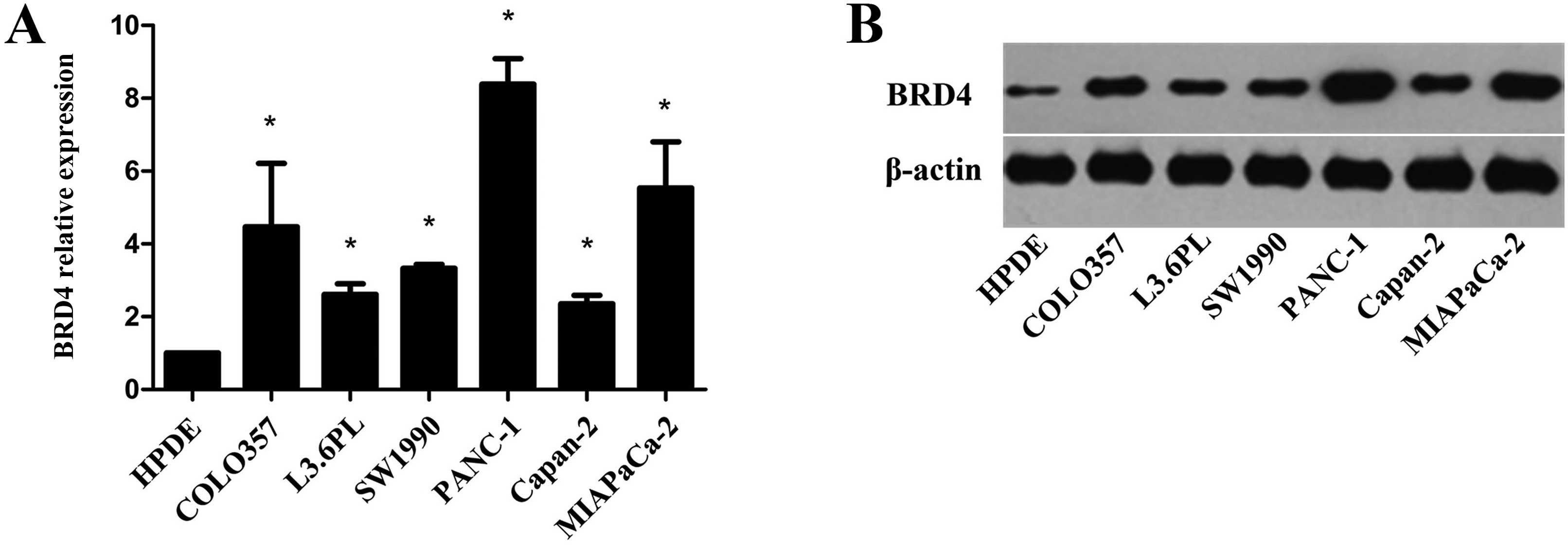
To investigate the potential role of BRD4 in PDAC,
we assessed the expression level of BRD4 in the PDAC cell lines
COLO357, L3.6PL, SW1990, PANC-1, Capan-2 and MIAPaCa-2, as well as
in the normal pancreatic epithelial cell line HPDE. qRT-PCR and
western blot analyses showed that the mRNA and protein expression
levels of BRD4 were significantly higher in the PDAC cells than
these levels in the HPDE cells (Fig. 1A
and B). The highest expression levels of BRD4 were detected in
the PANC-1 and MIAPaCa-2 cells. The increased level of BRD4 in PDAC
suggests that BRD4 promotes pancreatic tumorigenesis.
BRD4 promotes the viability and
proliferation of pancreatic cancer cells in vitro
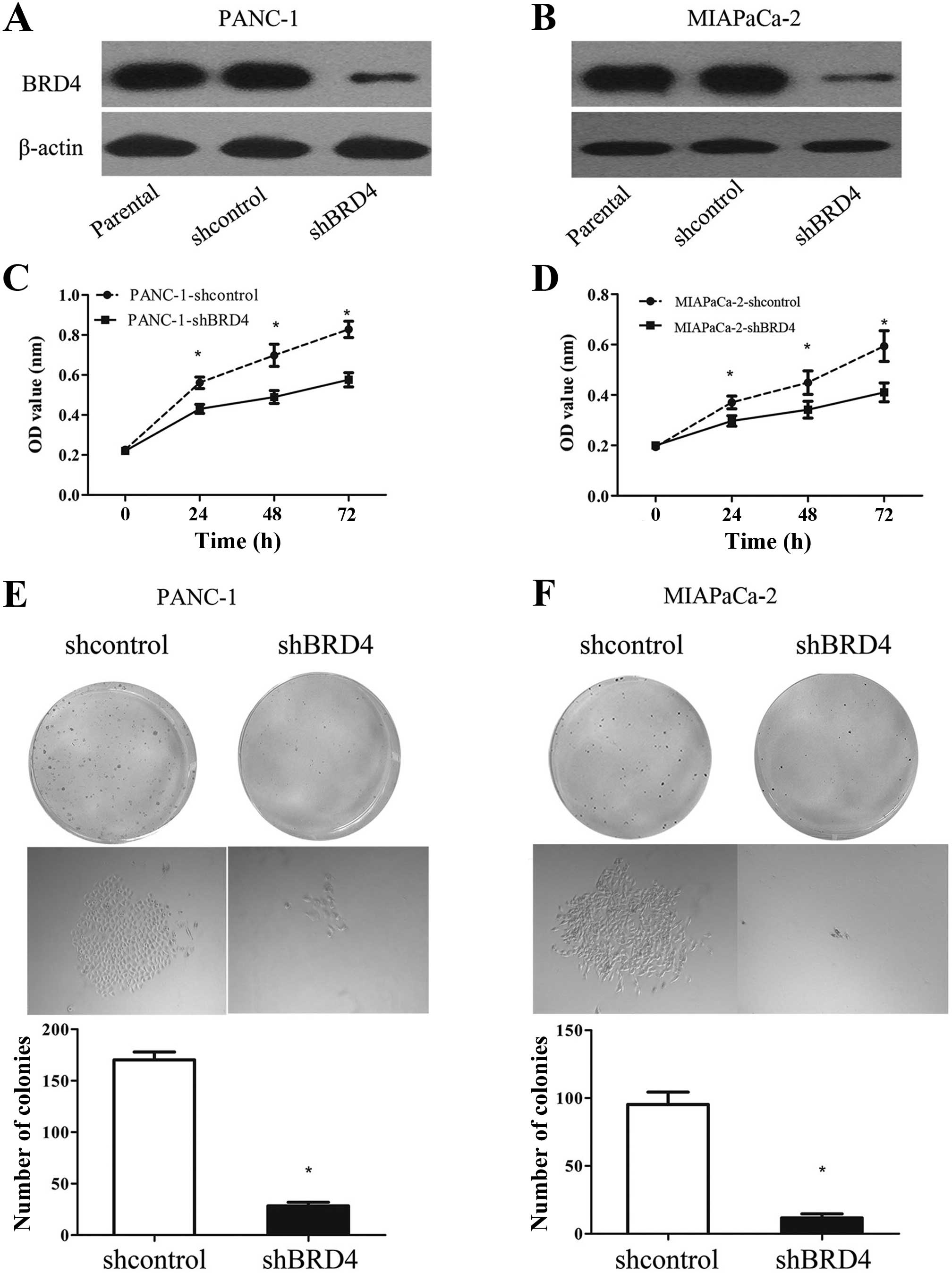
We downregulated BRD4 in the PANC-1 and MIAPaCa-2
cells to examine the influence of this protein on the biological
behavior of PDAC cells. The expression level of BRD4 was confirmed
by western blotting (Fig. 2A and
B). PANC-1 and MIAPaCa-2 cells were selected for further
studies due to their high expression levels of BRD4. The viability
of the PANC-1 and MIAPaCa-2 cells was investigated using the CCK-8
assay after 24, 48 and 72 h of incubation. BRD4 knockdown evidently
inhibited the viability of both cell lines (Fig. 2C and D). Furthermore, the colony
formation assay showed that the proliferation of stable
BRD4-knockdown cells was severely inhibited (Fig. 2E and F). These results suggest that
BRD4 is involved in the growth of pancreatic cancer cells.
BRD4 is essential for pancreatic cancer
progression in vivo
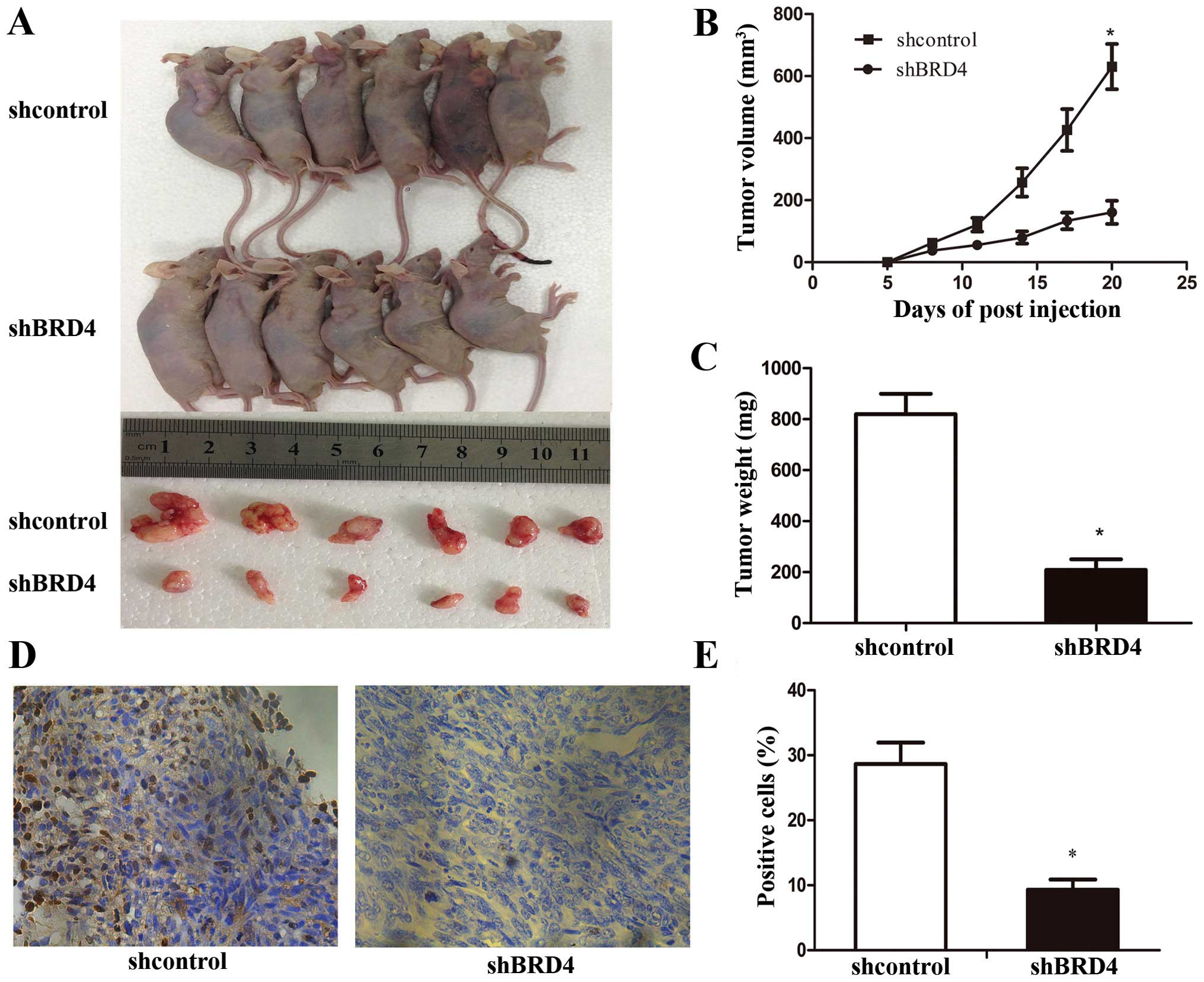
To study the relevance of BRD4 to PDAC progression
in vivo, we subcutaneously transplanted shBRD4 and shcontrol
PANC-1 cells into nude mice (n=6/group). At 20 days after cell
implantation, the tumors were removed and photographed (Fig. 3A). Consistent with a previous study,
the present study showed that BRD4 silencing did not affect tumor
initiation (15). However, the
periodic measurements of tumor volume and the final weight of the
excised tumors demonstrated that BRD4 shRNA-PANC-1 cells had
significantly retarded tumor growth at termination compared with
the control cells (Fig. 3B and C).
Histologic analysis of tumor proliferation revealed that shBRD4
tumors had significantly fewer Ki-67-positive cells than the
shcontrol tumors (Fig. 3D and E).
These results indicate that BRD4 participates in PDAC proliferation
and tumor maintenance in vivo.
BRD4 knockdown causes GEM
sensitization
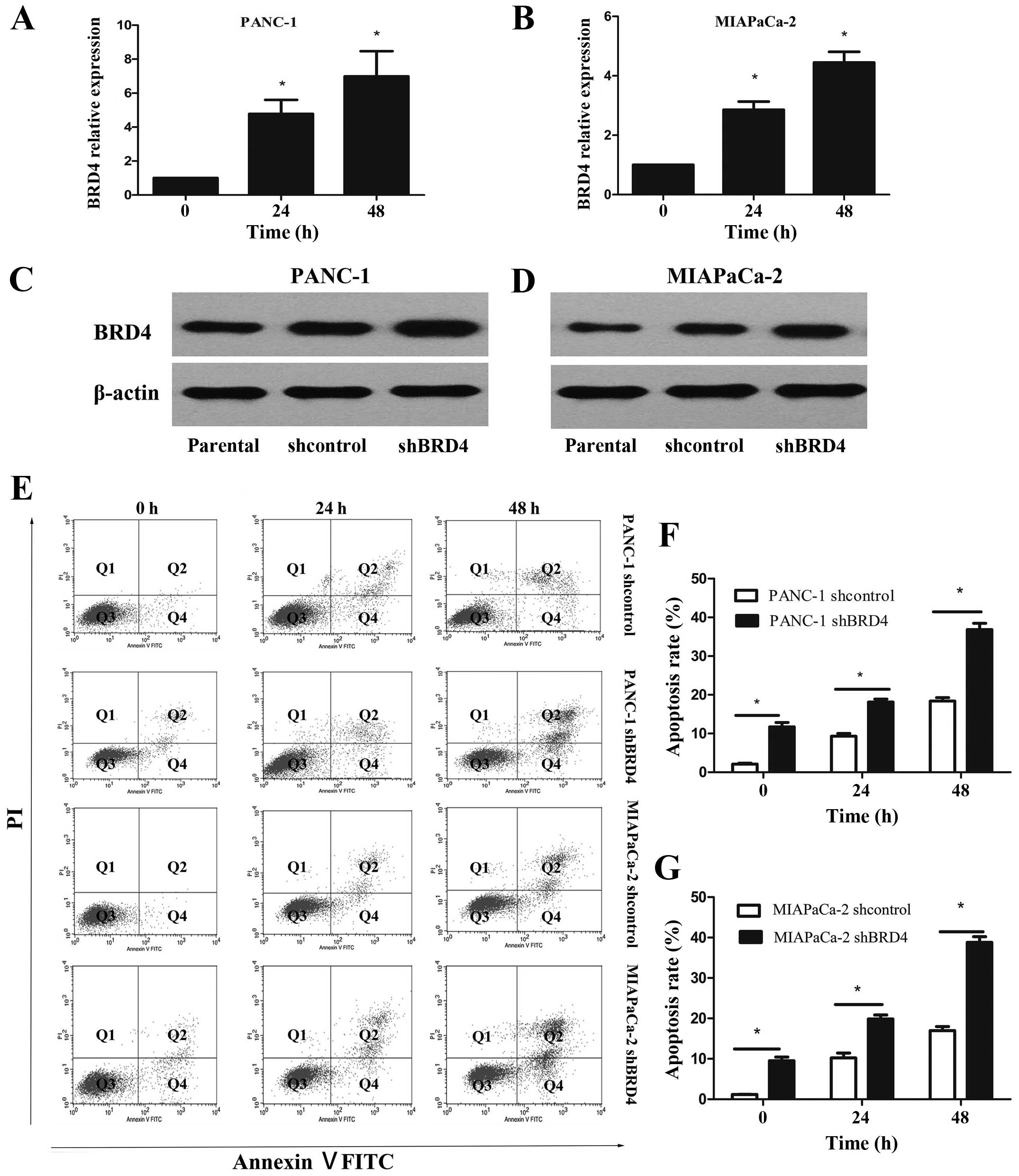
PANC-1 and MIAPaCa-2 cells were treated with 2
μmol/l GEM to investigate the role of BRD4 in drug resistance. GEM
significantly increased the mRNA and protein expression levels of
BRD4 in the PANC-1 and MIAPaCa-2 cells (Fig. 4A–D). Then, we examined the cell
apoptosis rate of PANC-1 and MIAPaCa-2 cells after treatment with 2
μmol/l GEM for 24 or 48 h to further elucidate the relationship
between BRD4 and drug resistance in pancreatic cancer cells. BRD4
knockdown alone induced cell apoptosis in the PANC-1 and MIAPaCa-2
cells; however, treatment with GEM significantly promoted cell
apoptosis (Fig. 4E–G). These
results revealed that BRD4 decreased the GEM sensitivity of
pancreatic cancer cells. In addition, BRD4 downregulation and GEM
treatment synergistically influenced the chemotherapeutic efficacy
in the PDAC cells.
BRD4 regulates the Shh signaling pathway
in pancreatic cancer cells
Recent study has shown that the Shh signaling
pathway is regulated by BRD4 (16).
The Shh signaling pathway plays a critical role in the genesis of
PDAC, and maintenance of hedgehog signaling is important for
aberrant proliferation and tumorigenesis (12,13).
We analyzed the expression of the principal components of the Shh
signaling pathway in BRD4-knockdown PANC-1 and MIAPaCa-2 cells to
further explore the mechanism by which BRD4 influences the
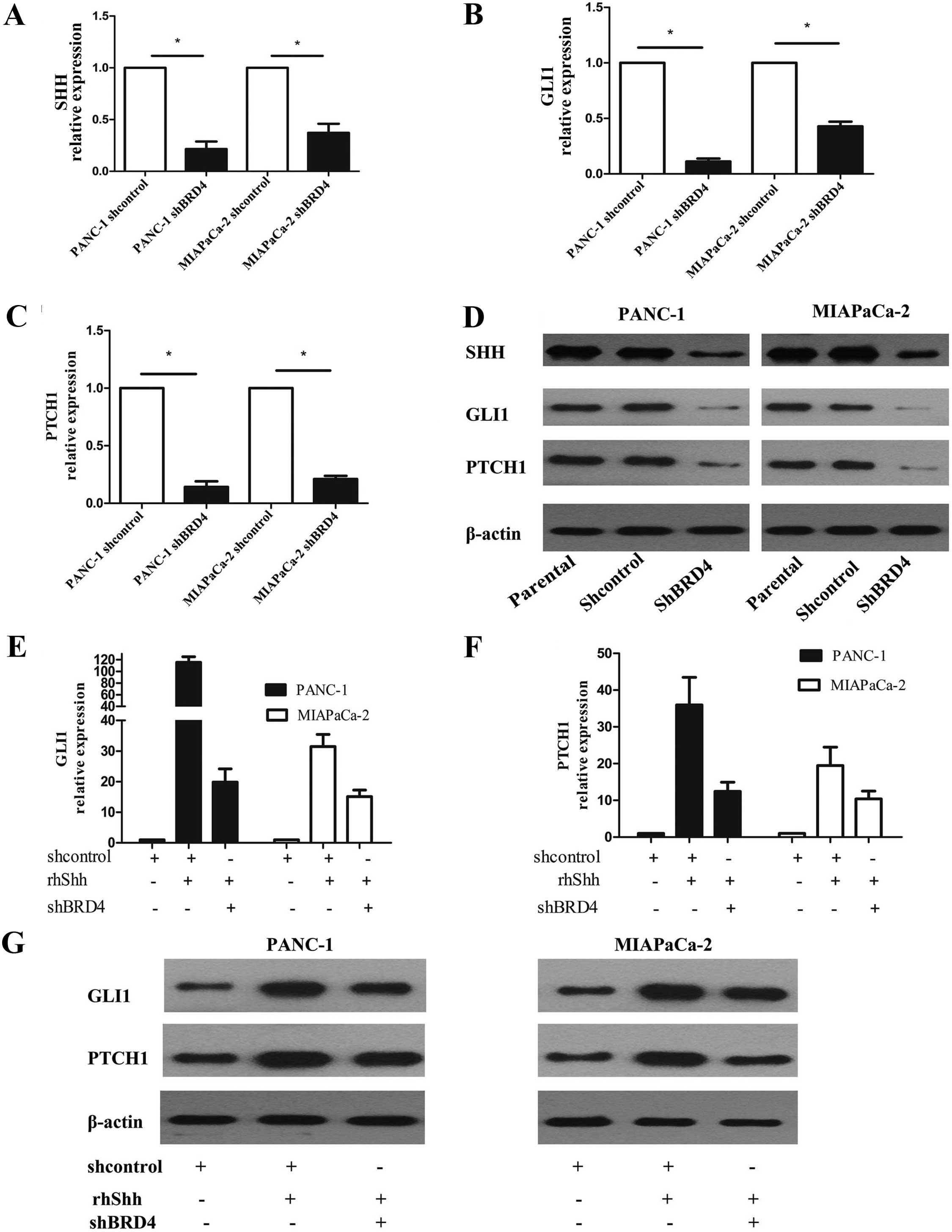
proliferation and viability of pancreatic cancer cells. BRD4
knockdown decreased the mRNA and protein levels of Shh
pathway-related genes, including SHH, PTCH1 and GLI1, in both cell
lines (Fig. 5A–D). These results
indicate that BRD4 promotes the Shh signaling pathway in pancreatic
cancer cells in a ligand-dependent manner. Several in vitro
experiments using cell lines indicated that the Shh signaling
pathway is partially activated in PDAC in a ligand-independent
manner (17,18). Therefore, we investigated whether or
not BRD4-induced Shh contributes to Shh signaling pathway
activation. BRD4 knockdown in PANC-1 and MIAPaCa-2 cells followed
by rhShh stimulation markedly inhibited Shh target gene expression
(Fig. 5E–G). This result supports
the essential role of BRD4 in the Shh signaling pathway. These data
suggest that the BRD4-induced increase in Shh transcription does
not contribute to Shh pathway activation. In other words,
BRD4-induced Shh pathway activation is essentially independent of
Shh.
Discussion
Epigenetic modifications in cancer initiation and
progression have attracted increased attention. Targeting
epigenetic modulators known as ‘readers’ is a promising cancer
treatment strategy. As an epigenome reader, the BRD4 protein plays
a critical role at the interface between chromatin remodeling and
transcriptional regulation. Previous studies have described the
therapeutic potential of targeting BRD4 in models of acute myeloid
leukemia, glioblastoma, multiple myeloma, lung adenocarcinoma and
osteosarcoma (8–10,19,20).
By contrast, BRD4 is downregulated in breast and colon cancers;
thus, this protein may serve as a tumor suppressor in these cancers
(21,22). Whether or not BRD4 is a tumor
promoter or suppressor remains controversial. These contradictory
results reveal that the function of BRD4 in different tissues
depends on the type of cancer, the mechanism of tumorigenesis, and
the abundance of different BRD4 downstream targets. Therefore,
future research must demonstrate the conclusive function of BDR4 in
different types of cancers. In the present study, BRD4 was
overexpressed in PDAC cells. Consistent with a recent study
(11), we also showed that BRD4
promoted the viability and proliferation of pancreatic cancer cells
in vitro and this was further illustrated in vivo.
These results suggest that BRD4 is crucial in pancreatic cancer
tumorigenesis.
PDAC is a fatal disease due to its aggressiveness
and resistance to chemotherapy. Selective inhibition targeting
small-molecule proteins, together with effective conventional
chemotherapeutic agents, is a promising approach to combat cancer
chemoresistance. In the present study, GEM significantly
upregulated BRD4 expression in PDAC cell lines. This upregulation
may be involved in the adaptive response to DNA damage in PDAC
chemotherapy. We also found that BRD4 knockdown in PDAC cells
enhanced the apoptotic effect of GEM, further verifying our
hypothesis regarding the involvement of BRD4 in chemoresistance.
These data suggest that BRD4 inhibition and GEM treatment
synergistically influence pancreatic cancer treatment.
The epigenetic state of a cell is markably
influenced by the cell line. Thus, chromatin-binding proteins
possibly have different transcriptional targets in diverse types of
cancer. BET family members induce specific subsets of gene
expression in different cell types. For example, the BET protein in
cell lineage derived from hematologic malignancies promotes cell
cycle progression by affecting MYC gene expression (23). In acute lymphoblastic leukemia, this
protein induces the same effect by influencing c-Myc and IL7R gene
expression (8). However, a recent
study has reported that cyclin D1 is the principal effector of BET
protein-induced cell-cycle progression in malignant peripheral
nerve sheath tumors (19). In lung
adenocarcinoma, instead of MYC or cyclin D1, FOSL1 plays the same
role in cell cycle progression (10). BRD4 silencing represses not only
FOSL1 and c-Myc expression in PDAC yet also HMGA2, an architectural
protein that we previously identified to mediate chemoresistance in
pancreatic cancer cells (11). The
present study demonstrated that BRD4 induces PDAC cell viability
and proliferation by regulating the Shh signaling pathway, which
enhances PDAC cell proliferation and GEM resistance (12–14).
Mammals have three known paralogous genes, namely, Shh, Indian
hedgehog and Desert hedgehog (24).
Among these genes, Shh is specifically overexpressed in PDAC
(14). The Shh signaling pathway is
initiated by the binding of the secreted Shh ligands to PTCH1 that
relieves the PTCH1-mediated repression of SMO. SMO triggers the
downstream signaling cascades that induce the nuclear translocation
of Gli and consequently activates the transcription of Shh target
genes, including PTCH1, Gli1, cyclin D and E and MYC. Aberrant Shh
signaling activity is transduced through three signal transduction
models: i) ligand-independent signaling; ii) ligand-dependent
autocrine/juxtacrine signaling; and iii) ligand-dependent paracrine
signaling (25). In the present
study, BRD4 activated the Shh signaling pathway of PDAC in a
ligand-independent manner. This finding suggests that BRD4 promotes
the malignant transformation and drug resistance of PDAC cells by
activating the Shh signaling pathway.
In conclusion, BRD4 promotes the proliferation and
GEM resistance of PDAC possibly by regulating the Shh signaling
pathway. Therefore, BRD4 is a novel target for the development of
alternative therapeutic approaches for PDAC.
Acknowledgements
This study was financially supported by China’s
National Natural Science Foundation (81071967 and 30872500).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davis JL, Pandalai PK, Ripley RT, Langan
RC and Avital I: Expanding surgical treatment of pancreatic cancer:
the role of regional chemotherapy. Pancreas. 41:678–684. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang Z, He N and Zhou Q: Brd4 recruits
P-TEFb to chromosomes at late mitosis to promote G1 gene
expression and cell cycle progression. Mol Cell Biol. 28:967–976.
2008. View Article : Google Scholar :
|
|
5
|
Nicodeme E, Jeffrey KL, Schaefer U, et al:
Suppression of inflammation by a synthetic histone mimic. Nature.
468:1119–1123. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Loven J, Hoke HA, Lin CY, et al: Selective
inhibition of tumor oncogenes by disruption of super-enhancers.
Cell. 153:320–334. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chapuy B, McKeown MR, Lin CY, et al:
Discovery and characterization of super-enhancer-associated
dependencies in diffuse large B cell lymphoma. Cancer Cell.
24:777–790. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ott CJ, Kopp N, Bird L, et al: BET
bromodomain inhibition targets both c-Myc and IL7R in high-risk
acute lymphoblastic leukemia. Blood. 120:2843–2852. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pastori C, Daniel M, Penas C, et al: BET
bromodomain proteins are required for glioblastoma cell
proliferation. Epigenetics. 9:611–620. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lockwood WW, Zejnullahu K, Bradner JE and
Varmus H: Sensitivity of human lung adenocarcinoma cell lines to
targeted inhibition of BET epigenetic signaling proteins. Proc Natl
Acad Sci USA. 109:19408–19413. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sahai V, Kumar K, Knab LM, et al: BET
bromodomain inhibitors block growth of pancreatic cancer cells in
three-dimensional collagen. Mol Cancer Ther. 13:1907–1917. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Berman DM, Karhadkar SS, Maitra A, et al:
Widespread requirement for Hedgehog ligand stimulation in growth of
digestive tract tumours. Nature. 425:846–851. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thayer SP, di Magliano MP, Heiser PW, et
al: Hedgehog is an early and late mediator of pancreatic cancer
tumorigenesis. Nature. 425:851–856. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu M, Li L, Liu Z, et al: ABCB2 (TAP1) as
the downstream target of SHH signaling enhances pancreatic ductal
adenocarcinoma drug resistance. Cancer Lett. 333:152–158. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Segura MF, Fontanals-Cirera B,
Gaziel-Sovran A, et al: BRD4 sustains melanoma proliferation and
represents a new target for epigenetic therapy. Cancer Res.
73:6264–6276. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang Y, Gholamin S, Schubert S, et al:
Epigenetic targeting of Hedgehog pathway transcriptional output
through BET bromodomain inhibition. Nat Med. 20:732–740. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Onishi H, Kai M, Odate S, et al: Hypoxia
activates the hedgehog signaling pathway in a ligand-independent
manner by upregulation of Smo transcription in pancreatic cancer.
Cancer Sci. 102:1144–1150. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nolan-Stevaux O, Lau J, Truitt ML, et al:
GLI1 is regulated through Smoothened-independent mechanisms in
neoplastic pancreatic ducts and mediates PDAC cell survival and
transformation. Genes Dev. 23:24–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Patel AJ, Liao CP, Chen Z, Liu C, Wang Y
and Le LQ: BET bromodomain inhibition triggers apoptosis of
NF1-associated malignant peripheral nerve sheath tumors through Bim
induction. Cell Rep. 6:81–92. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lamoureux F, Baud’huin M, Rodriguez
Calleja L, et al: Selective inhibition of BET bromodomain
epigenetic signalling interferes with the bone-associated tumour
vicious cycle. Nat Commun. 5:35112014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rodriguez RM, Huidobro C, Urdinguio RG, et
al: Aberrant epigenetic regulation of bromodomain BRD4 in human
colon cancer. J Mol Med. 90:587–595. 2012. View Article : Google Scholar
|
|
22
|
Crawford NP, Alsarraj J, Lukes L, et al:
Bromodomain 4 activation predicts breast cancer survival. Proc Natl
Acad Sci USA. 105:6380–6385. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mertz JA, Conery AR, Bryant BM, et al:
Targeting MYC dependence in cancer by inhibiting BET bromodomains.
Proc Natl Acad Sci USA. 108:16669–16674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Geissler K and Zach O: Pathways involved
in Drosophila and human cancer development: the Notch, Hedgehog,
Wingless, Runt, and Trithorax pathway. Ann Hematol. 91:645–669.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Teglund S and Toftgård R: Hedgehog beyond
medulloblastoma and basal cell carcinoma. Biochim Biophys Acta.
1805:181–208. 2010.PubMed/NCBI
|