Introduction
Many studies have proven that the process of
carcinogenesis not only depends on genetic alterations but also on
abnormal cellular memory, or epigenetic changes which are
associated with a heritable gene expression profile critical for
cancer development (1–4). An emerging perspective in therapeutic
approaches for various types of cancers is to target epigenetic
alterations due to their reversible nature, and these epigenetic
alterations are manifested in both global changes in nucleosome
packaging and in localized gene promoter changes of various
tumor-suppressor genes (TSGs) which influence the transcription of
genes during neoplastic initiation and progression (1,2,4–7).
Epigenetic alterations are mainly mediated by DNA
methyltransferases (DNMTs), involved in DNA methylation, and
histone deacetylases (HDACs), which play a pivotal role in histone
deacetylation. However, there are several other classes of enzymes
which are involved in post-translational modifications of histone
tails and could be considered to represent an additional facet for
the development of new anticancer therapies (5,8,9).
Available epigenetic targeting modalities which are
currently being investigated include HDAC and DNMT inhibitors.
However, certain disadvantages that currently restrict the general
use of these synthetic epigenetic drugs are that they exhibit a
lack of specificity, have a short duration of action and may cause
normal cells to change their functional and structural patterns
with unforeseen effects (8–10). Burgeoning evidence in the last
decade has provided unprecedented clues that diet and environmental
factors directly influence epigenetic mechanisms in humans. Dietary
polyphenols from green tea, turmeric, soybeans, broccoli and others
have been shown to possess multiple cell regulatory activities
within cancer cells. More recently, it has been noted that various
dietary polyphenols may exert their chemopreventive effects in part
by modulating various components of the epigenetic machinery, and
therefore the drugs that target these alterations are being studied
in various human towards a better cancer treatment strategy
(11,12).
In green tea, among the most abundant chemical
compounds, catechins which include (−)-epigallocatechin-3-gallate
(EGCG), have been shown to possess antioxidant, antiproliferative,
anti-inflammatory, anti-angiogenic and antimetastatic activities,
induce differentiation or apoptosis, arrest the cell cycle, inhibit
telomerase activity and inhibit DNA adduct formation (13–18).
Contemplating the potential role of EGCG as an epigenetic modifier,
the present study was designed to investigate the inhibition of
DNMTs and HDACs by EGCG and its effect on the expression of
epigenetically modified TSGs including retinoic acid receptor-β
(RARβ), cadherin 1 (CDH1), death-associated protein kinase-1
(DAPK1) and MGMT in human cervical cancer cell line HeLa. To
correlate the inhibition of DNMT and HDAC activity induced by EGCG,
in silico molecular modeling and docking studies on DNMT3B
and HDAC1 were performed.
Materials and methods
Cell culture
The human cervical carcinoma cell line HeLa was
generously provided by Dr Tahir A Rizvi, UAE University, Al-Ain,
United Arab Emirates (UAE). It was maintained in Dulbecco’s
modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine
serum (FBS) and 100X Pen Strep (all from Sigma, USA) in a
humidified atmosphere of 5% CO2 in air at 37°C.
Preparation of drugs
EGCG was obtained from Sigma. A stock solution of
EGCG (10 mM) was prepared in water, sterile filtered with 0.2-μm
filters and stored at −20°C in aliquots. Fresh EGCG solution was
used in each experiment and further dilutions were made in complete
medium to required concentrations of 25 μM for the treatment of
HeLa cells. A sub-stock of 500 μM TSA was prepared from 5 mM stock
solution. A working concentration of 0.025 μM was further used for
the experiments. A stock solution of 219 mM 5-aza-2′-deoxycytidine
(5-Aza-dC) (Sigma) was prepared and further working concentration
of 1 μM was made from a 10 mM sub-stock.
DNMT activity assay
HeLa cells were treated with EGCG (25 μM) and
5-Aza-dC (1 μM) for 3 days. After the treatment at various time
points, the cells were harvested and nuclear extracts were prepared
using the EpiQuik™ nuclear extraction kit (Epigentek, USA) as per
the manufacturer’s protocol. Furthermore, the DNMT activity was
assayed using the EpiQuik™ DNMT activity assay kit (Epigentek) as
per the protocol instructions.
HDAC activity assay
The effect of EGCG on HDAC activity in the HeLa
cells was determined using the EpiQuik™HDAC activity assay kit
(Epigentek). Briefly, HeLa cells were treated with 25 μM EGCG and
0.025 μM TSA for 3 days, harvested and nuclear extracts were then
prepared using the EpiQuik™ nuclear extraction kit following the
manufacturer’s instructions. Furthermore, DNMT activity was assayed
using the EpiQuik™ DNMT activity assay kit as per protocol
instructions.
Molecular modeling studies of DNMT3B and
HDAC1 with EGCG
To address the interaction of EGCG with the
epigenetic modulator enzymes HDAC1 and DNMT3B, we required
validated 3D structures of the proteins. These structures were
prepared and validated as detailed in our previous study [HDAC1 PDB
ID: 4BKX (19); mDNMT3B was modeled
from the structure of DNMT3A PDB ID: 2QRV (20)]. The substrate binding pocket was
defined using the CASTp server as detailed in our previous study
(unpublished data). The residues that constitute the substrate
binding cavity of both proteins are listed in Table I.
 | Table IResidues defining the substrate
binding pocket of HDAC1 and mDNMT3B. |
Table I
Residues defining the substrate
binding pocket of HDAC1 and mDNMT3B.
| Protein | Residues lining the
substrate binding cavity |
|---|
| mDNMT3B | C-651, E-605, F-581, D-582,
G-583, T-586, S-604, E-605, V-606, C-607, V-628, G-648, S-649,
P-650, C-651, N-652, S-655, V-657, 658, N-658, P-659, L-671, E-697,
V-699, V-700, A-701, R-731, A-732, R-733, R-773, I-774, K-777,
S-778, N-779, S-780, I-781, R-823, G-824, Q-827, K-828, G-831,
R-832, S-833, W-834 |
| HDAC1 | H-140, H-141, D-176, H-178, D-264,
L-271, F-109, W-135, A-136, G-137, L-139, G-149, C-151, F-205 |
The substrate binding pocket of mDNMT3B is fairly
large and houses both the active residue (Cys-651) and the
co-factor binding site (Glu-605). In several studies, the active
site of class I HDACs has been paralleled to a tunnel that ends in
a catalytically vital zinc ion (21). The cavity predicted by CASTp was
similar to this description and includes the catalytic residue
His-141 and Zn ion. Binding of EGCG in the same cavity where
inhibitors 5-Aza-dC/TSA bind suggests that EGCG produces its
inhibitory effects by a similar mechanism.
Docking
3 Dimensional structures of EGCG, 5-Aza-dC and TSA
in mol2 format were retrieved from the ZINC database (22). The SwissDock server, which uses
EADock algorithm was used to perform blind docking of ligands (EGCG
and 5-Aza-dC) with protein mDNMT3B and ligand (EGCG) with protein
HDAC1 (23). All the residues of
the proteins were held fixed and a binding pocket was not defined
so as not to bias the docking towards the active site. The
parameter selected for docking on the SwissDock server was
‘accurate’ with no flexibility of side chain of any amino acid of
the target proteins. After calculating their energies using CHARMM,
the different predicted binding modes of the ligand are ranked
according to their FullFitness scores. A more favorable binding
mode is indicated by a more negative FullFitness score.
UCSF-Chimaera, a molecular visualization software was used to
perform the analyses of all docked poses (24).
Bisulfite modification and
methylation-specific PCR (MS-PCR)
DNA was extracted from the EGCG-treated HeLa cells
at various time points (0, 24, 48 and 72 h) using the GenElute™
Mammalian Genomic DNA Miniprep kit (Sigma) as per the
manufacturer’s instructions. After the DNA isolation, bisulphite
modification and purification of modified DNA samples were carried
out by the Imprint DNA Modification kit (Sigma) protocol. These
modified DNA samples were used as a template for
methylation-specific PCR (MSP), to distinguish between methylated
and unmethylated promoter regions of the RARβ, CDH1 and DAPK1 genes
using specific primer sets as previously described (methylated and
unmethylated respectively) (25–27).
MSP was performed on 50 ng of bisulfite-treated DNA under the
following conditions: initial denaturation at 95°C for 5 min,
followed by 35 amplification cycles (denaturation at 94°C for 30
sec, annealing Tm for RARβ, 56°C; CDH1, 56°C; DAPK1, 57°C; MGMT,
55°C; for 30 sec, and extension at 72°C for 45 sec) with a final
extension at 72°C for 7 min.
Reverse transcription-PCR
Total RNA isolation was carried out as per the
manufacturer’s protocol using the GenElute™ Mammalian Genomic Total
RNA kit (Sigma) from 25 μM EGCG-treated HeLa cells at various time
points (24, 48 and 72 h) including the untreated control. Reverse
transcription of RNA to synthesize cDNA was performed using the
ProtoScript M-MuLV Taq RT-PCR kit (New England Biolabs, USA) from 5
mg of total RNA (at 42°C for 60 min) followed by RT-PCR using
gene-specific primers for β-actin, RARβ, CDH1, DAPK1, DNMTB and
HDAC1. The PCR cycle was as follows: initial denaturation at 95°C
for 5 min, followed by 35 amplification cycles (denaturation at
94°C for 30 sec; annealing Tm for β-actin, 56°C; RARβ, 56°C; CDH1,
55.5°C; DAPK1, 56°C; GSTP1, 55°C; DNMTB, 56°C; HDAC1, 56°C for 30
sec and extension at 72°C for 45 sec), with a final extension at
72°C for 7 min. The primer sequences used were described previously
(28–32). Amplified products were visualized on
a 2% agarose gel containing ethidium bromide.
Results
EGCG inhibits the activity of DNMTs and
reduces the mRNA transcription level of DNMT3B in HeLa cells
EGCG treatment (2.5 μM) at various time points (24,
48 and 72 h) was found to exert significant inhibitory action on
the activity of DNMTs in HeLa cells treated in time-dependent
manner and inhibited the enzyme activity by 5% (24 h), 12% (48 h)
and 20% (72 h) in HeLa cells compared with the untreated control
(Fig. 1A). In contrast,
time-dependent (24, 48 and 72 h) exposure of HeLa cells to 1 μM
5-Aza-dC resulted in 8, 19 and 29% inhibition of DNMT activity in
comparison to the untreated control. Furthermore, to decipher the
probable reason for inhibition of enzyme activity, the findings
were correlated with the changes in the expression of DNMT3B at the
mRNA transcription level induced by EGCG in the HeLa cells.
Untreated HeLa cells showed increased levels of DNMT3B mRNA whereas
the cells treated with 25 μM EGCG showed a significant decrease in
the expression of DNMT3B in a time-dependent manner (24, 48 and 72
h) (Fig. 1B). In addition, an
almost similar effect was observed in the 5-Aza-dC-treated HeLa
cells and showed a time-dependent decrease in the expression of
DNMT3B. β-actin was used as an internal control for sample
comparison (Fig. 1B).
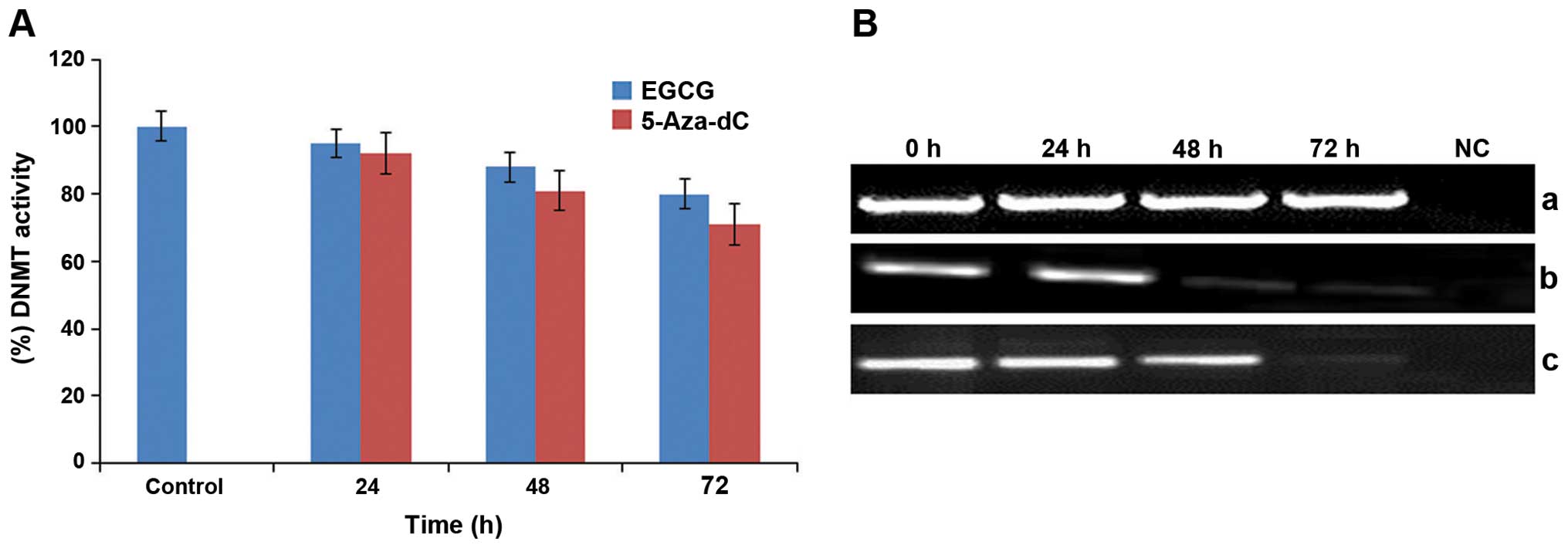 | Figure 1Effect of EGCG and 5-Aza-dC on DNA
methyltransferases (DNMTs) in human cervical cancer HeLa cells. (A)
HeLa cells treated with EGCG (25 μM) and 5-Aza-Dc (1.0 μM)
exhibited significant inhibition in the activity of DNMTs in a
time-dependent manner. Values are means ± SD of 3 independent
experiments. Each value for EGCG treatment differs from the control
value (P<0.05). (B) HeLa cells treated with EGCG (25 μM) and
5-Aza-Dc (1.0 μM) exhibited a significant time-dependent reduction
in the mRNA expression of DNMT3B in comparison to the untreated
cells. Panel a, β-actin expression as an internal control; panel b,
expression of DNMT3B following treatment with EGCG; and panel c,
expression of DNMT3B following treatment with 5-Aza-dC. Lane 1,
expression of DNMT3B gene in untreated HeLa cells; lanes 2–4,
time-dependent alterations in the expression of DNMT3B upon
treatment with EGCG and 5-Aza-Dc for 24, 48 and 72 h, respectively;
lane 5, negative control (NC) for RT-PCR. EGCG,
(−)-epigallocatechin-3-gallate; 5-Aza-dC,
5-aza-2′-deoxycytidine. |
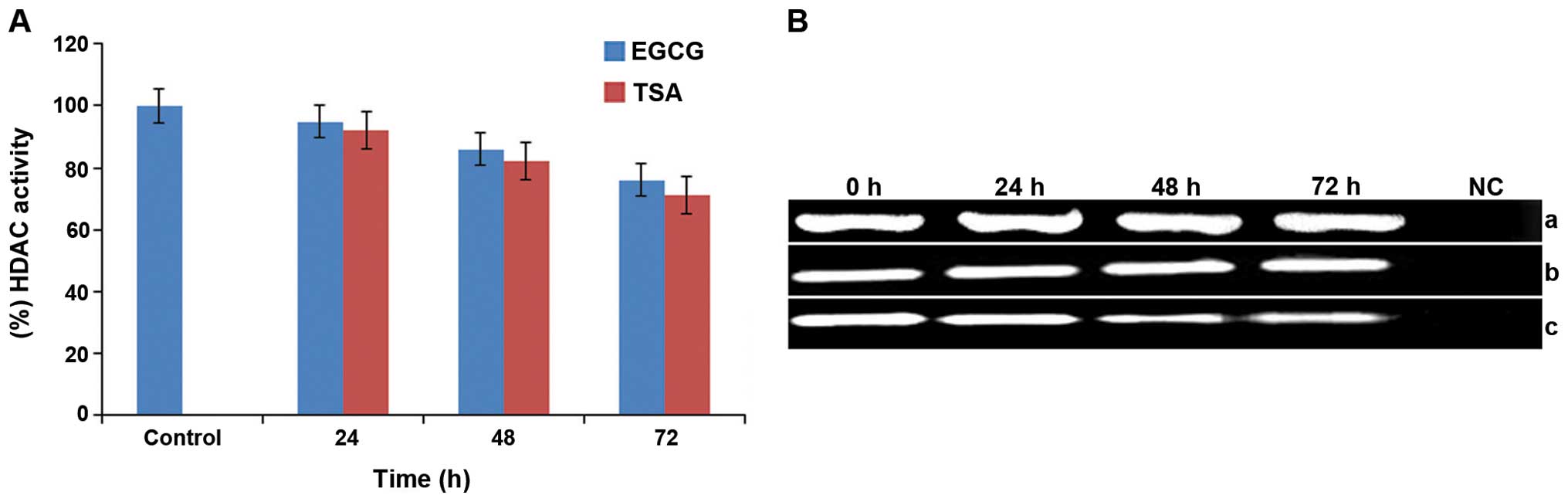
Effect of EGCG on HDAC activity and the
expression of HDAC1 in HeLa cells
Chromatin modification is primarily regulated by
HDAC enzymes and their high activity is linked with epigenetically
silenced genes. The activity of HDACs in HeLa cells was determined
by treating the cells with EGCG (25 μM) for 24, 48 and 72 h,
respectively. It was observed that HeLa cells treated with 25 μM
EGCG showed a time-dependent decrease of 5, 14 and 24% in HDAC
activity (Fig. 2A). It was also
observed that exposure to the HDAC inhibitor (0.025 μM TSA) caused
a time-dependent (24, 48 and 72 h) decrease in the activity of
HDACs in the HeLa cells and caused 8, 18 and 29% inhibition,
respectively (Fig. 2A).
Furthermore, it was also examined whether HDAC activity may or may
not be correlated with a decrease in HDAC1 expression. It was found
that EGCG (25 μM)-treated HeLa cells did not show any significant
changes in the expression of HDAC1 in comparison to the untreated
cells (Fig. 2B). However,
TSA-treated HeLa cells showed a significant reduction in the
expression of HDAC1 at 24, 48 and 72 h, respectively (Fig. 2B).
EGCG interacts with DNMT3B and HDAC1: An
in silico molecular model
In order to investigate the possible mechanism by
which EGCG inhibits HDAC1 and DNMT3B, an in silico
theoretical molecular modeling approach was used. The cavity
predicted by CASTp which included the HDAC1 residues expected to
interact with ligand TSA as well as the active site His-141 and Zn
ion was taken to be the substrate binding site of HDAC1. The CASTp
predicted cavity that included the active site residue Cys-651 and
co-factor binding residue Glu-605 was defined as the
substrate-binding cavity for DNMT3B. Previously, we evaluated the
structure and the predicted cavity of DNMT3B by docking its
well-known inhibitor 5-Aza-dC, using the SwissDock server. The same
methodology was used to dock EGCG on HDAC1 and mDNMT3B. FullFitness
and Gibbs free energy (ΔG) of each run (256 runs) of the docking
experiment were evaluated. Favorable binding modes were scored
based on FullFitness and cluster formation. The value of
FullFitness was used to rank clusters for further analysis.
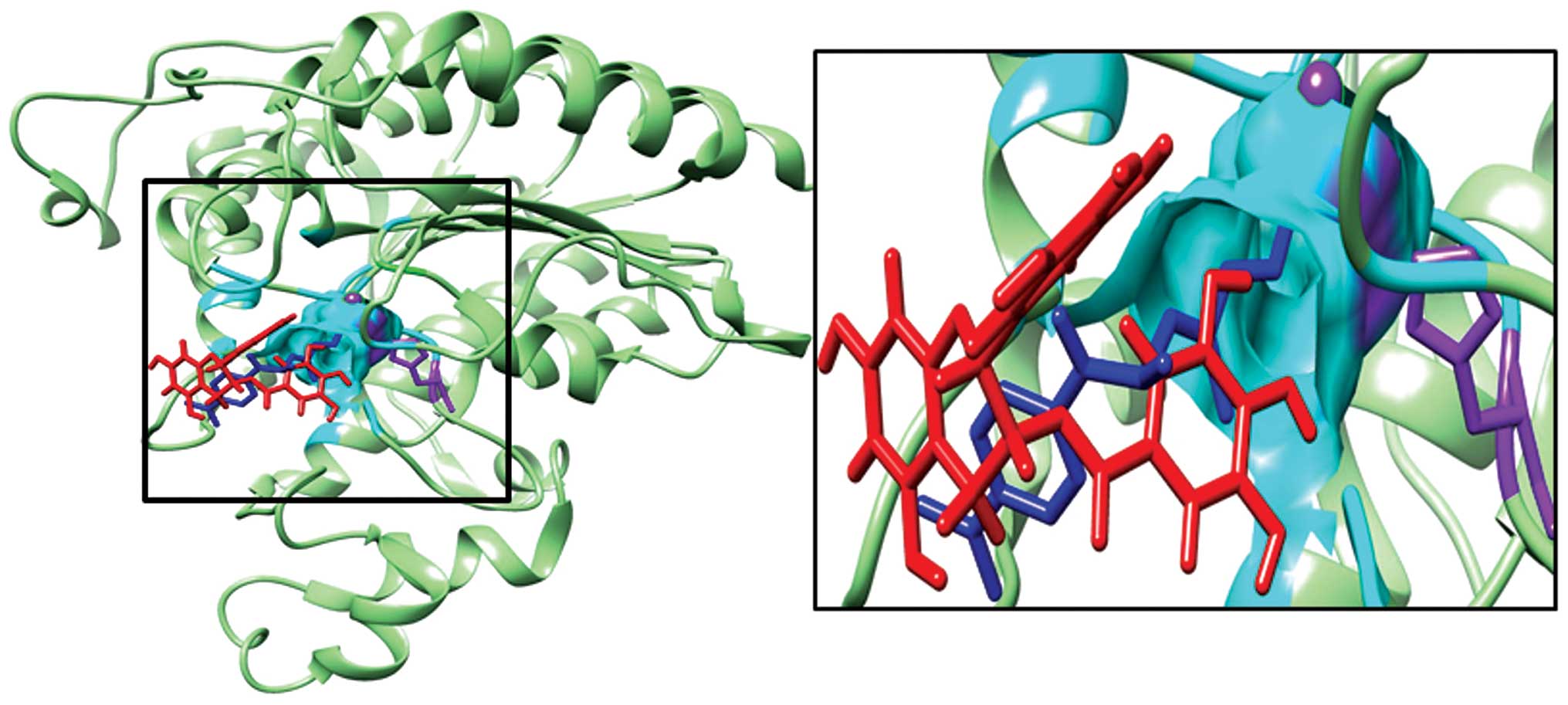
Interaction of EGCG with mDNMT3B
The docking of 5-Aza-dC, a well-known inhibitor of
DNMT3B, was performed first to define the substrate binding site of
the protein. The docking results produced 43 clusters of ligand
around the modeled catalytic domain of DNMT3B. Fifteen clusters
were found to bind in the CASTp predicted cavity of which the
ligand models in the top 2 clusters (0 and 1) were found to be in
close proximity to the active site Cys-651 and cofactor (S-adenosyl
methionine) binding residue Glu-605. The distance of the closest
atoms of Cys-651 and Glu-605 from the most favorable docking model
of 5-Aza-dC was found to be 1.86 and 2.34 Å, respectively. The
docking result of 5-Aza-dC on mDNMT3B established the validity of
the substrate binding cavity predicted by CASTp. Following this,
the binding of the ligand EGCG on mDNMT3B was probed by performing
their docking using the SwissDock server. The docking results
produced 34 clusters of ligand EGCG around the modeled catalytic
domain of DNMT3B. When the clusters were analyzed it was found that
15 of them could bind in the substrate binding cavity constituting
a total of 120 predicted binding elements out of a total of 256. It
is striking that almost half of the total predicted elements are
docked in the substrate binding cavity. The predicted clusters
include top ranked cluster 1 with 16 elements as well as several
other clusters. While cluster rank 0 is energetically lower it was
not considered as the predicted binding of EGCG was on the outer
edge of the catalytic domain, a region which may not represent a
pocket or be accessible to the ligand in the complete protein.
Notably, it was observed that the gallate moiety of EGCG was
consistently positioned in close proximity to the important
residues in the substrate binding cavity. This may be interpreted
as an indication of its important role in the binding of EGCG to
the protein. The amino acids, Arg 832, Arg 823, Ser 778 and Asn
652, were repeatedly observed to be involved in hydrogen bond
formation with EGCG. Table II
shows the summary result of SwissDock docking and the FullFittness
and estimated ΔG values for the most favorable interaction.
Observation of the majority of clusters, including the top ranked
ones, in the cavity strongly suggests that the preferred binding of
EGCG on mDNMT3B is within the substrate binding cavity. Fig. 3 shows a representative image of the
binding of EGCG and 5-Aza-dC on the protein mDNMT3B. Table III lists all of the mDNMT3B
residues within 5 Å of the most energetically favorable docking
model of EGCG.
| Table IIDocking results of ligands (EGCG, TSA
and 5-Aza-dC) on receptors (HDAC1 and mDNMT3B). |
Table II
Docking results of ligands (EGCG, TSA
and 5-Aza-dC) on receptors (HDAC1 and mDNMT3B).
| Receptor | Ligand | Clusters | Cluster ranks | Total elements | FullFitness
(kcal/mol) | Estimated ΔG
(kcal/mol) |
|---|
| mDNMT3B | 5-Aza-dC | 15/43 | 0–1, 3–5, 7, 17,
19, 24, 34, 36–37, 39, 42, 43 | 73/256 | −2071.4 | −9.5 |
| mDNMT3B | EGCG | 21/44 | 8, 10, 11, 14–16,
18, 19, 24–26, 29–30, 35–37, 39, 40, 41, 43, 44 | 120/256 | −1866.53 | −6.94 |
| HDAC1 | EGCG | 3/31 | 0, 2, 9 | 23/256 | −2042.02 | −7.48 |
| Table IIIResidues of HDAC1 and mDNMT3B within
5 Å of EGCG. |
Table III
Residues of HDAC1 and mDNMT3B within
5 Å of EGCG.
| Protein | Residues within 5 Å
of EGCG |
|---|
| mDNMT3B | C-651, E-605, V-606, C-607
F-581, D-582, G-583, S-610, D-627, V-628, R-629, G-648, S-649,
P-650, N-652, T-668, L-671, K-828, R-832, S-833 |
| HDAC1 | H-141, G-27, H-28, P-29, D-99,
G-149, S-148, F-150, H-178, E-203, Y-204, F-205, R-270, L271,
Y-303 |
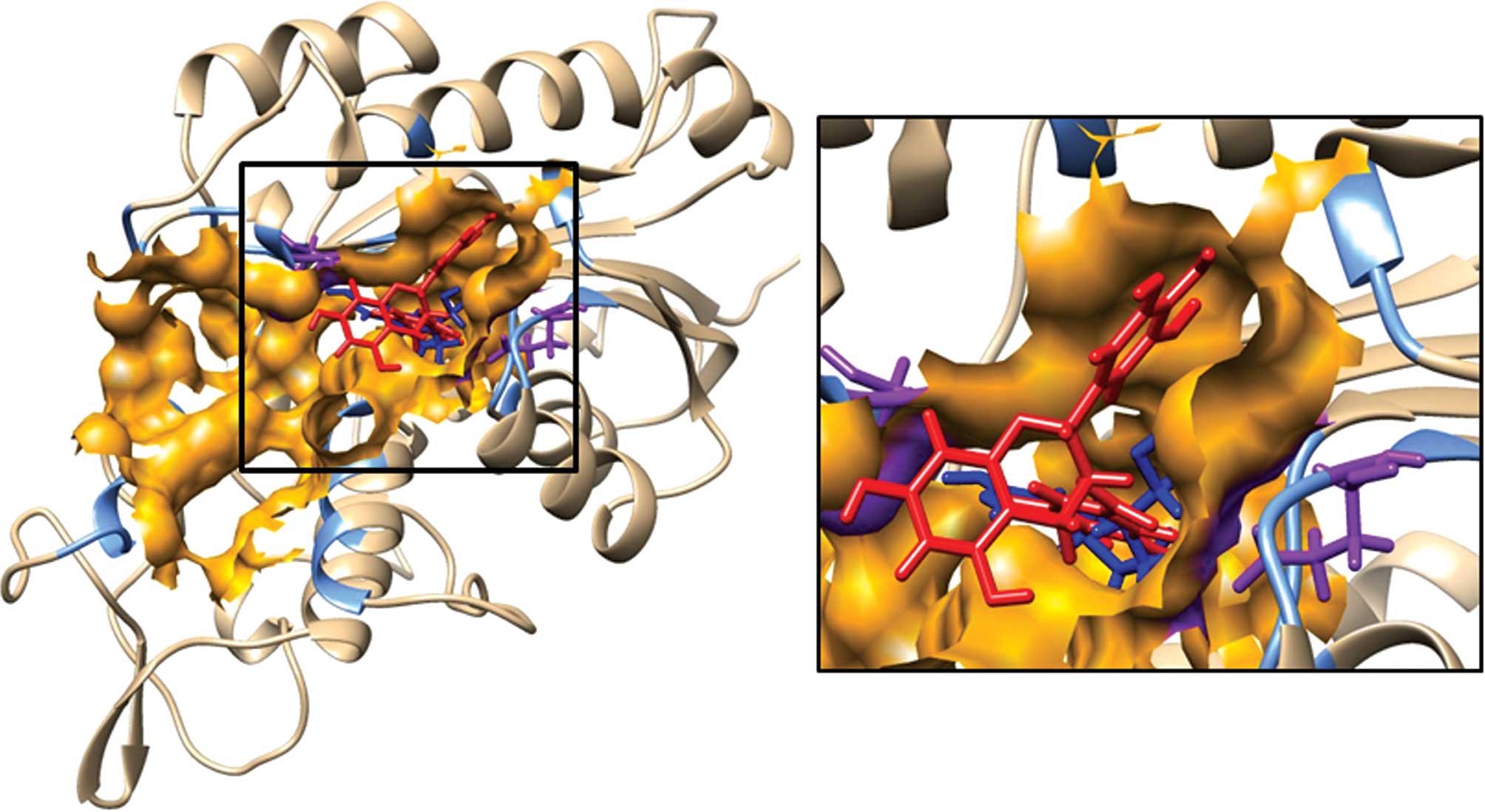
Interaction of EGCG with HDAC1
The docking results produced 31 clusters of ligand
EGCG around the complete protein HDAC1. Analysis of these clusters
showed that 3 of these clusters were able to bind in the substrate
binding cavity. These clusters together contained a total of 23
elements out of 256 predicted binding modes. Notably, these
clusters included the top ranked clusters 0 and 2 in addition to
cluster 9. Table II shows the
SwissDock docking results as well as the FullFittness and estimated
ΔG values for the most favorable interaction. The lowest energy
model of cluster rank zero was considered to be the most favorable
interaction. The presence of two high ranking clusters strongly
suggests that the preferred binding of EGCG on HDAC1 is within the
substrate binding cavity. The gallate moiety of EGCG seems to play
an important role as it is oriented towards the active residue in
the least energy model. Fig. 4
shows the most energetically favorable binding of EGCG on HDAC1 as
well as TSA on HDAC1 which was achieved by superimposing the
crystal structure of HDAC8 bound to TSA on HDAC1. The image visibly
records that the predicted binding orientation of EGCG and
transposed TSA overlap the same region on HDAC1. It is noteworthy
that the key catalytic residues such as the active site His-141 and
Zn ion are within 5 Å of the ligands. Table III lists all the HDAC1 residues
within 5 Å of the most energetically favorable docking model of
EGCG.
EGCG reactivates RARβ, CDH1 and DAPK1
genes through reversal of hypermethylation in HeLa cells
Many TSGs are inactivated via aberrant epigenetic
changes at their promoter region during cancer development, and due
to the reversible nature of these changes epigenetic-based cancer
treatment strategy is attracting more research interest. To
determine whether EGCG-induced inhibition of DNMT3B and HDAC1
results in epigenetic changes leading to reactivation of various
TSGs such as RARβ, CDH1 and DAPK1, expression of these genes was
further correlated with the changes in the methylation of their
promoter regions. It was observed that HeLa cells treated with 25
μM EGCG exhibited a time-dependent (24, 48 and 72 h) significant
increase in the expression of RARβ, CDH1 and DAPK1 at the mRNA
level in comparison to the untreated cells (Fig. 5). Furthermore, it was observed that
the changes in the expression of these genes were associated with
the reversal of 5′ CpG island methylation of their promoter
regions. Treated HeLa cells were subjected to MSP with
methylation-specific and unmethylation-specific sets of primers.
EGCG treated HeLa cells showing amplification only after using
methylated and unmethylated primer set, were considered as
hypermethylated and unmethylated respectively. From the MSP
results, it was observed that RARβ, CDH1 and DAPK1 genes were found
to be hypermethylated in the untreated HeLa cells. However, after
treatment with EGCG, the 5′ CpG island methylation of the promoters
of the these genes was reversed as evident by the decreased level
of amplimer intensity with methylation-specific primers whereas it
was significantly increased with the unmethylated set of primers in
a time-dependent manner (Fig. 5B).
These results confirm that EGCG restores the expression of RARβ,
CDH1 and DAPK1 genes via the reversal of 5′ CpG island methylation
and inhibition of epigenetic modulators such as DNMTs and HDACs in
HeLa cells.
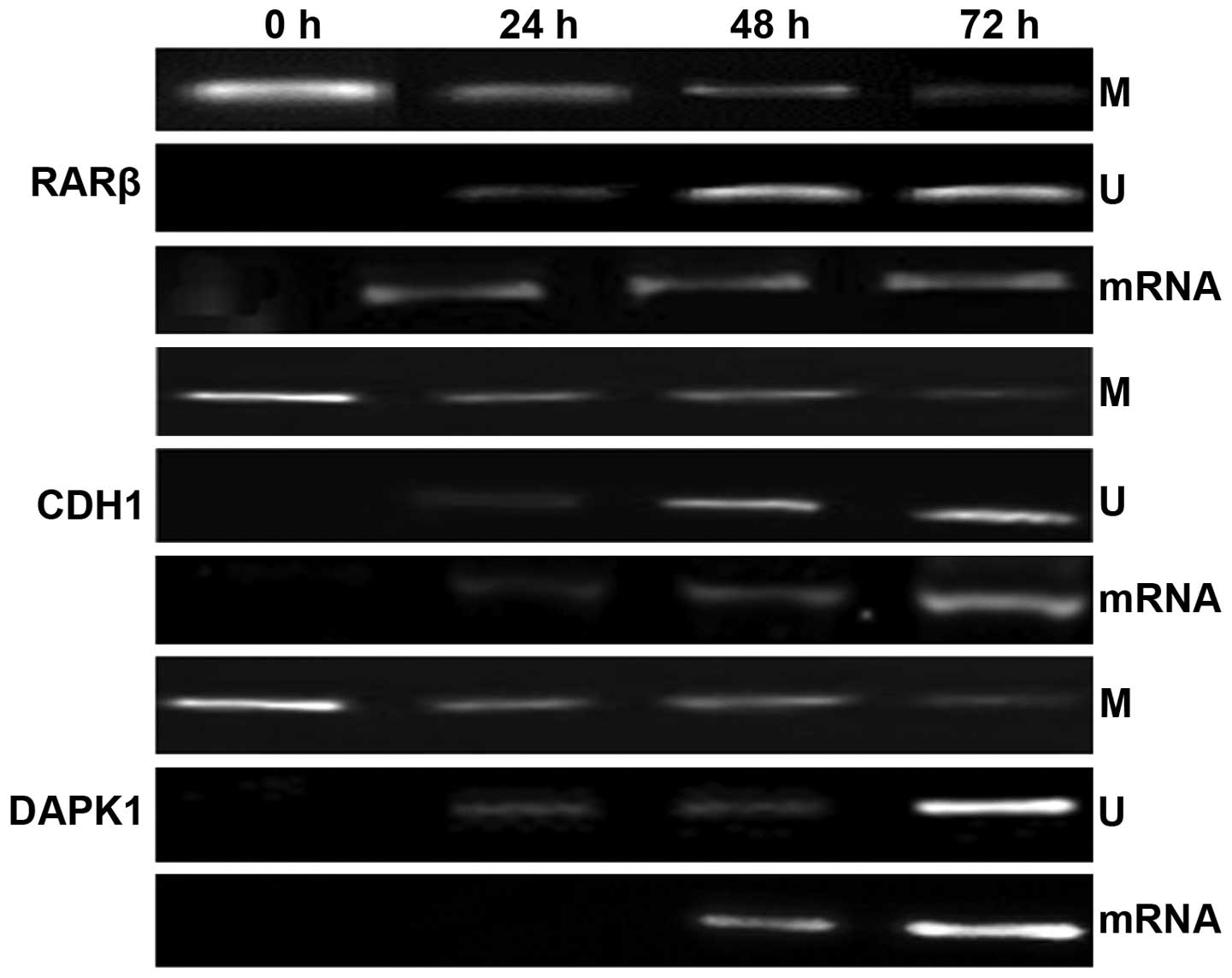 | Figure 5Alterations in the methylation status
and mRNA expression levels of RARβ, CDH1 and DAPK1 genes after
treatment with EGCG. Panels M and U show the methylation-specific
bands (M) and unmethylation-specific bands (U): lane 1, methylation
status of these genes in untreated HeLa cells; lane 2–4,
time-dependent modulation in the methylation status of RARβ, CDH1
and DAPK1 genes for 24, 48 and 72 h, respectively. Panel mRNA shows
the expression of these genes after treatment with EGCG: lane 2–4,
time-dependent modulation in the expression of HDAC1 upon treatment
for 24, 48 and 72 h, respectively, whereas lane 1 shows the
expression of these genes in untreated HeLa cells. |
Discussion
In recent years, researchers in the field of
epigenetics have made great strides in understanding the many
molecular sequences and patterns that determine which genes can be
turned on and off. The present study has made it increasingly clear
that in addition to genetic changes, epigenetic alterations are
also very critical for cancer development (1,2).
Unlike genetic alterations, certain epigenetic alterations i.e.
aberrant DNA methylation and histone acetylation which lead to gene
inactivation of various tumor-suppressor genes (TSGs) in cancer
cells, can be reversed using epigenetic drugs, making them valuable
therapeutic targets for cancer therapy. The drugs that target these
epigenetic alterations are termed epigenetic drugs. Currently, two
classes of synthetic epigenetic drugs based on nucleoside and
non-nucleoside nature are being investigated as DNA
methyltransferase (DNMT) and histone deacetylase (HDAC) inhibitors
(33,34). However, there are many drawbacks of
these drugs such as lack of specificity, short duration of action
and cytotoxicity towards normal cells that currently restrict the
general use of these drugs (8–10).
More importantly, a wide gamut of reports indicates
that dietary phytochemicals can modulate epigenetic alterations and
alter susceptibility to various diseases including cancer. In the
development of epigenetic drugs as inhibitors of DNMTs and HDACs,
dietary phytochemicals are gaining more interest due to their safe
therapeutic profile (11,12,35).
One such promising agent is (−)-epigallocatechin-3-gallate (EGCG).
Its anticarcinogenic effects via various molecular pathways have
been established in previous studies (13–18).
Previous studies in our laboratory also found that EGCG was
selectively cytotoxic towards cancer cells and inhibited the growth
of cancer cells in a dose- and time-dependent manner and the
EC50 was found to be 100 μM for a duration of 24 h and
50 μM for 48 h (13). From these
results, we selected a sublethal dose (25 μM EGCG) for the present
study.
DNA methylation and histone acetylation and
de-acetylation are most commonly occurring epigenetic events taking
place in the mammalian genome and control the expression of various
genes including TSGs (36–38). A critical step in DNA methylation
involves DNMTs, the enzymes that catalyze the methylation process.
It has been established that inactivation of many TSGs via DNA
methylation and histone modification is due to the high enzymatic
activity of DNMTs and HDACs. Various studies have indicated the
importance of DNMTs and HDACs as epigenetic targets leading to the
development of many synthetic drugs that function as their
inhibitors (7,39–41).
During our screening study to determine whether EGCG affects
epigenetic modulation via DNMT and HDAC enzymes, it was
demonstrated that EGCG-treated HeLa cells showed significant
inhibition in the enzymatic activity of these enzymes in a
time-dependent manner (Figs. 1A and
2A). Almost similar effects were
observed in HeLa cells after treatment with their respective well
characterized inhibitors (Figs. 1A
and 2A). These results are
consistent with other studies in which many dietary agents along
with EGCG have shown their inhibitory action on the enzymatic
activity of DNMT and HDAC enzymes.
Several scientific studies have demonstrated that
DNMT3B, one of the important enzymes of the DNMT family and HDAC1,
one of the members of the HDAC enzyme family, are overexpressed in
various types of cancers (42–48).
To determine whether EGCG has any effect on the mRNA transcription
level of these enzymes, the mRNA expression level was studied in
EGCG-treated HeLa cells. It was found that treated Hela cells
showed a time-dependent decrease in the expression of DNMT3B
whereas no significant changes in the expression of HDAC1 in the
EGCG-treated HeLa cells were observed (Figs. 1B and 2B). However, time-dependent exposure of
5-Aza-dC and TSA decreased the expression of DNMT3B and HDAC1,
respectively, in the HeLa cells. The present study is in line with
various studies in which various dietary agents act as epigenetic
modifiers and modulate the epigenetic changes via targeting DNMTs
and HDACs (49–52).
Furthermore, in silico molecular modeling
studies were performed to ascertain whether the direct interaction
of EGCG with modeled DNMT3B and HDAC1 explains the EGCG-mediated
enzymatic inhibition of DNMTs and HDACs. Our analyses of the
predicted docking results emphatically indicate that EGCG directly
binds in the substrate binding pocket of these two enzymes.
The substrate binding pocket of DNMT3B was
delineated using computational and knowledge-based approaches.
Docking of 5-Aza-dC in concurrence with knowledge concerning the
active sites and cofactor binding sites helped us to identify with
certainty the substrate binding pocket for DNMT3B. Table III lists the residues lining the
pocket including active site C-651 and E-605 essential for binding
to SAM, which is responsible for methyltransferase activity.
Fig. 4 illustrates the docking of
EGCG and 5-Aza-dC on mDNMT3B. Notably, a very high proportion (15
out of 34, containing 120 out of 256 independent docking runs) of
predicted top ranked clusters for EGCG was observed in the defined
substrate binding pocket of mDNMT3B. These results show that EGCG
and 5-Aza-dC dock within the substrate binding cavity of the
protein and therefore may have a similar mechanism of protein
inhibition by preventing the entry of the natural ligand into the
active site. Remarkably, we observed that the gallate moiety
frequently binds in close proximity to the active residue or the
other key residues such as Arg 832, Arg 823, Ser 778 and Asn 652.
Docking studies on DNMT1 have proposed that the D ring/gallate
moiety of EGCG occupies a binding pocket analogous to the pyrimidyl
ring of cytosine and could be significant for its direct inhibition
of the enzyme (53,54). The recurrent involvement of the
amino acids Arg 832, Arg 823, Ser 778 and Asn 652, in hydrogen bond
formation with EGCG underscores their importance in the catalytic
pocket. Further emphasis supporting this hypothesis is lent by a
report on the inhibition of DNMT3B by nanomycin following key
interactions with Arg 731, Arg 733, Arg 832 and Asn 652 (55,56).
These residues have been reported to be involved in the DNA
methylation mechanism (56). Our
docking study also reinforces the role of this residue. Hence, we
can expect that the residue Arg 832 is important for inhibitor
binding and stability and when involved in binding to the inhibitor
will result in enzyme inactivity. In Table III we list those residues of
DNMT3B which may be involved in interactions with EGCG.
In Fig. 3, the best
energetically favored model of EGCG docking on HDAC1 overlaps with
the binding site of TSA. As a result of the binding of EGCG to
HDAC1, we can expect that it would impede the entry and catalysis
of the actual substrate. This leads us to propose that EGCG may be
functionally similar to TSA, as an HDAC inhibitor. The residues of
HDAC1 that interact with the most energetically favorable docking
model of EGCG are listed in Table
III.
These indications are strong enough for us to
recommend that given the better safety profile of EGCG in
comparison to 5-Aza-dC and TSA, EGCG is a better candidate as a
similarly functioning epigenetic modulator. Our molecular modeling
and docking studies not only successfully explain the mechanism of
action of EGCG in inhibiting epigenetic modulating enzymes but also
paves the way to embark upon further possibilities such as
structure-guided optimization studies and pharmacophore
modeling.
Many reports have shown that inactivation of
tumor-suppressor genes is associated with promoter hypermethylation
in various types of cancers (57–59).
It is now well established that methylation of DNA and
modifications of histones are intimately interconnected. For
example, DNMTs can bind to HDACs, thereby repressing gene
transcription through histone deacetylase activity. There is a
family of proteins collectively referred to as methyl CpG binding
proteins (MBDs) that share a methyl-CpG binding domain. These
proteins are thought to recruit HDACs to methyl-CpG-enriched
regions of the genome to repress transcription (4–7). In
the present study, the possible epigenetic modulatory effects of
EGCG on various epigenetically silenced TSGs including RARβ, CDH1
and DAPK1 in HeLa cells were studied and were also correlated with
its ability to inhibit DNMTB and HDAC1 activity. Possibly,
EGCG-mediated inhibition of DNMTs and HDACs favors to change the
DNA methylation and histone de-acetylation status which results in
the reactivation of these genes. Extensive studies have found that
the RARβ, CDH1 and DAPK1 genes are transcriptionally silenced due
to promoter hypermethylation during the development of various
human cancers (60–64). In the present study, it was observed
that the RARβ, CDH1 and DAPK1 genes were hypermethylated and this
was correlated with their respective expression which was found to
be undetectable in the untreated HeLa cells (Fig. 5). However, after treatment with
EGCG, HeLa cells showed time-dependent changes in the methylation
status as the bands in the methylated panel were reduced and
significantly detectable in the unmethylated panel which was
further linked with restoration of the expression of RARβ, CDH1 and
DAPK1 genes during the time exposure (Fig. 5). The present study is similar to
several other studies which have shown that various dietary agents
including EGCG reactivate many TSGs via modulation of various
epigenetic pathways (49,51,52,59,65,66).
The present study demonstrated that EGCG functions
as a potential epigenetic modifier and acts via inhibitory action
on the activity of DNMTs and HDACs and reactivates epigenetically
silenced TSGs by altering the methylation status of the promoters
of these genes. EGCG can be used as an effective inhibitor of DNMTs
and HDACs to prevent cancer. However, further mechanistic studies
as well as in vivo studies and clinical trials are needed to
determine the efficacy of EGCG for therapeutic purposes as an
epigenetic drug.
Acknowledgements
The authors are grateful to Dr Kota Reddy, Academic
President and Dr Firdos Alam Khan, Chairperson, School of Life
Sciences, Manipal University, Dubai, for their constant support and
encouragement. The present study was financially supported by Zayed
University Research Incentive Fund (RIF) (activity code: R13052)
and the Intramural Research Program at Manipal University, Dubai,
United Arab Emirates (UAE).
References
|
1
|
Ting AH, McGarvey KM and Baylin SB: The
cancer epigenome - components and functional correlates. Genes Dev.
20:3215–3231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Choi JD and Lee JS: Interplay between
epigenetics and genetics in cancer. Genomics Inf. 11:164–173. 2013.
View Article : Google Scholar
|
|
3
|
Flis S, Gnyszka A and Flis K: DNA
methyltransferase inhibitors improve the effect of chemotherapeutic
agents in SW48 and HT-29 colorectal cancer cells. PLoS One.
27:e923052014. View Article : Google Scholar
|
|
4
|
Geurts van Kessel A: The cancer genome:
from structure to function. Cell Oncol. 37:155–165. 2014.
View Article : Google Scholar
|
|
5
|
Tallen G and Riabowol K: Keep-ING balance:
Tumor suppression by epigenetic regulation. FEBS Lett.
588:2728–2742. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wee S, Dhanak D, Li H, Armstrong SA,
Copeland RA, Sims R, Baylin SB, Liu XS and Schweizer L: Targeting
epigenetic regulators for cancer therapy. Ann NY Acad Sci.
1309:30–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jeong HM, Kwon MJ and Shin YK:
Overexpression of cancer-associated genes via epigenetic
derepression mechanisms in gynecologic cancer. Front Oncol.
4:122014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Subramaniam D, Thombre R, Dhar A and Anant
S: DNA methyltransferases: A novel target for prevention and
therapy. Front Oncol. 4:802014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Campbell RM and Tummino PJ: Cancer
epigenetics drug discovery and development: The challenge of
hitting the mark. J Clin Invest. 124:64–69. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cantley MD and Haynes DR: Epigenetic
regulation of inflammation: Progressing from broad acting histone
deacetylase (HDAC) inhibitors to targeting specific HDACs.
Inflammopharmacology. 21:301–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Henning SM, Wang P, Carpenter CL and Heber
D: Epigenetic effects of green tea polyphenols in cancer.
Epigenomics. 5:729–741. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang P, He X and Malhotra A: Epigenetic
targets of polyphenols in cancer. J Environ Pathol Toxicol Oncol.
33:159–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sharma C, Nusri Q-A, Begum S, Javed E,
Rizvi TA and Hussain A: (−)-Epigallocatechin-3-gallate induces
apoptosis and inhibits invasion and migration of human cervical
cancer cells. Asian Pac J Cancer Prev. 13:4815–4822. 2012.
View Article : Google Scholar
|
|
14
|
Lin CH, Chao LK, Hung PH and Chen YJ: EGCG
inhibits the growth and tumorigenicity of nasopharyngeal
tumor-initiating cells through attenuation of STAT3 activation. Int
J Clin Exp Pathol. 7:2372–2381. 2014.PubMed/NCBI
|
|
15
|
Siddiqui IA, Bharali DJ, Nihal M, Adhami
VM, Khan N, Chamcheu JC, Khan MI, Shabana S, Mousa SA and Mukhtar
H: Excellent anti-proliferative and pro-apoptotic effects of
(−)-epigallocatechin-3-gallate encapsulated in chitosan
nanoparticles on human melanoma cell growth both in vitro and in
vivo. Nanomedicine. 10:1619–1626. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luo HQ, Xu M, Zhong WT, Cui ZY, Liu FM,
Zhou KY and Li XY: EGCG decreases the expression of HIF-1α and VEGF
and cell growth in MCF-7 breast cancer cells. J BUON. 19:435–439.
2014.PubMed/NCBI
|
|
17
|
Butt MS, Ahmad RS, Sultan MT, Nasir Qayyum
MM and Naz A: Green tea and anticancer perspectives: Updates from
last decade. Crit Rev Food Sci Nutr. 55:792–805. 2015. View Article : Google Scholar
|
|
18
|
Yokoyama M, Noguchi M, Nakao Y, Ysunaga M,
Yamasaki F and Iwasaka T: Antiproliferative effects of the major
tea polyphenol, (−)-epigallocatechin gallate and retinoic acid in
cervical adenocarcinoma. Gynecol Oncol. 108:326–331. 2008.
View Article : Google Scholar
|
|
19
|
Millard CJ, Watson PJ, Celardo I,
Gordiyenko Y, Cowley SM, Robinson CV, Fairall L and Schwabe JW:
Class I HDACs share a common mechanism of regulation by inositol
phosphates. Mol Cell. 51:57–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jia D, Jurkowska RZ, Zhang X, Jeltsch A
and Cheng X: Structure of Dnmt3a bound to Dnmt3L suggests a model
for de novo DNA methylation. Nature. 449:248–251. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thangapandian S, John S, Lee Y,
Arulalapperumal V and Lee KW: Molecular modeling study on tunnel
behavior in different histone deacetylase isoforms. PLoS One.
7:e493272012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Irwin JJ, Sterling T, Mysinger MM, Bolstad
ES and Coleman RG: ZINC: A free tool to discover chemistry for
biology. J Chem Inf Model. 52:1757–1768. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grosdidier A, Zoete V and Michielin O:
SwissDock, a protein-small molecule docking web service based on
EADock DSS. Nucleic Acids Res. 39:W270–W277. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pettersen EF, Goddard TD, Huang CC, Couch
GS, Greenblatt DM, Meng EC and Ferrin TE: UCSF Chimera - a
visualization system for exploratory research and analysis. J
Comput Chem. 25:1605–1612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
El-Shakankiry NH and Mossallam GI: p15
(INK4B) and E-cadherin CpG island methylation is frequent in
Egyptian acute myeloid leukemia. J Egypt Natl Canc Inst.
18:227–232. 2006.
|
|
26
|
Suzuki M, Shigematsu H, Iizasa T,
Hiroshima K, Nakatani Y, Minna JD, Gazdar AF and Fujisawa T:
Exclusive mutation in epidermal growth factor receptor gene, HER-2,
and KRAS, and synchronous methylation of nonsmall cell lung cancer.
Cancer. 106:2200–2207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yaqinuddin A, Qureshi SA, Qazi R and Abbas
F: Down-regulation of DNMT3b in PC3 cells effects locus-specific
DNA methylation, and represses cellular growth and migration.
Cancer Cell Int. 8:132008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Saito Y, Kanai Y, Sakamoto M, Saito H,
Ishii H and Hirohashi S: Overexpression of a splice variant of DNA
methyltransferase 3b, DNMT3b4, associated with DNA hypomethylation
on pericentromeric satellite regions during human
hepatocarcinogenesis. Proc Natl Acad Sci USA. 99:10060–10065. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Goelden U, Ukena SN, Pfoertner S, Hofmann
R, Buer J and Schrader AJ: RAR-beta(1) overexpression in
chromophobe renal cell carcinoma: A novel target for therapeutic
intervention? Exp Oncol. 27:220–224. 2005.PubMed/NCBI
|
|
30
|
Wang AG, Kim SU, Lee SH, Kim SK, Seo SB,
Yu DY and Lee DS: Histone deacetylase 1 contributes to cell cycle
and apoptosis. Biol Pharm Bull. 28:1966–1970. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jin Y, Blue EK and Gallagher PJ: Control
of death-associated protein kinase (DAPK) activity by
phosphorylation and proteasomal degradation. J Biol Chem.
281:39033–39040. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chang J, Nicolau MM, Cox TR, Wetterskog D,
Martens JW, Barker HE and Erler JT: LOXL2 induces aberrant acinar
morphogenesis via ErbB2 signaling. Breast Cancer Res. 15:R672013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Capobianco E, Mora A, La Sala D, Roberti
A, Zaki N, Badidi E, Taranta M and Cinti C: Separate and combined
effects of DNMT and HDAC inhibitors in treating human multi-drug
resistant osteosarcoma HosDXR150 cell line. PLoS One. 9:e955962014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Al-Rayyan N, Litchfield LM, Ivanova MM,
Radde BN, Cheng A, Elbedewy A and Klinge CM: 5-Aza-2-deoxycytidine
and trichostatin A increase COUP-TFII expression in
antiestrogen-resistant breast cancer cell lines. Cancer Lett.
347:139–150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thakur VS, Deb G, Babcook MA and Gupta S:
Plant phytochemicals as epigenetic modulators: Role in cancer
chemoprevention. AAPS J. 16:151–163. 2014. View Article : Google Scholar :
|
|
36
|
Jiao F, Wang X, Yan Z, Liu C, Yue Z, Li Z,
Ma Y, Li Y and Wang J: Effect of dynamic DNA methylation and
histone acetylation on cPouV expression in differentiation of chick
embryonic germ cells. Stem Cells Dev. 22:2725–2735. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Herranz M and Esteller M: DNA methylation
and histone modifications in patients with cancer: Potential
prognostic and therapeutic targets. Methods Mol Biol. 361:25–62.
2007.
|
|
38
|
Oyer JA, Chu A, Brar S and Turker MS:
Aberrant epigenetic silencing is triggered by a transient reduction
in gene expression. PLoS One. 4:e48322009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zopf S, Ocker M, Neureiter D, Alinger B,
Gahr S, Neurath MF and Di Fazio P: Inhibition of DNA
methyltransferase activity and expression by treatment with the
pan-deacetylase inhibitor panobinostat in hepatocellular carcinoma
cell lines. BMC Cancer. 12:3862012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu WG and Otterson GA: The interaction of
histone deacetylase inhibitors and DNA methyltransferase inhibitors
in the treatment of human cancer cells. Curr Med Chem Anticancer
Agents. 3:187–199. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vendetti FP and Rudin CM: Epigenetic
therapy in non-small-cell lung cancer: Targeting DNA
methyltransferases and histone deacetylases. Expert Opin Biol Ther.
13:1273–1285. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ben Gacem R, Hachana M, Ziadi S, Ben
Abdelkarim S, Hidar S and Trimeche M: Clinicopathologic
significance of DNA methyltransferase 1, 3a, and 3b overexpression
in Tunisian breast cancers. Hum Pathol. 43:1731–1738. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Girault I, Tozlu S, Lidereau R and Bièche
I: Expression analysis of DNA methyltransferases 1, 3A, and 3B in
sporadic breast carcinomas. Clin Cancer Res. 9:4415–4422.
2003.PubMed/NCBI
|
|
44
|
Mizuno S, Chijiwa T, Okamura T, Akashi K,
Fukumaki Y, Niho Y and Sasaki H: Expression of DNA
methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in
acute and chronic myelogenous leukemia. Blood. 97:1172–1179. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qu Y, Mu G, Wu Y, Dai X, Zhou F, Xu X,
Wang Y and Wei F: Overexpression of DNA methyltransferases 1, 3a,
and 3b significantly correlates with retinoblastoma tumorigenesis.
Am J Clin Pathol. 134:826–834. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Müller BM, Jana L, Kasajima A, Lehmann A,
Prinzler J, Budczies J, Winzer KJ, Dietel M, Weichert W and Denkert
C: Differential expression of histone deacetylases HDAC1, 2 and 3
in human breast cancer - overexpression of HDAC2 and HDAC3 is
associated with clinicopathological indicators of disease
progression. BMC Cancer. 13:2152013. View Article : Google Scholar :
|
|
47
|
Santoro F, Botrugno OA, Dal Zuffo R, et
al: A dual role for Hdac1: Oncosuppressor in tumorigenesis,
oncogene in tumor maintenance. Blood. 121:3459–3468. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang H, Salz T, Zajac-Kaye M, Liao D,
Huang S and Qiu Y: Overexpression of histone deacetylases in cancer
cells is controlled by interplay of transcription factors and
epigenetic modulators. FASEB J. 28:4265–4279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fazi F, Travaglini L, Carotti D, et al:
Retinoic acid targets DNA-methyltransferases and histone
deacetylases during APL blast differentiation in vitro and in vivo.
Oncogene. 24:1820–1830. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shu L, Khor TO, Lee JH, Boyanapalli SS,
Huang Y, Wu TY, Saw CL, Cheung KL and Kong AN: Epigenetic CpG
demethylation of the promoter and reactivation of the expression of
Neurog1 by curcumin in prostate LNCaP cells. AAPS J. 13:606–614.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Moseley VR, Morris J, Knackstedt RW and
Wargovich MJ: Green tea polyphenol epigallocatechin 3-gallate,
contributes to the degradation of DNMT3A and HDAC3 in HCT 116 human
colon cancer cells. Anticancer Res. 33:5325–5333. 2013.PubMed/NCBI
|
|
52
|
Fan H, Zhang R, Tesfaye D, Tholen E, Looft
C, Hölker M, Schellander K and Cinar MU: Sulforaphane causes a
major epigenetic repression of myostatin in porcine satellite
cells. Epigenetics. 7:1379–1390. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H,
Welsh W and Yang CS: Tea polyphenol (−)-epigallocatechin-3-gallate
inhibits DNA methyltransferase and reactivates methylation-silenced
genes in cancer cell lines. Cancer Res. 63:7563–7570.
2003.PubMed/NCBI
|
|
54
|
Lee WJ, Shim JY and Zhu BT: Mechanisms for
the inhibition of DNA methyltransferases by tea catechins and
bioflavonoids. Mol Pharmacol. 68:1018–1030. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kuck D, Caulfield T, Lyko F and
Medina-Franco JL: Nanaomycin A selectively inhibits DNMT3B and
reactivates silenced tumor suppressor genes in human cancer cells.
Mol Cancer Ther. 9:3015–3023. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Caulfield T and Medina-Franco JL:
Molecular dynamics simulations of human DNA methyltransferase 3B
with selective inhibitor nanaomycin A. J Struct Biol. 176:185–191.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Akhavan-Niaki H and Samadani AA: DNA
methylation and cancer development: Molecular mechanism. Cell
Biochem Biophys. 67:501–513. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gokul G and Khosla S: DNA methylation and
cancer. Subcell Biochem. 61:597–625. 2013. View Article : Google Scholar
|
|
59
|
Shu XS, Li L and Tao Q: Chromatin
regulators with tumor suppressor properties and their alterations
in human cancers. Epigenomics. 4:537–549. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Narayan G, Arias-Pulido H, Koul S, Vargas
H, Zhang FF, Villella J, Schneider A, Terry MB, Mansukhani M and
Murty VV: Frequent promoter methylation of CDH1, DAPK, RARB, and
HIC1 genes in carcinoma of cervix uteri: Its relationship to
clinical outcome. Mol Cancer. 2:242003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kang S, Kim JW, Kang GH, Lee S, Park NH,
Song YS, Park SY, Kang SB and Lee HP: Comparison of DNA
hypermethylation patterns in different types of uterine cancer:
Cervical squamous cell carcinoma, cervical adenocarcinoma and
endometrial adenocarcinoma. Int J Cancer. 118:2168–2171. 2006.
View Article : Google Scholar
|
|
62
|
Braggio E, Maiolino A, Gouveia ME,
Magalhães R, Souto Filho JT, Garnica M, Nucci M and Renault IZ:
Methylation status of nine tumor suppressor genes in multiple
myeloma. Int J Hematol. 91:87–96. 2010. View Article : Google Scholar
|
|
63
|
Liu S, Ren S, Howell P, Fodstad O and
Riker AI: Identification of novel epigenetically modified genes in
human melanoma via promoter methylation gene profiling. Pigment
Cell Melanoma Res. 21:545–558. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Xu XL, Yu J, Zhang HY, Sun MH, Gu J, Du X,
Shi DR, Wang P, Yang ZH and Zhu JD: Methylation profile of the
promoter CpG islands of 31 genes that may contribute to colorectal
carcinogenesis. World J Gastroenterol. 10:3441–3454.
2004.PubMed/NCBI
|
|
65
|
Ho AS, Turcan S and Chan TA: Epigenetic
therapy: Use of agents targeting deacetylation and methylation in
cancer management. Onco Targets Ther. 6:223–232. 2013.PubMed/NCBI
|
|
66
|
Wu CC, Chuang HY, Lin CY, et al:
Inhibition of Epstein-Barr virus reactivation in nasopharyngeal
carcinoma cells by dietary sulforaphane. Mol Carcinog. 52:946–958.
2013. View Article : Google Scholar
|