Introduction
Cancer cachexia, a condition of advanced protein
calorie malnutrition, is characterized by loss of skeletal muscle
protein, depletion of lipid storage, anorexia, poor performance and
ultimately death (1–3). Cancer cachexia occurs in ~80% of
cancer patients and is the primary cause of death for 22–30% of all
cancer patients (3,4). However, cancer cachexia is difficult
to diagnose until patients have lost more than 5–7% of their body
weight (5,6). Furthermore, it was reported that 80%
of patients with upper gastrointestinal cancers and 60% of patients
with lung cancer had already experienced substantial weight loss at
the moment of diagnosis (7).
Therefore, it is essential to illuminate the early mechanisms in
the development of cancer cachexia.
The pathogenesis of cancer cachexia including
alterations in energy and substrate metabolisms is multifunctional.
Skeletal muscle protein depletion (i.e. muscle atrophy) due to
hyper-catabolism is the most common characteristic of cancer
cachexia. Cancer cachexia is a complex process that occurs as a
consequence of a variety of stressors including neural inactivity,
mechanical unloading, inflammation, metabolic stress and elevated
glucocorticoids (1,8,9).
Several proteolytic systems, such as the lysosome, the
calcium-dependent systems and the ubiquitin-dependent systems are
involved in degrading skeletal muscle (8). However, it was reported that ubiquitin
dependent proteolysis is the main mechanism for the enhanced muscle
protein degradation in cancer cachexia (10). In addition, cytokines such as tumor
necrosis factor-α (TNF-α), interleukin-1 (IL-1), interleukin-6
(IL-6) and interferon-γ (IFN-γ) have been proposed as mediators for
the cancer cachectic process (3,11,12).
The ubiquitin-dependent proteasome pathway includes
a series of reactions involving three classes of proteins:
ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes
(E2), and ubiquitin protein ligases (E3) (13). Muscle atrophy F-Box
(MAFbx)/atrogin-1 and muscle ring-finger-1 (MuRF-1), identified
over 10 years ago, are two muscle-specific E3 ubiquitin ligases
that are increased in skeletal muscle under atrophy-inducing
conditions such as humans under voluntary bed rest (14) or with spinal cord injury (15), amyotrophic lateral sclerosis
(16), chronic obstructive
pulmonary disease (COPD) (17) and
in animal models of aging (18),
diabetes (19), starvation
(20), making them excellent
markers for muscle atrophy. However, little is known concerning the
role of muscle-specific E3 ubiquitin ligase genes during the
process of cancer cachexia.
The goal of the present study was to elucidate the
role of muscle-specific E3 ubiquitin ligases during cancer cachexia
in vitro and in vivo. Firstly, we determined the
expression of muscle-specific E3 ubiquitin ligases both in patients
with malignant and benign disease. Furthermore, in a mouse model of
cancer cachexia, we analyzed the expression of muscle-specific E3
ubiquitin ligases and other ubiquitin proteasome pathway-associated
genes. Finally, we illustrated the protective effect of atrogin-1
small interfering RNA (siRNA) in an atrophy model of muscle cells
induced by TNF-α. Our results showed that muscle-specific E3
ubiquitin ligases were upregulated both in humans with malignant
disease and mice with cancer cachexia. Atrogin-1 gene silencing
protected myotubes from atrophy induced by TNF-α. We believe that
such a comprehensive analysis of muscle-specific E3 during cancer
cachexia in clinical patients to an animal model and muscle cells
illuminated the importance of muscle-specific E3 during cancer
cachexia and may provide novel targets for drug discovery in the
intervention of the progression of cancer cachexia.
Materials and methods
Patients and muscle samples
Twenty-one patients with malignant disease admitted
to Zhongshan Hospital of Fudan University between August 2011 and
December 2013 were enrolled in the present study. Diagnoses of
malignant disease were confirmed by postoperative pathological
examinations. Twenty-three patients undergoing surgery for benign
abdominal diseases served as the control group. Exclusion criteria
were patients with acute or chronic renal and liver failure, acute
and chronic hepatitis, diabetes, metabolic acidosis, sepsis, AIDS,
inflammatory bowel disease, autoimmune disorders, chronic heart
failure, hyperthyroidism and chronic obstructive pulmonary disease.
Written informed consent was obtained from all patients, and
permission for the present study was obtained from the Ethics
Committee of the Zhongshan Hospital, Fudan University.
The nutritional assessments of patients before
surgery included height, actual body weight, body mass index (BMI),
total lymphocyte count, serum albumin and prealbumin. A biopsy
specimen was obtained from the rectus abdominis muscles according
to the methods described by Bossola et al (21). The biopsy specimen was immediately
frozen in liquid nitrogen and then stored at −70°C until further
analysis. No complications occurred during the biopsy
procedure.
Animals and treatments
BALB/c male mice (6–8 week of age) weighing 16–20 g
were purchased from the Shanghai Laboratory Animal Center, Chinese
Academy of Sciences. Mice were housed at 22±1°C, with a 12-h
light/dark cycle, free access to water and conventional diet. Mice
were acclimated to the environment for one week before the study
started. All animal manipulations were carried out according to the
guidelines of regulations for the use of experimental animals of
the Chinese Academy of Science. All efforts were made to minimize
animal suffering and to use only the number of animals necessary to
produce reliable scientific data.
Colon 26/clone 20 cells, which have been reported to
induce severe cachexia in BALB/c mice by subcutaneous inoculation,
were purchased from the Shanghai Institute of Pharmaceutical
Industry. The cells were cultured in RPMI-1640 supplemented with 5%
fetal bovine serum and 1% penicillin-streptomycin at 37°C in 5%
CO2. On study day 0, 1.0×106 cells suspended
in 100 ml phosphate-buffered saline (PBS) were injected
subcutaneously into the armpits of the mice in the tumor group. An
equal volume of PBS without tumor cells was injected into the
control group mice. The body weight of each mouse was recorded
every two days after tumor cell inoculation. On day 16, the mice
were sacrificed via cervical dislocation. Non-tumor body weights
and gastrocnemius muscle weights were recorded. Muscle specimens
were frozen at −70°C for further analysis.
Cell lines and treatment in vitro
Mouse skeletal muscle C2C12 cells were propagated
and maintained in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with antibiotics and 10% fetal bovine serum at 37°C in
the presence of 5% CO2. To induce differentiation, C2C12
cells were propagated in DMEM supplemented with antibiotics and 2%
horse serum for the indicated period. After 3 days of
differentiation, according to a previous report (22), C2C12 cells were treated with 10
ng/ml recombinant mouse TNF-α (R&D Systems, USA) for 96 h to
extract proteins, and C2C12 cells were treated with 6 ng/ml
recombinant mouse TNF-α for 2 h to extract RNAs.
Construction and infection of the
lentivirus containing siRNA targeting the human atrogin-1 gene
Five pairs of siRNAs targeting the human atrogin-1
gene (Table I) were designed using
Ambion siRNA Target Finder and were synthesized by Invitrogen
Biotechnology Co., Ltd. (Shanghai, China). In addition, negative
control siRNA was also synthesized for monitoring the influence of
the exogenous genes. These siRNAs were cloned into vector pBS-hU6-I
and then into vector FG12. The length and accuracy of the sequences
were confirmed by DNA sequencing. The recombinant FG12 vectors were
cotransfected with pRSVREV, pMDLg/pRRE and pHCMV-G into 293T cells
to package lentivirus particles, with which C2C12 cells were
infected. The infected C2C12 cells were cultured and differentiated
to form myotubes before TNF-α was added to induce atrophy.
Atrogin-1 mRNA and protein were identified by real-time PCR
(RT-qPCR) and western blotting, respectively. Myotube morphology
was observed and photographed directly in the culture plate without
fixation.
 | Table IPrimers of atrogin-1 siRNAs. |
Table I
Primers of atrogin-1 siRNAs.
| No. | Sense Antisense | Sequences |
|---|
| 1 | Sense |
5′ACCGGAGGTATACAGTAAGGAGTTCAAGAGACTCCTTACTGTATACCTCCTTTTTGGATCCC3′ |
| Antisense |
5′TCGAGGGATCCAAAAAGGAGGTATACAGTAAGGAGTCTCTTGAACTCCTTACTGTATACCTC3′ |
| 2 | Sense |
5′ACCGCTTTCAACAGACTGGACTTTCAAGAGAAGTCCAGTCTGTTGAAAGCTTTTTGGATCCC3′ |
| Antisense |
5′TCGAGGGATCCAAAAAGCTTTCAACAGACTGGACTTCTCTTGAAAGTCCAGTCTGTTGAAAG3′ |
| 3 | Sense |
5′ACCGGCTGTTGGAGCTGATAGCTTCAAGAGAGCTATCAGCTCCAACAGCCTTTTTGGATCCC3′ |
| Antisense |
5′TCGAGGGATCCAAAAAGTCTGTGCTGGTGGGCAACTCTCTTGAAGTTGCCCACCAGCACAGA3′ |
| 4 | Sense |
5′ACCGTCTGTGCTGGTGGGCAACTTCAAGAGAGTTGCCCACCAGCACAGACTTTTTGGATCCC3′ |
| Antisense |
5′TCGAGGGATCCAAAAAGTCTGTGCTGGTGGGCAACTCTCTTGAAGTTGCCCACCAGCACAGA3′ |
| 5 | Sense |
5′ACCGCGTTTGATCTTGTCTGACTTCAAGAGAGTCAGACAAGATCAAACGCTTTTTGGATCCC3′ |
| Antisense |
5′TCGAGGGATCCAAAAAGCGTTTGATCTTGTCTGACTCTCTTGAAGTCAGACAAGATCAAACG3′ |
| Ctrl | Sense |
5′ACCGATGTTGTCAACGACTAGTTTCAAGAGAACTAGTCGTTGACAACATCTTTTTGGATCCC3′ |
| Antisense |
5′TCGAGGGATCCAAAAAGATGTTGTCAACGACTAGTTCTCTTGAAACTAGTCGTTGACAACAT3′ |
Western blot analysis
Total proteins were extracted from cells or muscle
tissues and quantified by the BCA method. Western blotting was
performed to determine the protein expression of atrogin-1. The
protocol used for western blotting was previously described
(23). Briefly, 50 mg total
proteins was separated by SDS-PAGE, transferred to PVDF membranes,
and analyzed by western blotting with a goat polyclonal anti-human
atrogin-1 antibody (Santa Cruz Biotechnology, USA). An antibody
against α-tubulin (Santa Cruz Biotechnology) served as an
endogenous control. Finally, the membrane was visualized by an
imaging system in the dark to quantify the protein level.
RNA extraction and RT-qPCR analysis
Total RNAs were isolated from the cells or tissues
with TRIzol (Invitrogen, Carlsbad, CA, USA). RNA integrity was
detected by an agarose gel method, and the RNA concentration was
quantified by measuring A260 and A280 absorbance in a NanoDrop
spectrophotometer (Thermo Scientific, USA). cDNA synthesis was
carried out with 2 μg of total RNAs by reverse transcription
(RT) (Promega, Madison, WI, USA). RT-qPCR was performed using
7900HT Fast Real-Time PCR System (Applied Biosystems, Inc., Foster
City, CA, USA). Real-time Master Mix (Toyobo, Osaka, Japan) was
used to detect and quantify the expression levels of the target
genes. The PCR reaction consisted of an initial denaturation at
95°C for 10 min followed by 40 cycles of 30 sec at 95°C, 10 sec at
60°C and 30 sec at 72°C, and finally 10 min at 72°C. Amplification
of the target cDNA was normalized to β-actin expression. Relative
levels of target mRNA expression were calculated using the
2−ΔΔCt method. The primers used are listed in Table II.
| Table IIPrimer sequences of the target genes
and β-actin. |
Table II
Primer sequences of the target genes
and β-actin.
| Species | Genes | Forward primers | Reverse primers |
|---|
| Human | Atrogin-1 |
GACTTCTCAACTGCCATTC |
TCGTCTCCATCCGATACAC |
| Human | MuRF-1 |
GCTGAGCCAGAAGTTTGA |
CAGGGCGTCTGCTATGTG |
| Human | β-actin |
GATCATTGCTCCTCCTGAGC |
ACTCCTGCTTGCTGATCCAC |
| Mouse | Atrogin-1 |
CTGGATTGGAAGAAGATGTA |
CTTGAGGGGAAAGTGAGACG |
| Mouse | MuRF-1 |
CCTACTTGCTCCTTGTGC |
TCCTGCTCCTGCGTGAT |
| Mouse | Ubiquitin |
GACAGGCAAGACCATCACC |
CACCCAAGAACAAGCACAA |
| Mouse | E2-14K |
TGGACCAGAAGGGACACC |
TGGCTGGACTGTTTGGATT |
| Mouse | β-actin |
AGGTGTGCACCTTTTATTGGTCTCAA |
TCCCTCTGGTTTGGAAGTATGT |
Immunohistochemical staining
Immunohistochemical staining (IHC) of the tissues
was performed as previously described (23). Briefly, muscle tissues were fixed in
4% formalin for 24 h and paraffin-embedded using a conventional
method. Sections (3-μm) were dehydrated in xylene and graded
alcohols. Antigen retrieval was performed with 0.01 M citrate
buffer at pH 6.0 at 95°C for 20 min. The slides were then incubated
with atrogin-1 antibody at 4°C overnight and then with the
fluorescein isothiocyanate (FITC)-conjugated secondary antibodies
for 60 min at room temperature in the dark.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 5.0 software (GraphPad Software, USA). Continuous variables
were compared using the Student’s t-test and are expressed as the
mean and standard deviation (SD). P<0.05 was considered to
indicate a statistically significant result.
Results
Atrogin-1 and MuRF1 mRNA are highly
expressed in the patients with malignant disease
The clinical characteristics of the patients with
malignant disease and benign disease are shown in Table III. Nutrition statuses such as
BMI, total lymphocyte count, serum albumin and pre-albumin were
significantly different in the two groups (P<0.05), while other
characteristics showed no significant differences.
| Table IIICharacteristics of the included
patients. |
Table III
Characteristics of the included
patients.
| Malignant disease
group (n=21) | Benign disease
group (n=23) | P-value |
|---|
| Mean age,
years | 59.4±11.2 | 53.8±10.9 | NS |
| Gender (M:F) | 14:7 | 17:6 | NS |
| Weight, kg | 61.1±9.2 | 63.4±10.4 | NS |
| BMI | 20.5±2.9 | 22.5±3.1 | 0.03 |
| Weight loss,
n/total | 6/21 | 0/23 | <0.001 |
| Total lymphocyte
count, ×109/l | 1.5±0.7 | 1.6±0.6 | NS |
| Serum albumin,
g/l | 39.9±5.3 | 44.2±4.3 | 0.004 |
| Serum pre-albumin,
g/l | 0.21±0.06 | 0.28±0.04 | <0.001 |
The mRNA levels of atrogin-1 and MuRF-1 in the
rectus abdominis muscles were analyzed by RT-qPCR. Considering the
mean ΔCt of the benign disease group as control, we determined the
ΔCt of atrogin-1 and MuRF-1 in the malignant disease group. As
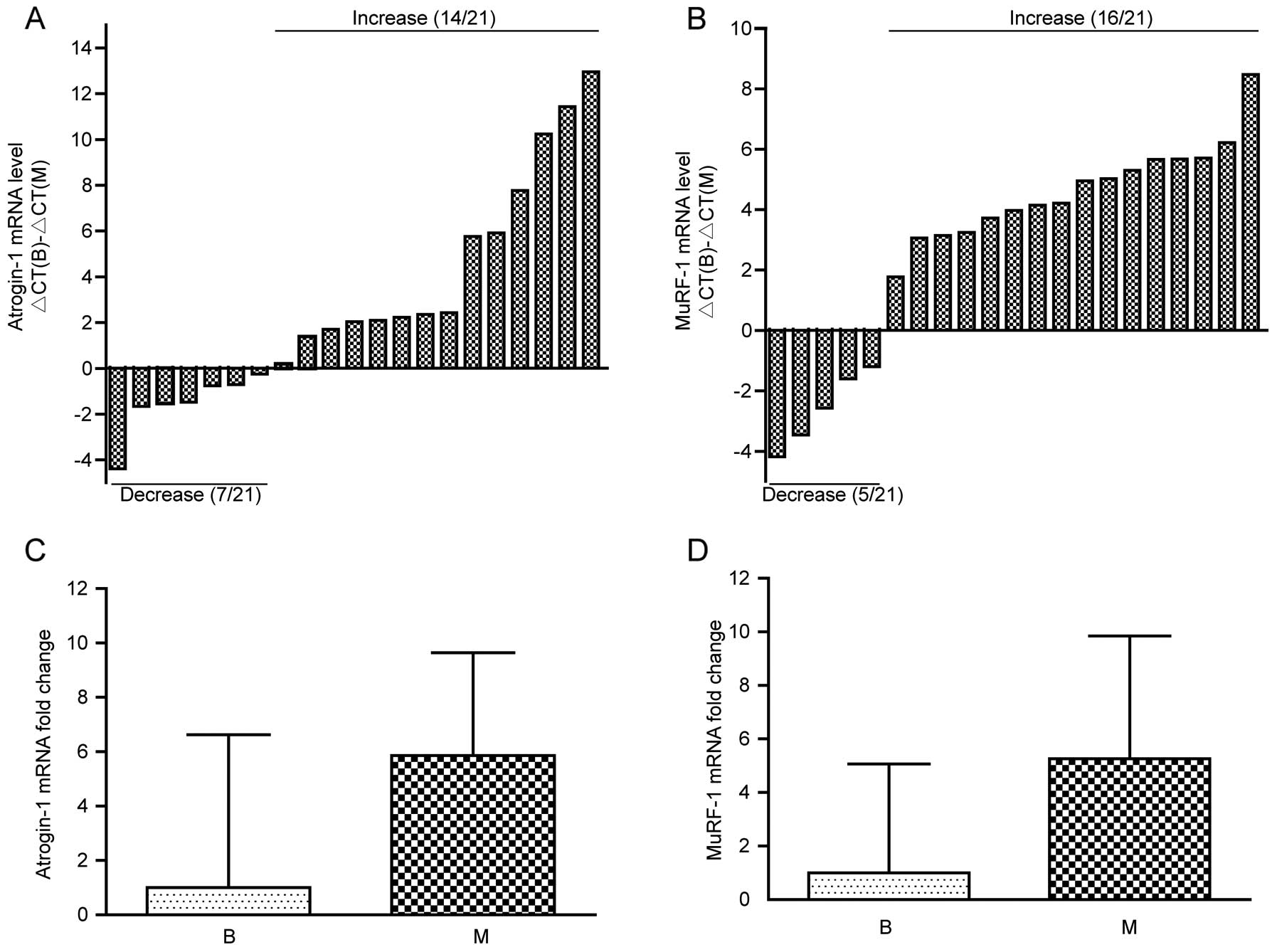
shown in Fig. 1A and B, atrogin-1
and MuRF-1 mRNA levels were increased in 66.7% (14/21) and 76.2%
(16/21) of the malignant disease patients, respectively. Univariate
analysis indicated that the atrogin-1 and MuRF-1 mRNA levels were
significantly highly expressed in the malignant disease patients
(P<0.05, data not shown). In addition, to determine whether the
expression of atrogin-1 and MuRF-1 mRNA increased before weight
loss, we compared the mRNA levels of atrogin-1 and MuRF-1 in 16
malignant disease patients and 23 benign disease patients without
weight loss. The results showed that both atrogin-1 and MuRF-1 mRNA
levels tended to increase in the malignant disease patients but
without statistical difference (P=0.099, P=0.132, respectively,
Fig. 1C and D).
Successful construction of the colon-26
adenocarcinoma cell-induced cachexia mouse model
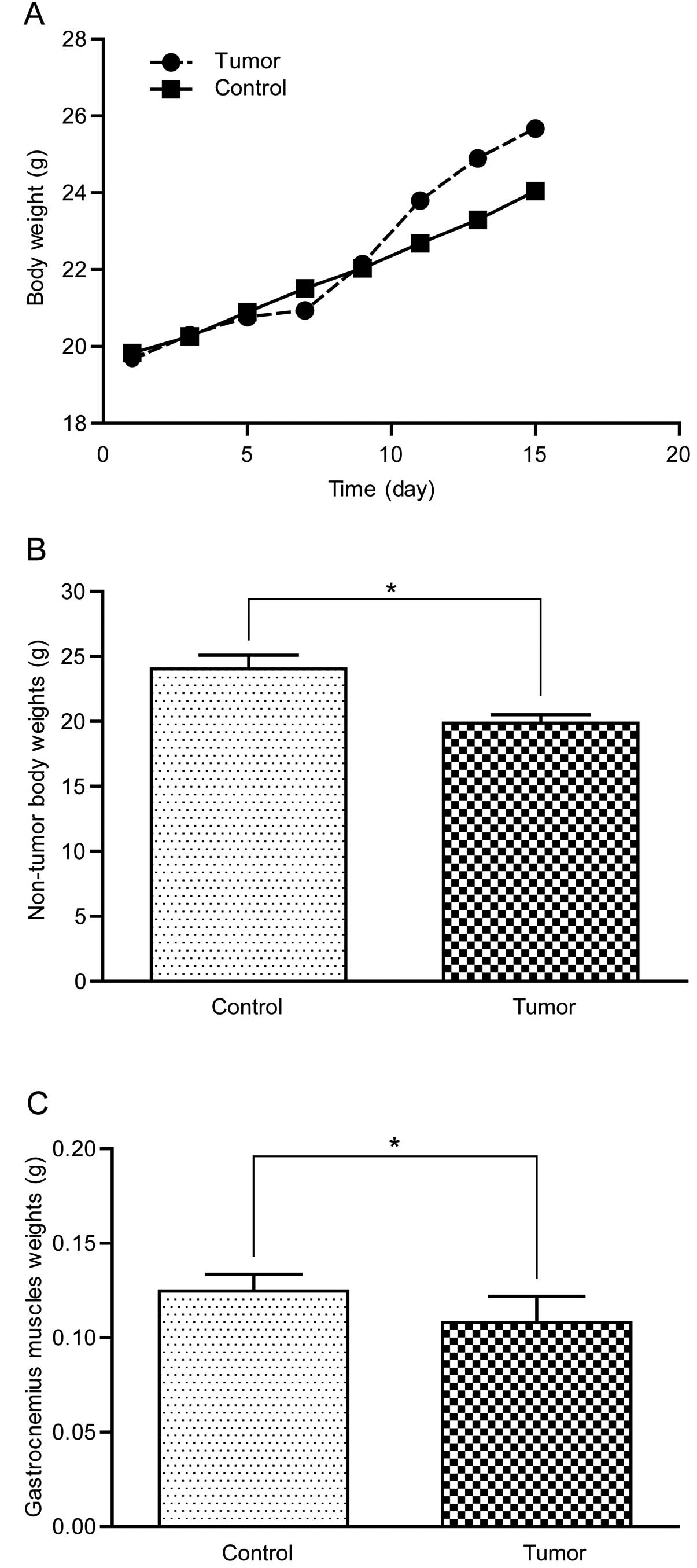
Subcutaneous tumors could be touched by hand four
days after colon-26 adenocarcinoma cell inoculation and reached ~8
cm3 when the mice were sacrificed. As shown in Fig. 2A, the whole body weight of the
tumor-bearing mice increased slowly at the beginning yet had a
rapid growth afterwards due to the rapid growth of the subcutaneous
tumors. Non-tumor body weight and gastrocnemius muscle weight of
the tumor-bearing mice were significantly lower than the control
group (Fig. 2B and C), indicating
that cancer cachexia was effectively induced in this model.
Ubiquitin proteasome pathway-associated
genes are highly expressed in the cachexia mice
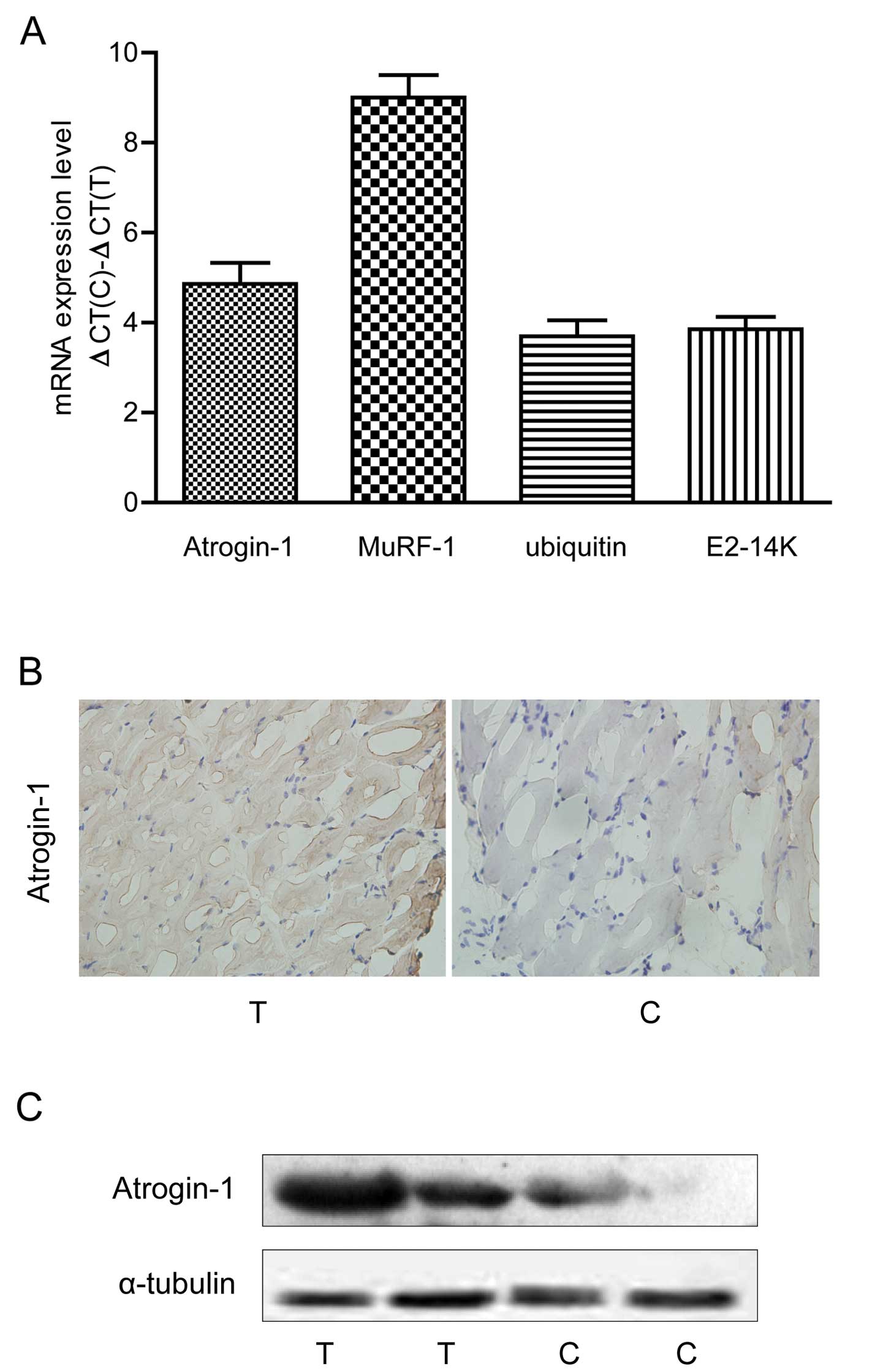
We determined the expression of four ubiquitin
proteasome pathway-associated genes (atrogin-1, MuRF-1, ubiquitin
and E2-14K) in the muscles of colon-26-induced cachexia mice.
Compared to the non-tumor mice, atrogin-1, MuRF-1, ubiquitin and
E2-14K mRNA levels were significantly increased in the cachexia
mice (P<0.05, Fig. 3A). Western
blotting and IHC confirmed that the atrogin-1 protein was
overexpressed in the cachexia mice (Fig. 3B and C).
Optimization of the lentiviral vector
system for siRNAs targeting atrogin-1
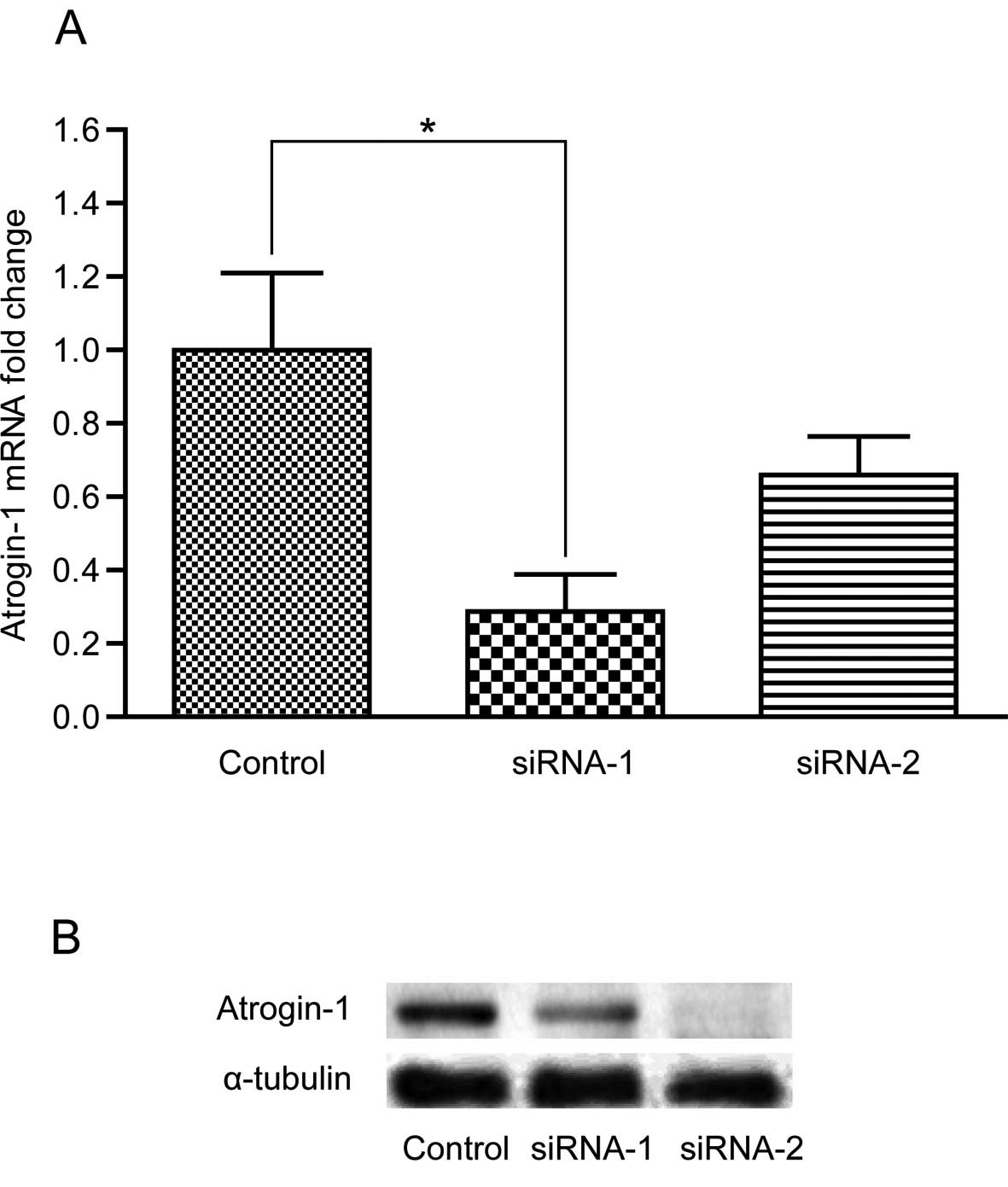
The accuracy of the recombinant plasmids was
confirmed by DNA sequencing. Two correct recombinant plasmids
PBSU6I-atrogin-1 siRNA-1 and -2 were chosen for further analysis,
while three incorrect recombinant plasmids PBSU6I-atrogin-1
siRNA-3, -4 and -5 were discarded. After transfection with the
recombinant lentivirus vector expressing atrogin-1 siRNA-1 and -2,
atrogin-1 was downregulated in the C2C12 cells. However, only
atrogin-1 siRNA-1 was identified to be the most effective siRNA in
inhibiting atrogin-1 expression. As shown in Fig. 4, atrogin-1 was downregulated in the
C2C12 cells transfected with atrogin-1 siRNA-1 and -2 compared to
the non-specific siRNA control, yet only siRNA-1 significantly
downregulated the expression of atrogin-1 (P<0.05). Thus, we
chose siRNA-1 for subsequent study.
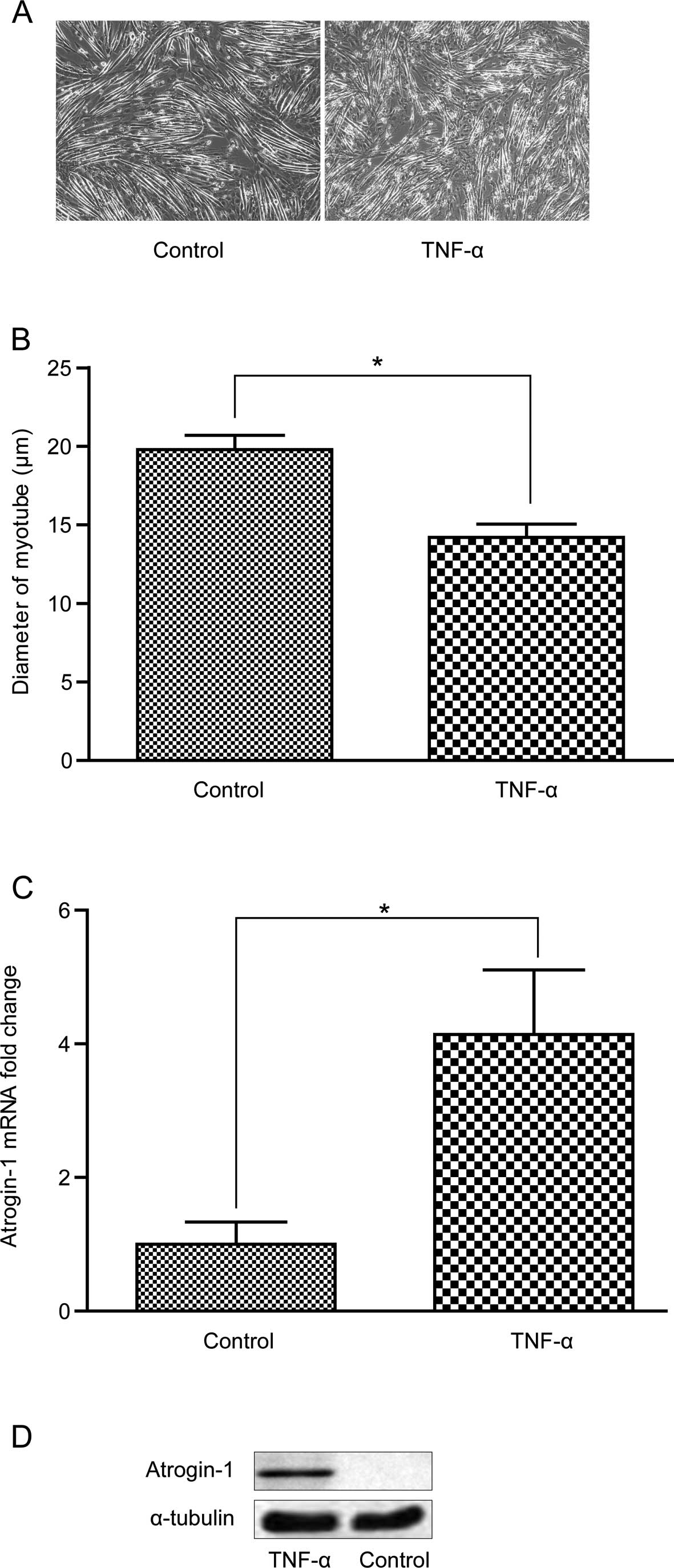
TNF-α induces myotube atrophy and
atrogin-1 is overexpressed in the C2C12 cells
After treatment with 10 ng/ml TNF-α, the C2C12 cells
grew slowly and myotubes atrophied with a significantly decreased
diameter (Fig. 5A and B). Atrogin-1
was significantly increased at the mRNA and protein levels after
treatment with 6 ng/ml TNF-α for 2 h and 10 ng/ml TNF-α for 96 h,
respectively (Fig. 5C and D).
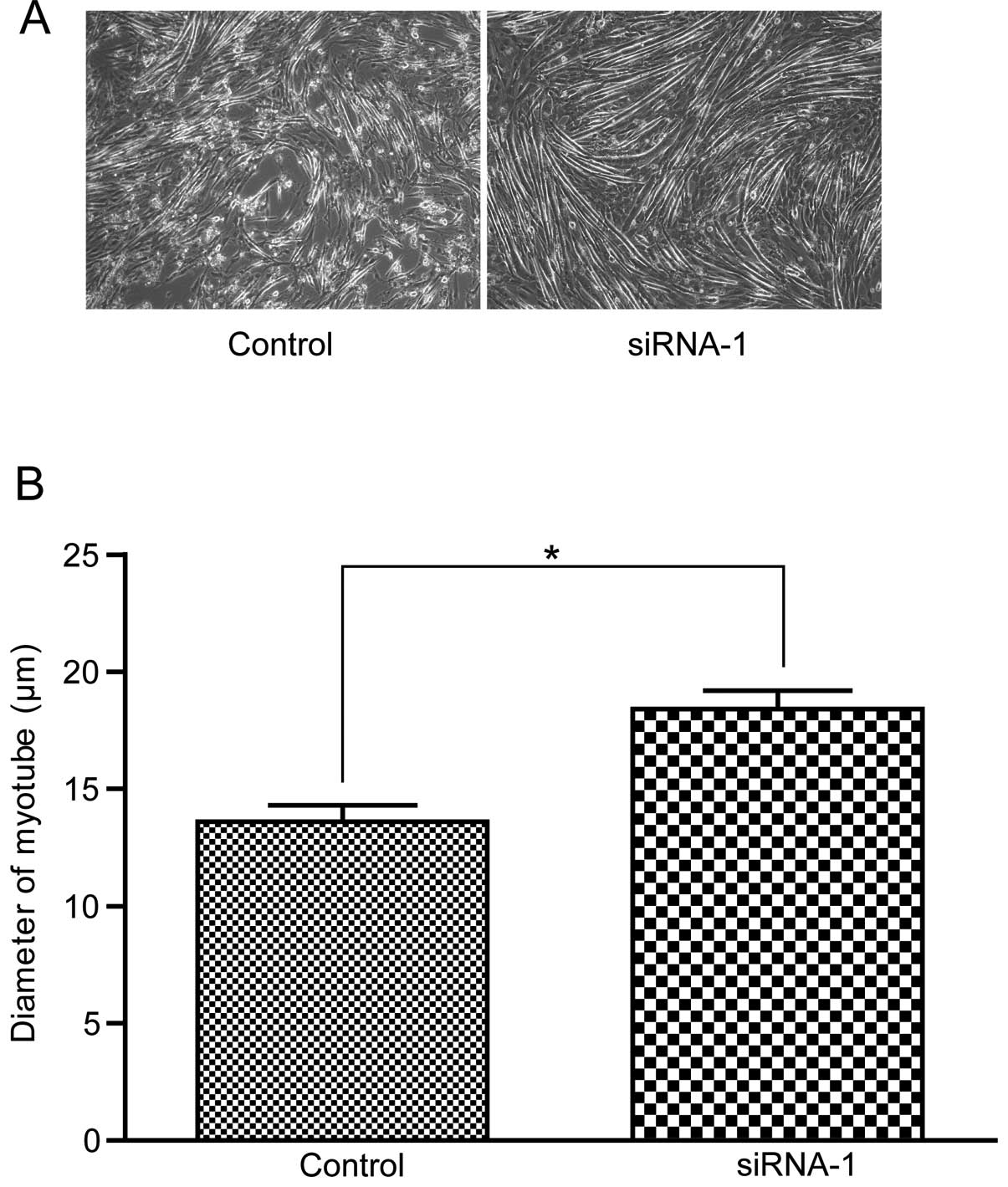
Atrogin-1 siRNA-1 reduces myotube atrophy
induced by TNF-α in the C2C12 cells
To investigate whether inhibition of atrogin-l
affects myotube atrophy induced by TNF-α, we compared the myotube
morphology and myotube diameter in the C2C12 cells in the atrogin-1
siRNA-1 group and the non-specific siRNA control group. No
differences were observed in the two groups before incubation with
TNF-α. However, after treatment with TNF-α, the myotubes underwent
atrophy with a significant decrease in myotube diameter in the
control group, while no obvious changes were observed in the
atrogin-1 siRNA-1 group (Fig.
6).
Discussion
In the present study, we demonstrated the
involvement of the muscle-specific E3 ubiquitin ligases in muscle
atrophy of cancer cachexia in clinical patients, a mouse model and
C2C12 cells. To the best of our knowledge, we firstly report that
atrogin-1 and MuRF-1 mRNAs tend to be upregulated in malignant
disease patients even before body weight loss. Furthermore, we
found that four ubiquitin proteasome pathway-associated genes
(atrogin-1, MuRF-1, ubiquitin and E2-14K) were upregulated in
cancer cachexia mice, and knockdown of atrogin-l protected the
C2C12 cells from atrophy induced by TNF-α, implying the potential
mechanism underling the muscle wasting of cancer cachexia.
Atrogin-1 and MuRF-1 are elevated in human muscles
during various conditions that elicit muscle atrophy. In the
present study, atrogin-1 and MuRF-1 mRNA were found to be increased
in most of the malignant disease patients, which is consistent with
previous research (24). Increased
muscle proteasome activity was reported to be correlated with
disease severity in gastric cancer patients (21). However, a recent study reported that
the expression of atrogin-1 and MuRF-1 in gastric cancer patients
was not different from benign disease, and both genes were
unaffected by the stage of cancer or the severity of body weight
loss, except for a transient increase in mRNA expression of MuRF-1
in patients with stage IV disease relative to those with stage III
(25). The inconsistency suggests
that atrogin-1 and MuRF-1 expression may be affected by various
factors, and studies enrolling more cancer patients are urgently
needed to reduce individual differences. In addition, we found that
atrogin-1 and MuRF-1 tended to be upregulated even before a visible
decline in body weight, suggesting that high expression of
atrogin-1 and MuRF-1 may not be an outcome of body weight loss in
cancer cachexia patients but may contribute to this process.
Murine adenocarcinoma colon-26-bearing mice exhibit
progressive weight loss of skeletal muscle and adipose tissues
similar to cancer cachexia; thus, colon-26-induced cachexia in mice
has been extensively used to elucidate the mechanisms of cancer
cachexia (26,27). In this model, we found that four
ubiquitin proteasome pathway-associated genes (atrogin-1, MuRF-1,
ubiquitin and E2-14K) were significantly upregulated in the
tumor-bearing cancer cachexia mice. These results indicate that the
ubiquitin proteasome pathway may be involved in the course of
skeletal muscle loss in cancer cachexia. However, whether this
pathway plays a key role in this process needs further
analysis.
Previous studies indicate that TNF-α plays a
critical role in regulating muscle mass and protein degradation
during cachexia (28). Atrogin-1
mRNA has been shown to increase with TNF-α exposure in C2C12
myotubes (29). However, rare
studies have reported the effect of siRNA targeting atrogin-1 in
resisting the myotube atrophy from the adverse effect of TNF-α. In
the present study, an effective atrogin-1 siRNA was successfully
constructed and an obvious protective effect of atrogin-1 siRNA was
observed. These results suggest that knockdown of atrogin-1 may be
an effective method to treat muscle atrophy induced by TNF-α in
cancer cachexia.
Although our results suggest that muscle-specific
ubiquitin ligase E3 may contribute to the muscle atrophy of cancer
cachexia, there are several limitations to the present study.
Firstly, due to the rare number of cancer cachexia patients, we did
not analyze the atrogin-1 and MuRF-1 expression restricted to
cancer cachexia patients, but in several types of malignant disease
patients. Secondly, due to tissue limitation, the protein levels of
either atrogin-1 or MuRF-1 could not be determined in clinical
patients. Thirdly, in the mechanistic study, we only focused on the
effect of atrogin-1 but not of all associated genes. Studies to
investigate the effect and mechanism of other associated genes
should be conducted in the future.
In conclusion, the present study demonstrated that
atrogin-1 and MuRF-1 are highly expressed in malignant disease
patients and cancer cachexia mice. Additionally, knockdown of
atrogin-1 protected- myotubes from atrophy induced by TNF-α in
C2C12 cells. Given the fact that atrogin-1 and MuRF-1 tend to be
upregulated even before visible body weight loss in malignant
disease patients, future research may focus on the role of
atrogin-1 and MuRF-1 in the early stage of muscle atrophy in cancer
cachexia and how to exert the anti-cachectic effects of atrogin-1
and MuRF-1 antagonists. Taken together, our data suggest that
targeting atrogin-1 at an early stage of muscle atrophy may be an
effective strategy for molecular and clinical intervention in the
muscle wasting pathological process in cancer cachexia.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China grants (no 30571819).
References
|
1
|
Inui A: Cancer anorexia-cachexia syndrome:
Current issues in research and management. CA Cancer J Clin.
52:72–91. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Costelli P and Baccino FM: Cancer
cachexia: From experimental models to patient management. Curr Opin
Clin Nutr Metab Care. 3:177–181. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fearon KC, Glass DJ and Guttridge DC:
Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell
Metab. 16:153–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Onesti JK and Guttridge DC: Inflammation
based regulation of cancer cachexia. Biomed Res Int.
2014:1684072014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ando K, Takahashi F, Motojima S, Nakashima
K, Kaneko N, Hoshi K and Takahashi K: Possible role for
tocilizumab, an anti-interleukin-6 receptor antibody, in treating
cancer cachexia. J Clin Oncol. 31:e69–e72. 2013. View Article : Google Scholar
|
|
6
|
Gallagher IJ, Stephens NA, MacDonald AJ,
Skipworth RJ, Husi H, Greig CA, Ross JA, Timmons JA and Fearon KC:
Suppression of skeletal muscle turnover in cancer cachexia:
Evidence from the transcriptome in sequential human muscle
biopsies. Clin Cancer Res. 18:2817–2827. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bruera E: ABC of palliative care.
Anorexia, cachexia, and nutrition. BMJ. 315:1219–1222. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nicolini A, Ferrari P, Masoni MC, Fini M,
Pagani S, Giampietro O and Carpi A: Malnutrition, anorexia and
cachexia in cancer patients: A mini-review on pathogenesis and
treatment. Biomed Pharmacother. 67:807–817. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Argilés JM, Costelli P, Carbó N,
Pallarés-Trujillo J and López-Soriano FJ: Tumour growth and
nitrogen metabolism in the host (Review). Int J Oncol. 14:479–486.
1999.PubMed/NCBI
|
|
10
|
Jagoe RT and Goldberg AL: What do we
really know about the ubiquitin-proteasome pathway in muscle
atrophy? Curr Opin Clin Nutr Metab Care. 4:183–190. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan
R, Puzis L, Koniaris LG and Zimmers TA: JAK/STAT3 pathway
inhibition blocks skeletal muscle wasting downstream of IL-6 and in
experimental cancer cachexia. Am J Physiol Endocrinol Metab.
303:E410–E421. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Narsale AA and Carson JA: Role of
interleukin-6 in cachexia: Therapeutic implications. Curr Opin
Support Palliat Care. 8:321–327. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bodine SC and Baehr LM: Skeletal muscle
atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am
J Physiol Endocrinol Metab. 307:E469–E484. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ogawa T, Furochi H, Mameoka M, et al:
Ubiquitin ligase gene expression in healthy volunteers with 20-day
bedrest. Muscle Nerve. 34:463–469. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Urso ML, Chen YW, Scrimgeour AG, Lee PC,
Lee KF and Clarkson PM: Alterations in mRNA expression and protein
products following spinal cord injury in humans. J Physiol.
579:877–892. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Léger B, Vergani L, Sorarù G, Hespel P,
Derave W, Gobelet C, D’Ascenzio C, Angelini C and Russell AP: Human
skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a
reduction in Akt and an increase in atrogin-1. FASEB J. 20:583–585.
2006.PubMed/NCBI
|
|
17
|
Doucet M, Dubé A, Joanisse DR, Debigaré R,
Michaud A, Paré MÈ, Vaillancourt R, Fréchette E and Maltais F:
Atrophy and hypertrophy signalling of the quadriceps and diaphragm
in COPD. Thorax. 65:963–970. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Clavel S, Coldefy AS, Kurkdjian E, Salles
J, Margaritis I and Derijard B: Atrophy-related ubiquitin ligases,
atrogin-1 and MuRF1 are up-regulated in aged rat Tibialis Anterior
muscle. Mech Ageing Dev. 127:794–801. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dehoux M, Van Beneden R, Pasko N, Lause P,
Verniers J, Underwood L, Ketelslegers JM and Thissen JP: Role of
the insulin-like growth factor I decline in the induction of
atrogin-1/MAFbx during fasting and diabetes. Endocrinology.
145:4806–4812. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lecker SH, Jagoe RT, Gilbert A, Gomes M,
Baracos V, Bailey J, Price SR, Mitch WE and Goldberg AL: Multiple
types of skeletal muscle atrophy involve a common program of
changes in gene expression. FASEB J. 18:39–51. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bossola M, Muscaritoli M, Costelli P,
Grieco G, Bonelli G, Pacelli F, Rossi Fanelli F, Doglietto GB and
Baccino FM: Increased muscle proteasome activity correlates with
disease severity in gastric cancer patients. Ann Surg. 237:384–389.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li YP, Schwartz RJ, Waddell ID, Holloway
BR and Reid MB: Skeletal muscle myocytes undergo protein loss and
reactive oxygen-mediated NF-kappaB activation in response to tumor
necrosis factor alpha. FASEB J. 12:871–880. 1998.PubMed/NCBI
|
|
23
|
Zhou CF, Li XB, Sun H, Zhang B, Han YS,
Jiang Y, Zhuang QL, Fang J and Wu GH: Pyruvate kinase type M2 is
upregulated in colorectal cancer and promotes proliferation and
migration of colon cancer cells. IUBMB Life. 64:775–782. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Khal J, Hine AV, Fearon KC, Dejong CH and
Tisdale MJ: Increased expression of proteasome subunits in skeletal
muscle of cancer patients with weight loss. Int J Biochem Cell
Biol. 37:2196–2206. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
D’Orlando C, Marzetti E, François S, et
al: Gastric cancer does not affect the expression of
atrophy-related genes in human skeletal muscle. Muscle Nerve.
49:528–533. 2014. View Article : Google Scholar
|
|
26
|
Yasumoto K, Mukaida N, Harada A, et al:
Molecular analysis of the cytokine network involved in cachexia in
colon 26 adenocarcinoma-bearing mice. Cancer Res. 55:921–927.
1995.PubMed/NCBI
|
|
27
|
Iizuka N, Hazama S, Yoshimura K, Yoshino
S, Tangoku A, Miyamoto K, Okita K and Oka M: Anticachectic effects
of the natural herb Coptidis rhizoma and berberine on mice bearing
colon 26/clone 20 adenocarcinoma. Int J Cancer. 99:286–291. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pajak B, Orzechowska S, Pijet B, Pijet M,
Pogorzelska A, Gajkowska B and Orzechowski A: Crossroads of
cytokine signaling - the chase to stop muscle cachexia. J Physiol
Pharmacol. 59(Suppl 9): S251–S264. 2008.
|
|
29
|
Li YP, Chen Y, John J, Moylan J, Jin B,
Mann DL and Reid MB: TNF-alpha acts via p38 MAPK to stimulate
expression of the ubiquitin ligase atrogin1/MAFbx in skeletal
muscle. FASEB J. 19:362–370. 2005. View Article : Google Scholar : PubMed/NCBI
|