Introduction
Cardiac myxoma is the most common type of heart
tumor, and is associated with the risk of arterial embolism and
sudden death (1). However, the
molecular mechanism of myxoma initiation and progression remains
unexplored. Interestingly, it has been well established that the
genes associated with development always participate in the
progression of cancer (2). Thus,
the genes that are closely associated with heart development are
worth studying for a better understanding of myxoma pathology.
The myocyte enhancer factor 2 (MEF2) family of
transcription factors consists of 4 members in mammalian cells
(MEF2A, 2B, 2C and 2D). They have been well documented to play
important roles in the development of the heart (3). The expression levels of MEF2A, 2C and
2D were previously found to be elevated during cardiac development
(4). Mice with mutation of MEF2C
undergo looping morphogenesis failure during early cardiac
morphogenesis (5). In Xenopus
laevis, Mef2c and Mef2d both contribute to proper cardiac gene
expression (6).
More importantly, the MEF2 family (particularly
MEF2D) has been recently shown to be implicated in cancer biology.
Studies concerning leukemia reveal that the fusion between
MEF2D and DAZAP1 caused by t(1;19)(q23;p13.3)
chromosome translocation contributes to the formation and
progression of acute lymphoblastic leukemia (ALL) (7,8). The
role of MEF2D itself in cancer has also been reported in a recently
published study. Ma et al provided evidence that MEF2D
overexpression promotes the proliferation of liver cancer cells by
suppressing cell cycle arrest-associated genes; and MEF2D
overexpression may be due to the decline in tumor-suppressor
miR-122 expression (9).
Notably, the association between MEF2D and heart
disease has also been established in a study using a transgenic
mouse model. MEF2D null mutation was able to prevent mice from
stress-dependent cardiac hypertrophy, fetal gene activation and
fibrosis, while the forced expression of MEF2D promoted the above
pathological remodeling of the heart in mice (10). Atorvastatin was found to reverse
this pathological modeling of the heart by suppressing the activity
of MEF2D (11). These findings
suggest that MEF2D is closely implicated in heart diseases.
However, the link between MEF2D and heart tumors has
not yet been established. In the present study, we investigated the
expression profile of the MEF2 family and their possible roles in
cardiac myxoma. The underlying molecular mechanism was also
investigated.
Materials and methods
Cell line culture
The human normal lung fibroblast cell line MRC-5,
was purchased from the Shanghai Cell Collection (Shanghai, China).
The cells were cultured in Dulbecco’s modified Eagle’s medium
(DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco-BRL) at
37°C under a humidified 5% CO2 atmosphere.
Primary myxoma cell culture
The patient-derived primary cultures of cardiac
myxoma cells were obtained from fresh tumor specimens from patients
as previously described (12).
Briefly, the single-cell suspension was obtained by mechanical
manipulation of the cardiac myxoma specimen. The primary culture
was maintained in DMEM supplemented with 10% FBS. The established
primary myxoma cells were designated as CM001-CM006.
Quantitative PCR (qPCR) assay
The expression levels of mRNA of the MEF2 family and
miR-218 were determined by qPCR. To examine the levels of MEF2A,
MEF2C and MEF2D mRNA in cardiac myxoma, fresh tissues (cardiac
myxoma samples and the matched non-cancerous tissue, n=10) were
obtained with written informed consent from patients following the
protocols approved by the Ethics Review Board of Henan University
of Science and Technology (China). The patients underwent surgical
operation at the Department of Cardiovascular Surgery, The First
Affiliated Hospital of Henan University of Science and Technology.
Total RNA was extracted with TRIzol solution (Sigma-Aldrich)
according to the protocol provided by the manufacturer. The total
RNA was transcribed into cDNAs using ReverTra Ace quantitative PCR
(qPCR-RT) kit (Toyobo, Japan) according to the manufacturer’s
instructions. qPCR was performed using TaqMan® 2X
Universal PCR Master Mix (Applied Biosystems) on a CFX96™ Real-Time
PCR Detection System (Bio-Rad Laboratories Inc., Hercules, CA, USA)
supplied with analytical software. cDNA was obtained by PCR. The
sequences of the used primers were previously described by Ma et
al (9).
To detect the expression of miR-218, total RNA was
extracted with TRIzol solution according to the protocols provided
by the manufacturer. Reverse transcription reaction was carried out
using All-in-One™ First-Strand cDNA Synthesis kit (AORT-0020;
GeneCopoeia) following the manufacturer’s instructions. qPCR was
performed using All-in-One™ miRNA qRT-PCR Detection kit (AOMD-Q020;
GeneCopoeia) on a CFX96™ Real-Time PCR Detection System supplied
with analytical software. U6 was used as an endogenous reference.
The primers and probes for miR-218 were purchased from GeneCopoeia
(HmiRQP0327 and HmiRQP0326).
Immunoblotting assay
To examine the expression level of the indicated
proteins, the lysate containing the total proteins extracted using
M-PER® Mammalian Protein Extraction Reagent (Thermo
Scientific, Rockford, IL, USA) was separated by polyacrylamide gel
electrophoresis and transferred onto 0.45-μm nitrocellulose
membranes. After a 2-h blocking with 5% fat-free dry milk, the
membranes were then incubated with primary antibodies for 2 h. The
membranes were incubated with the corresponding secondary antibody
for 1 h and finally, visualized with SuperSignal West Dura Extended
Duration Substrate (Thermo Scientific). The involved antibodies
were all purchased from Cell Signaling Technology (Beverly, MA,
USA).
MEF2D siRNA treatment
The MEF2D-specific siRNA was purchased from the
Shanghai GenePharma Co. (Shanghai, China) as well as the control
siRNA. The cells were transfected with the indicated siRNA (50 nM)
24 h prior to the subsequent experiments.
Proliferation assay
Primary cardiac myxoma cells (5×103) were
planted into each well of 96-well plates. Overnight, the cells were
treated under the indicated conditions. At the indicated time
points, the cells were treated with 10 μl
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(5 mg/ml) for 4 h. MTT was then removed, and 150 μl DMSO was
replaced. The spectrophotometric absorbance was detected on a Model
550 microplate reader at 570 nm with a reference wavelength of 655
nm.
Animal experiments
All procedures for the animal experiments were
approved by the Committee on the Use and Care on Animals in Henan
University of Science and Technology and were performed following
institutional guidelines. After the indicated treatments, human
cardiac myxoma xenografts were established by subcutaneously
inoculating 5×105 cells into both flanks of 5-week-old
BALB/c nude mice (n=9). Tumor diameters were periodically measured
with calipers. The volumes were calculated following the formula:
Volume (mm3) = length (mm) x [width (mm)]2/2.
All animals received humane care according to the criteria outlined
in the ‘Guide for the Care and Use of Laboratory Animals’ prepared
by the National Academy.
Cell cycle analysis by flow
cytometry
The cells were exposed to the indicated treatments,
followed by being harvested, fixed in 70% ethanol and stained with
propidium podide (PI; 200 mg/ml) for flow cytometric analysis with
the Aria II sorter (BD Biosciences). For each group, 10,000 cells
were counted to determine the percentages of the
G0/G1, S and G2/M populations.
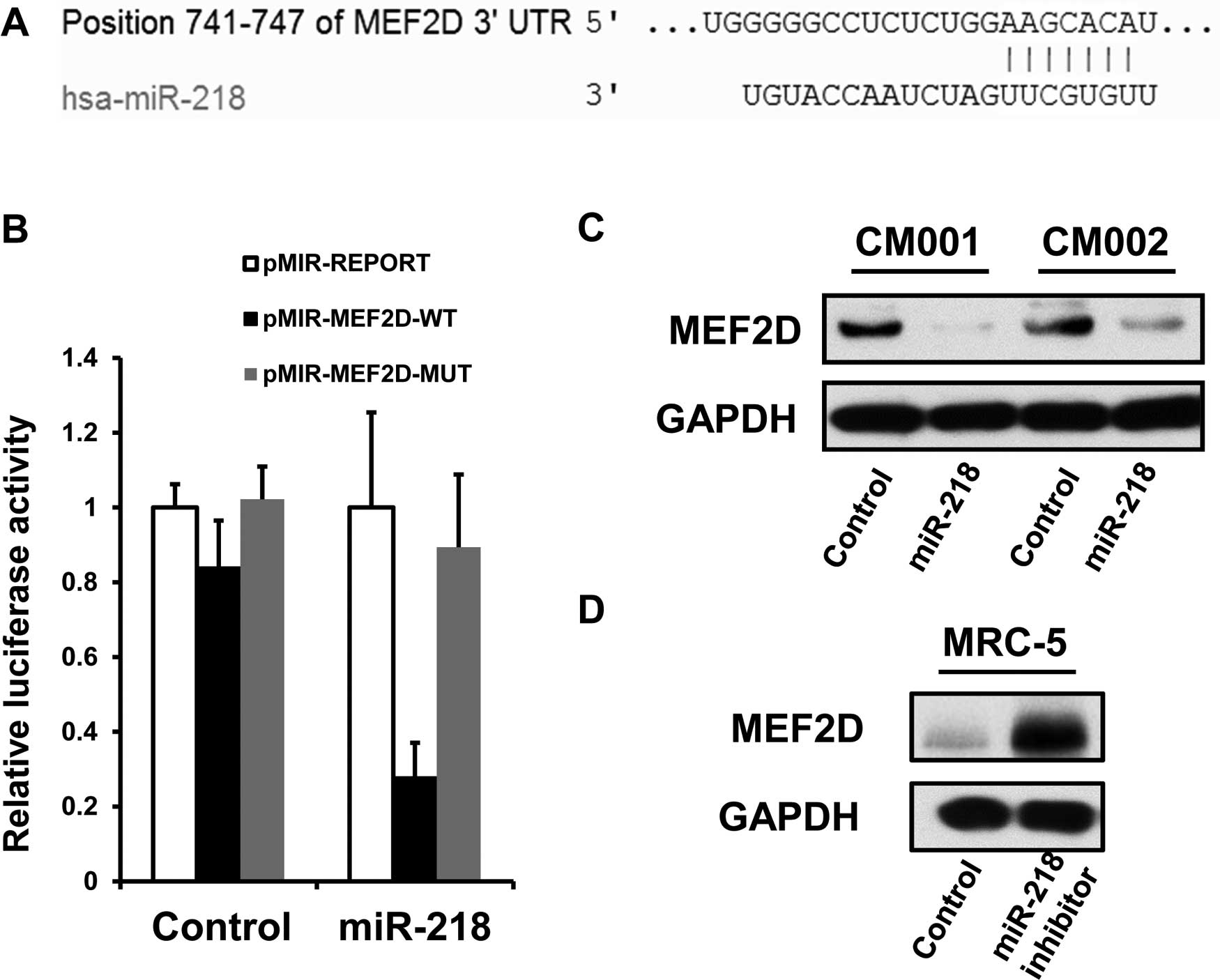
Identification of MEF2D as a target of
miR-218
The potential evolutionarily conserved miR-218
targets were predicted using the algorithm TargetScan (http://www.targetscan.org/). Luciferase assay was
applied to further confirm whether MEF2D is an authentic target of
miR-218. To generate the luciferase reporter vectors, a 251-bp
fully synthesized DNA construct identical to the region of the
MEF2D 3′UTR containing the predicted miR-218 binding site (AAGCACA)
was inserted into the SpeI and HindIII sites of the
pMIR-REPORT vector (Applied Biosystems), generating pMIR-MEF2D-WT.
To construct a control vector (pMIR-MEF2D-MUT), a mutation in the
miR-218 seed region was introduced into the Luc-m 3′UTR (AAGCTGA).
The mimics of miR-218 were purchased from GenePharma Co. Plasmids
for luciferase assays and mimics were cotransfected by
Lipofectamine 2000 (Invitrogen) according to the protocols provided
by the manufacturer. Subsequently, detection of luciferase activity
was performed with the Dual-Luciferase® Reporter Assay
kit (Promega, Madison, WI, USA) according to the manufacturer’s
instructions.
Statistical analysis
Each experiment was performed for at least three
times. All values are reported as means ± SD, and compared at a
given time point by the unpaired, two-tailed Student’s test. Data
were considered to indicate a statistically significant result at
P<0.05 and P<0.01.
Results
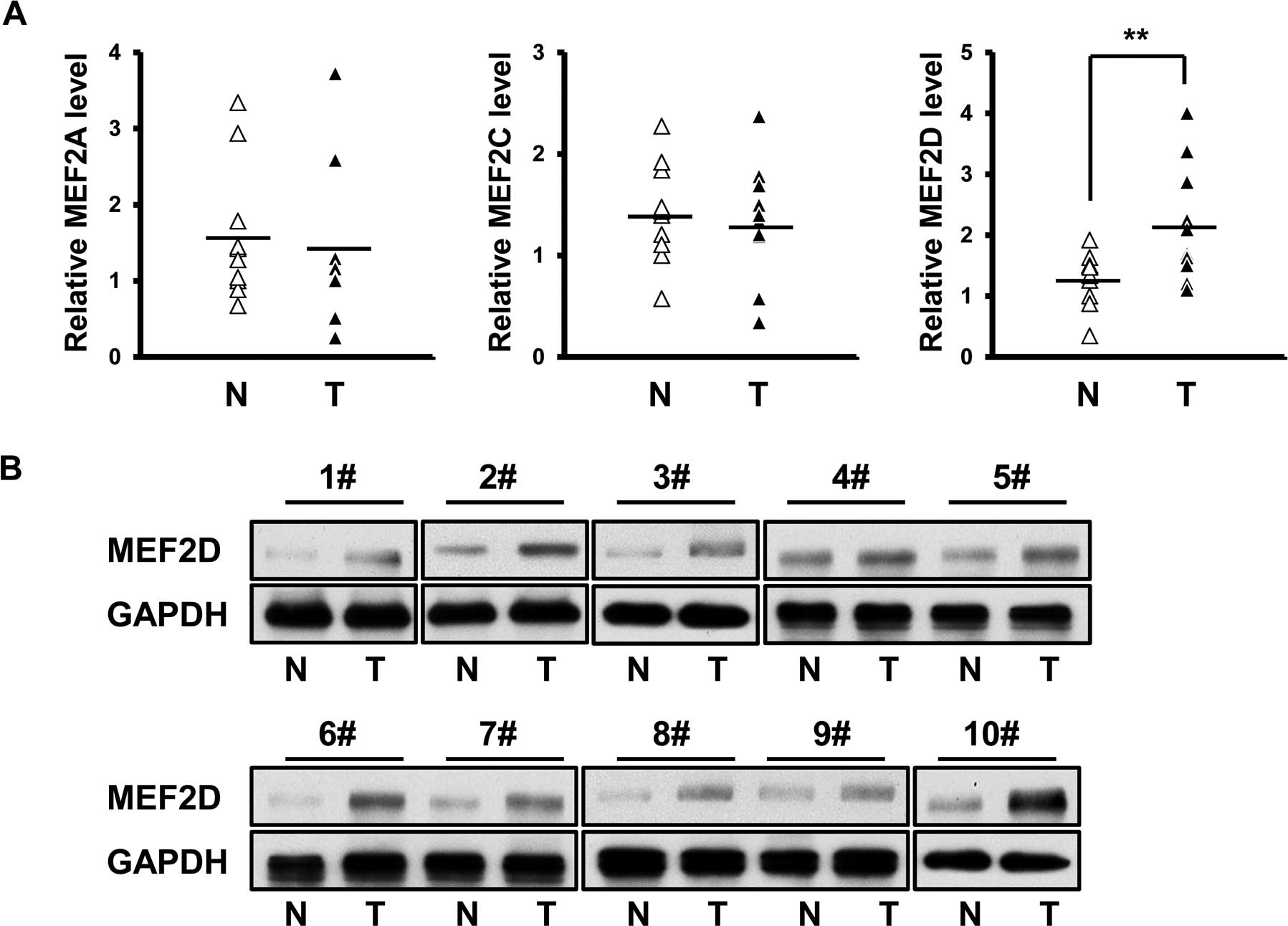
MEF2D is overexpressed in cardiac
myxoma
The expression profile of MEF2A, MEF2C and MEF2D
mRNA was investigated in fresh cardiac myxoma specimens and their
matched non-cancerous tissues (n=10). No significant difference was
detected in the mRNA level of MEF2A and MEF2C between the cancer
and normal tissues (Fig. 1A). In
contrast, MEF2D mRNA was found to be overexpressed in the cardiac
myxoma specimens (Fig. 1A).
Furthermore, immunoblot assay confirmed the differential expression
of MEF2D between the cancer and normal tissues (Fig. 1B).
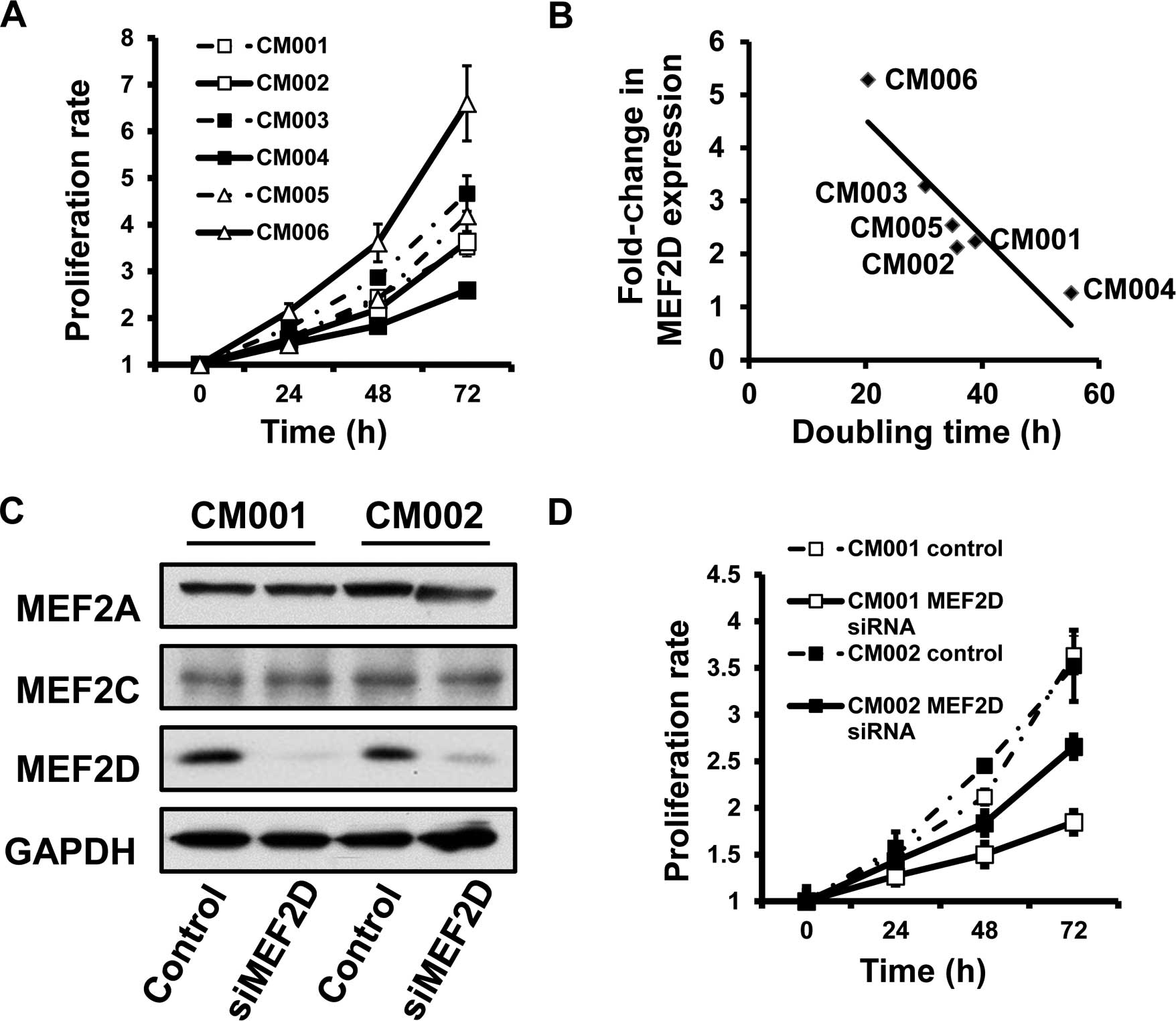
MEF2D expression is positively associated
with the proliferation of cardiac myxoma cells
Given the role of MEF2D in the proliferation of
hepatocellular carcinoma, we subsequently investigated whether
aberrant expression of MEF2D is also associated with the
proliferation of cardiac myxoma cells. MTT assays were employed to
determine the proliferation rate of the primary myxoma cells. The
data revealed that the levels of MEF2D protein were inversely
correlated with the doubling time of the myxoma cells (R=−0.943,
P=0.005) (Fig. 2A and B), revealing
that cancer cells with higher MEF2D expression proliferated more
rapidly.
MEF2D suppression leads to the reduction
in the proliferation rates of cardiac myxoma cells
We used small interfering RNA (siRNA) to
specifically silence MEF2D expression, followed by investigation of
the proliferation rate in myxoma cells. Our data showed that MEF2D
siRNA selectively inhibited MEF2D expression, yet not MEF2A and
MEF2C (Fig. 2C). We found that
MEF2D downregulation inhibited the growth of the tested primary
cardiac myxoma cells (Fig. 2D).
Collectively, MEF2D overexpression promoted the proliferation of
myxoma cells.
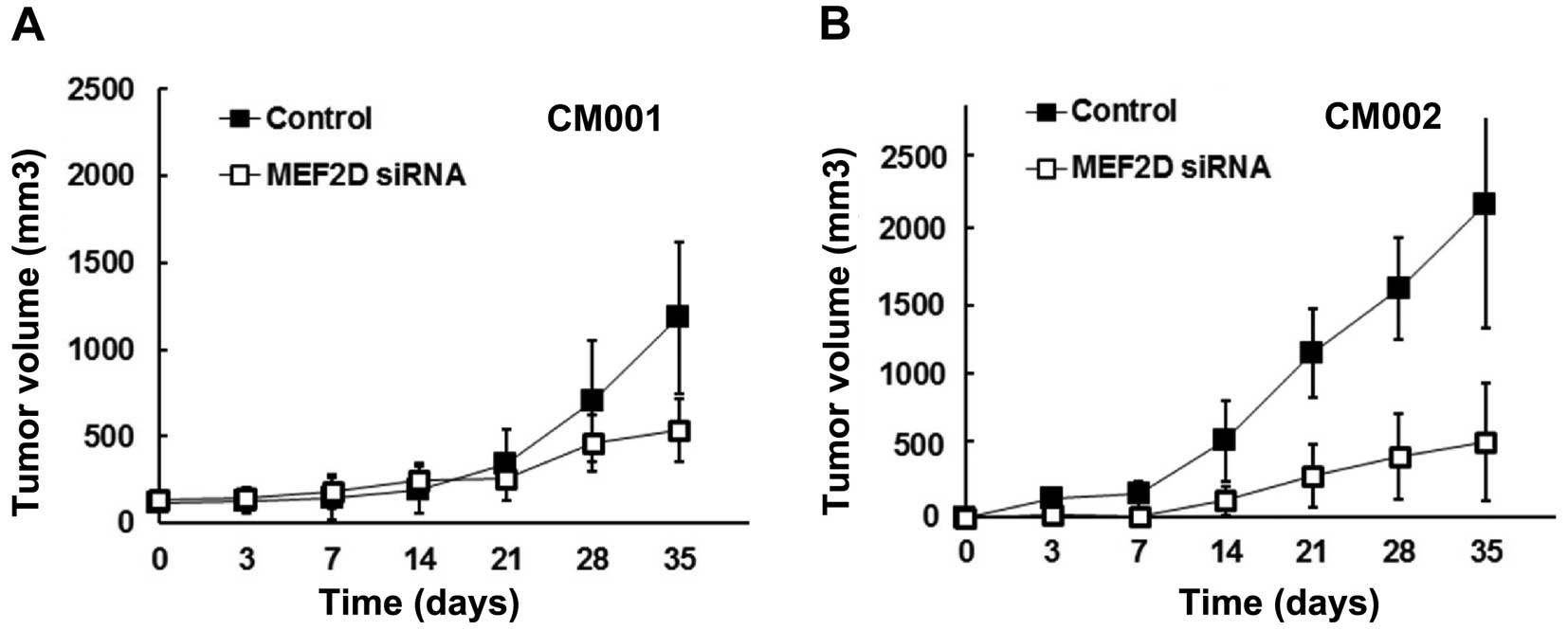
MEF2D downregulation impairs the
tumorigenicity of cardiac myxoma cells
The effect of MEF2D overexpression on the
tumorigenicity of myxoma cells was further studied in a mouse
model. The tumors from the MEF2D siRNA-treated CM001 and CM002
cells were found to grow slower than the control groups by 71 and
65%, respectively (Fig. 3A and
B).
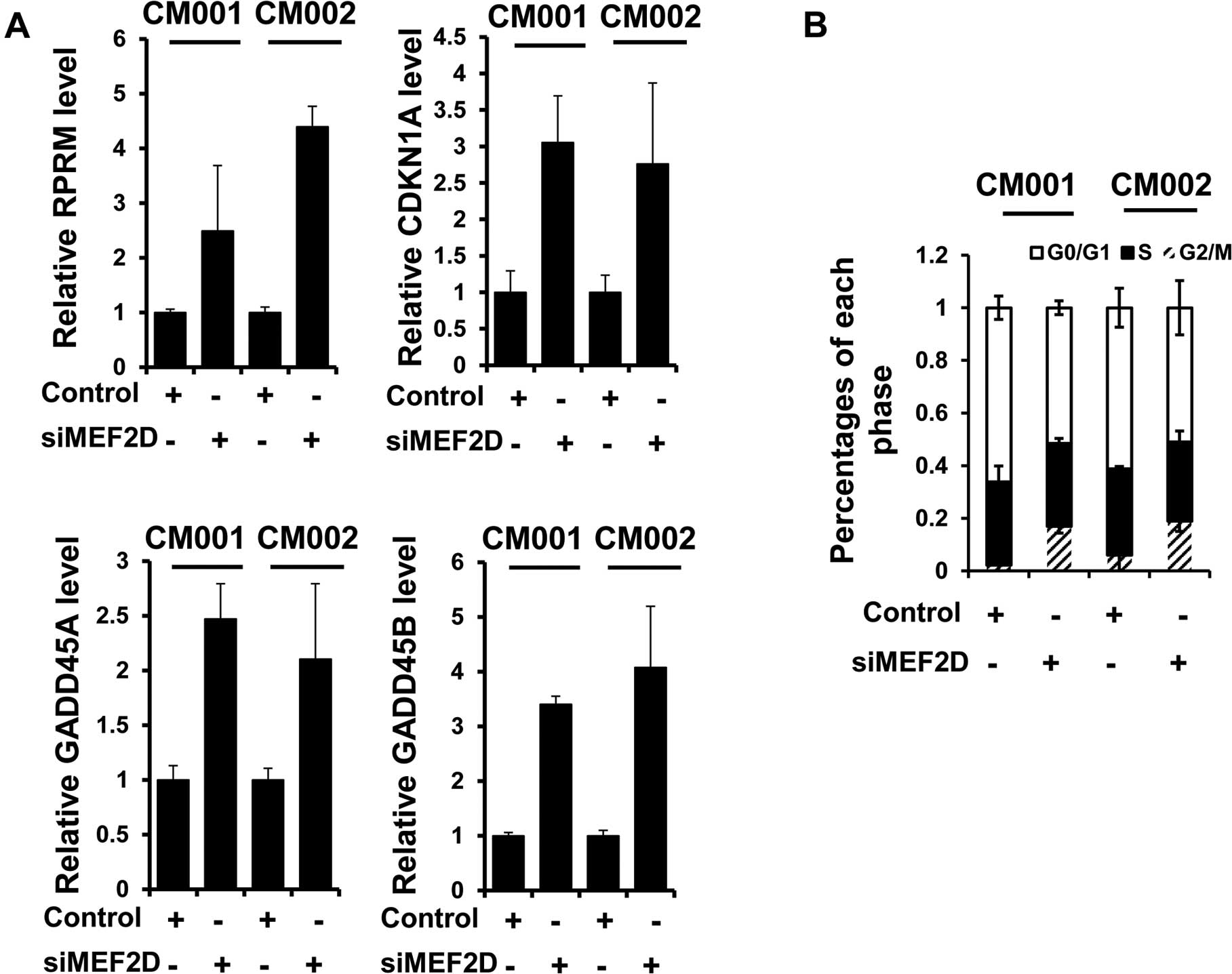
MEF2D facilitates cell cycle progression
in cardiac myxoma cells
To elucidate the mechanisms underlying the promotion
of proliferation and tumorigenesis of myxoma cells by MEF2D, we
examined the expression of known MEF2D targets, RPRM, GADD45A,
GADD45B and CDKN1A, following MEF2D siRNA treatment. Immunoblot
analysis revealed that the expression levels of the above mRNAs
were all increased when MEF2D expression was downregulated in the
myxoma cells (Fig. 4A).
Consistently, G2/M cell cycle arrest was also detected in both the
CM001 and CM002 cells treated with MEF2D siRNA, evidenced by flow
cytometric analysis (Fig. 4B).
These results indicated that MEF2D contributed to the proliferation
of myxoma cells by preventing G2/M cell cycle arrest.
miR-218 suppresses MEF2D expression in
myxoma cells by targeting the 3′UTR of its mRNA
The bioinformatic analysis showed that MEF2D is a
predictive target of miR-218 using an algorithm available online
(http://www.targetscan.org/) (Fig. 5A). Therefore, we employed luciferase
assays to confirm whether miR-218 negatively regulates the
expression of MEF2D by binding its putative recognition sites
within 3′UTR of MEF2D mRNA. Luciferase expression by pMIR-MEF2D-WT,
but not pMIR-MEF2D-MUT, was shown to be greatly reduced in the
myxoma cells treated with the miR-218 mimics (Fig. 5B). Furthermore, miR-218
overexpression was able to suppress the levels of endogenous MEF2D
proteins in the CM001 and CM002 cells (Fig. 5C), while miR-218 suppression
restored the expression of MEF2D in normal fibroblast MRC-5 cells
(Fig. 5D). The above data indicated
that MEF2D is an authentic miR-218 target.
miR-218 reduction promotes the
proliferation of myxoma cells
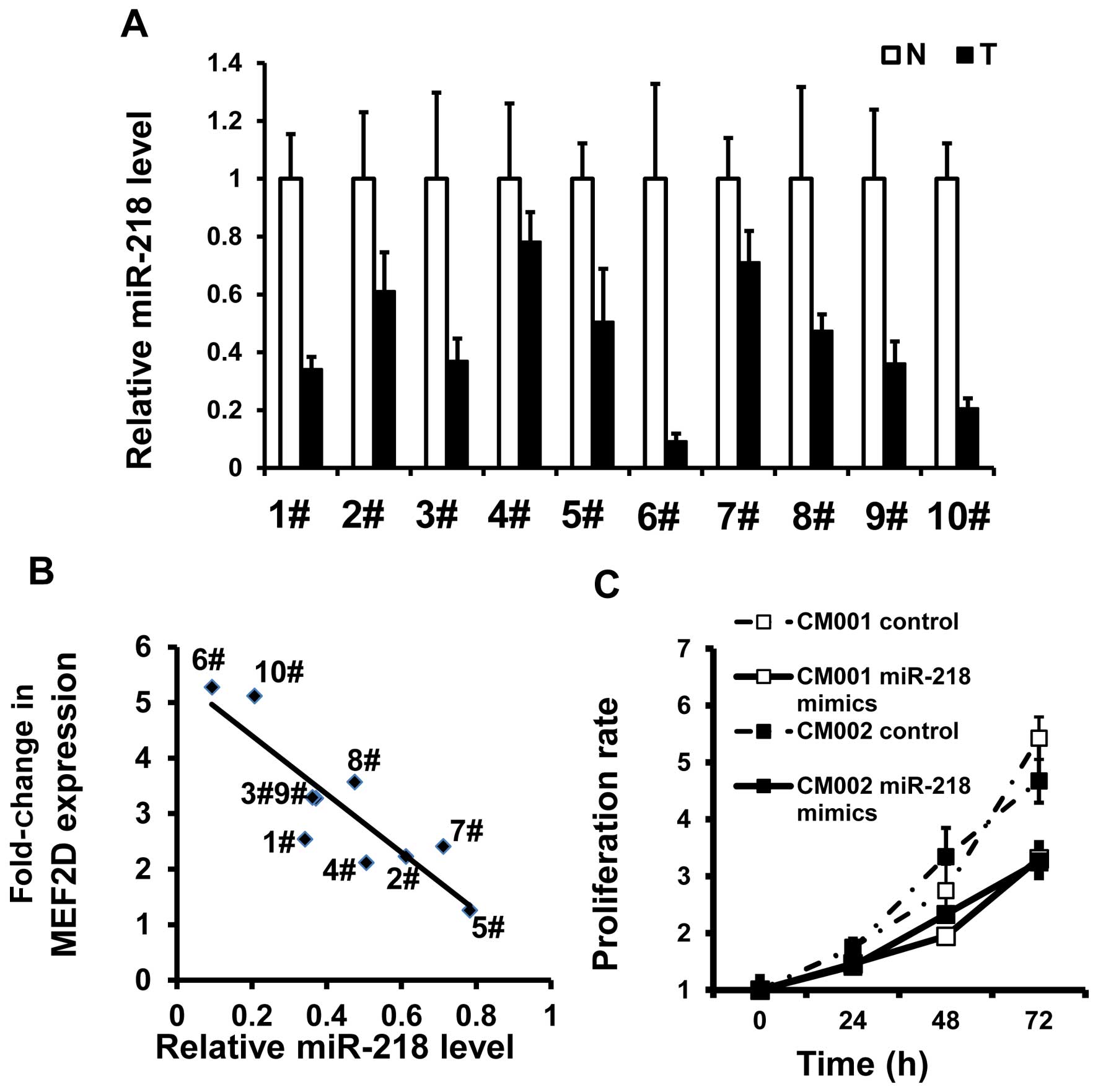
In the same tumor specimens, miR-218 was found to be
underexpressed (Fig. 6A). There was
an inverse association between MEF2D mRNA and miR-218 levels
(R=−0.881, P=0001) (Fig. 6B). MTT
assays also demonstrated that miR-218 restoration was able to
reduce the proliferation of the myxoma cells (Fig. 6C). These data indicate that
downregulation of miR-218 could be responsible for the elevated
expression of MEF2D and increased proliferation rates in the myxoma
cells.
Discussion
For the first time, the present study provides
evidence that development-associated regulators, MEF2 family
proteins, are implicated in cardiac myxoma. Specifically, MEF2D,
yet not other members, was shown to be differentially expressed in
the heart tumor tissues. Generally, the etiology of sporadic
cardiac myxomas remains elusive. Sauls et al reported that
valve interstitial cells regulate matrix organization during foetal
valve development through a serotonin, TG and filamin-A pathway.
Disrupting these key regulatory interactions can set the stage for
the generation of postnatal myxomatous valve disease (13). For familial (but not sporadic)
cardiac myxomas, germ-line mutations in the PRKAR1A gene were found
to be associated with this type of heart neoplasm (14,15).
However, the molecular mechanism of myxomagenesis is still far from
being completely elucidated.
The oncogenic role of MEF2D has been well
established in liver cancer and leukemia. The involved mechanism
appears to be primarily focused on the accelerated proliferation of
these cancerous cells (8,9). Our data further confirmed these
previous findings, since MEF2D suppression reduced cell
proliferation, impaired tumorigenicity, and induced cell cycle
arrest at the G2/M phase. Notably, MEF2D overexpression
was found to mediate stress-induced heart hypertrophy (10), implying this transcriptional factor
may also facilitate the proliferation of normal cardiac muscle
cells.
In addition, we confirmed that miR-218, a tumor
suppressor in many types of cancers (16–18),
suppressed the expression of MEF2D mRNA by targeting its 3′UTR. In
fact, miRNAs have been verified as major regulators of MEF2D
expression. miR-92 was shown to be overexpressed in hippocampal
neurons induced by fear and to downregulate the expression of MEF2D
(19). miR-122, a potent liver
cancer suppressor, was also identified as a negative regulator of
MEF2D expression (9).
Although miRNAs have been found to be closely
associated with the progression of various cancers, the link
between miRNAs and cardiac myxoma remains unexplored. Our study
provides evidence that miRNAs can also participate in the growth of
heart tumors, in line with their roles in other types of
tumors.
Collectively, the present study provides evidence
that MEF2D promotes the proliferation of human cardiac myxoma
cells. Our data also demonstrated that the miR-218/MEF2D pathway
may be an effective therapeutic target for patients with
myxoma.
References
|
1
|
Tatli S and Lipton MJ: CT for intracardiac
thrombi and tumors. Int J Cardiovasc Imaging. 21:115–131. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rubin P, Williams JP, Devesa SS, Travis LB
and Constine LS: Cancer genesis across the age spectrum:
Associations with tissue development, maintenance, and senescence.
Semin Radiat Oncol. 20:3–11. 2010. View Article : Google Scholar
|
|
3
|
Rana MS, Christoffels VM and Moorman AF: A
molecular and genetic outline of cardiac morphogenesis. Acta
Physiol. 207:588–615. 2013. View Article : Google Scholar
|
|
4
|
Edmondson DG, Lyons GE, Martin JF and
Olson EN: Mef2 gene expression marks the cardiac and skeletal
muscle lineages during mouse embryogenesis. Development.
120:1251–1263. 1994.PubMed/NCBI
|
|
5
|
Lin Q, Schwarz J, Bucana C and Olson EN:
Control of mouse cardiac morphogenesis and myogenesis by
transcription factor MEF2C. Science. 276:1404–1407. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guo Y, Kühl SJ, Pfister AS, Cizelsky W,
Denk S, Beer-Molz L and Kühl M: Comparative analysis reveals
distinct and overlapping functions of Mef2c and Mef2d during
cardiogenesis in Xenopus laevis. PLoS One. 9:e872942014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Prima V, Gore L, Caires A, Boomer T,
Yoshinari M, Imaizumi M, Varella-Garcia M and Hunger SP: Cloning
and functional characterization of MEF2D/DAZAP1 and DAZAP1/MEF2D
fusion proteins created by a variant t(1;19)(q23;p13.3) in acute
lymphoblastic leukemia. Leukemia. 19:806–813. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Prima V and Hunger SP: Cooperative
transformation by MEF2D/DAZAP1 and DAZAP1/MEF2D fusion proteins
generated by the variant t(1;19) in acute lymphoblastic leukemia.
Leukemia. 21:2470–2475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma L, Liu J, Liu L, et al: Overexpression
of the transcription factor MEF2D in hepatocellular carcinoma
sustains malignant character by suppressing G2-M
transition genes. Cancer Res. 74:1452–1462. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim Y, Phan D, van Rooij E, Wang DZ,
McAnally J, Qi X, Richardson JA, Hill JA, Bassel-Duby R and Olson
EN: The MEF2D transcription factor mediates stress-dependent
cardiac remodeling in mice. J Clin Invest. 118:124–132. 2008.
View Article : Google Scholar
|
|
11
|
Geng J, Zhao Z, Kang W, Wang W, Zhang Y
and Zhiming GE: Atorvastatin reverses cardiac remodeling possibly
through regulation of protein kinase D/myocyte enhancer factor 2D
activation in spontaneously hypertensive rats. Pharmacol Res.
61:40–47. 2010. View Article : Google Scholar
|
|
12
|
Negishi M, Sakamoto H, Sakamaki T,
Ishikawa O, Kanda T, Tamura J, Kurabayashi M and Nagai R:
Disaccharide analysis of glycosaminoglycans synthesized by cardiac
myxoma cells in tumor tissues and in cell culture. Life Sci.
73:849–856. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sauls K, de Vlaming A, Harris BS, et al:
Developmental basis for filamin-A-associated myxomatous mitral
valve disease. Cardiovasc Res. 96:109–119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mantovani G, Bondioni S, Corbetta S, et
al: Analysis of GNAS1 and PRKAR1A gene mutations in human cardiac
myxomas not associated with multiple endocrine disorders. J
Endocrinol Invest. 32:501–504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yin Z, Jones GN, Towns WH II, Zhang X,
Abel ED, Binkley PF, Jarjoura D and Kirschner LS: Heart-specific
ablation of Prkar1a causes failure of heart development and
myxomagenesis. Circulation. 117:1414–1422. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao X and Jin W: The emerging role of
tumor-suppressive microRNA-218 in targeting glioblastoma stemness.
Cancer Lett. 353:25–31. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wei Y, Du Y, Chen X, Li P, Wang Y, Zang W,
Zhao L, Li Z and Zhao G: Expression patterns of microRNA-218 and
its potential functions by targeting CIP2A and BMI1 genes in
melanoma. Tumour Biol. 35:8007–8015. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang C, Ge S, Hu C, Yang N and Zhang J:
miRNA-218, a new regulator of HMGB1, suppresses cell migration and
invasion in non-small cell lung cancer. Acta Biochim Biophys Sin.
45:1055–1061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vetere G, Barbato C, Pezzola S, Frisone P,
Aceti M, Ciotti M, Cogoni C, Ammassari-Teule M and Ruberti F:
Selective inhibition of miR-92 in hippocampal neurons alters
contextual fear memory. Hippocampus. 24:1458–1465. 2014. View Article : Google Scholar : PubMed/NCBI
|