Introduction
The proteasome is critical for the maintenance of
homeostasis of the majority of intracellular proteins and is
considered as a promising target for anticancer therapy. Bortezomib
is the first proteasome inhibitor used for the treatment of
relapsed or refractory multiple myeloma (1–3). In
vitro, the drug is active against a wide variety of tumor cell
types such as non-Hodgkin’s lymphoma, mantle cell lymphoma and
non-small cell lung cancer (4–7).
Bortezomib has an acceptable toxicity profile and has shown
clinical benefits, either alone or as part of a combination
therapy, in induction of chemo/radio-sensitization or by overcoming
drug resistance (6,8). It was been proven that bortezomib is
associated with apoptosis through upregulation of the pro-apoptotic
proteins and downregulation of the anti-apoptotic factors. The
latter includes the Bcl-2 subfamily proteins Bcl-xL, Bcl-2 and
Mcl-1 (9–12).
Mcl-1 is a short-lived protein which opposes several
apoptotic stimuli. These features distinguish it from other
anti-apoptotic Bcl-2 members (13).
Previous studies demonstrated that Mcl-1 is the essential survival
protein in multiple myeloma and can be accumulated as a result of
treatment with bortezomib (14).
Furthermore, RNA interference mediates Mcl-1 downregulation and
sensitizes human myeloma cell line to bortezomib, highlighting the
role of this protein in bortezomib-induced apoptosis (1).
Gliomas are the most common and highly invasive
primary brain tumors in humans. Despite some recent advances in
surgical, radiation and chemotherapy approaches, the prognosis
remains poor (15). Glioma cells
display extreme resistance to apoptotic stimuli which contributes
to the limited effectiveness of current therapies and the
difficulty in developing new efficient treatment regiments
(15,16). This resistance may be due to the
overexpression of anti-apoptotic Bcl-2-like proteins, such as
Mcl-1, Bcl-2 and Bcl-xL (16,17).
Bortezomib is successfully used for treating
hematological malignancies. Its reported benefits in the treatment
of solid tumors, however, are less than encouraging (18–22).
Resistance to bortezomib in solid tumors further stimulates
development of novel proteasome inhibitors, which would act
differently from bortezomib, as well as novel natural compounds
with proteasome-inhibitory activity that could be used as
chemo/radio-sensitizers. Current research efforts in solid tumors
are now focused on the use of bortezomib in combination with other
pro-apoptotic agents (1,8).
In the present study, we examined the effect of
Mcl-1 and/or the proteasome inhibitors on glioma cell growth and
survival. We aimed to assess the specific role of Mcl-1 in the
response to bortezomib.
Materials and methods
Cell lines and reagents
The glioblastoma cell lines U251 and U87 were
purchased from the Chinese Academy of Medical Sciences (CAMS) and
were grown as a monolayer in Dulbecco’s modified Eagle’s medium
(DMEM) with 10% heat-inactivated fetal calf serum, 100 U/ml
penicillin and 100 mg/ml streptomycin and were maintained in a
humidified incubator at 37°C and 5% CO2. Bortezomib was
obtained from Sigma (St. Louis, MO, USA). Antibodies against
caspase-3 (human mAb), PARP (F7 human mAb), cytochrome c
(human polyclonal), and Mcl-1 (S19, human mAb) were from Santa Cruz
Biotechnology (Santa Cruz, CA, USA).
MTT cell viability assay
U87 and U251 cells were plated on 96-well plates
(2,000 cells/well) and treated with bortezomib (100 ng/ml; Sigma)
for 24 h. Following the treatment, thiazolyl blue tetrazolium
bromide (MTT, 5 mg/ml; Sigma) was added to each well, and the cells
were incubated in the dark at 37°C. MTT produces a yellowish
solution that is converted to dark blue, water-insoluble MTT
formazan by mitochondrial dehydrogenases of the living cells. After
4 h, the medium was aspirated and the dark blue crystals were
dissolved in dimethyl sulfoxide (DMSO) (150 μl) and
incubated for 10 min. The absorbance of each sample was measured at
490 or 570 nm using a microplate reader (GENios; Tecan Schweiz AG,
Männedorf, Switzerland). The absorbance was proportional to the
number of the viable cells and was expressed relative to the
untreated negative control cultures.
Western blot analysis
Western blot analysis was performed on lysates
prepared from U251 and U87 cells treated as indicated. The cells
were homogenized in the lysis buffer containing 0.5 mM Tris-Cl (pH
6.8), 2% SDS (w/v), 10% glycerin (w/v) and protease inhibitor
cocktails (Sigma-Aldrich, Dublin, Ireland). After determining the
protein concentration of the samples using the BCA protein assay
(Pierce, Rockford, IL, USA), 20 μg samples were boiled in
gel-loading buffer and separated on 10–15% SDS-PAGE. Proteins were
transferred to the nitrocellulose membranes and analyzed following
the standard procedures. The membranes were incubated with
anti-human Mcl-1, anti-caspase-3 antibodies and anti-human PARP,
anti-human cytochrome c (Sigma) overnight at 4°C. The
membranes were then incubated with horseradish
peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch
Laboratories, Plymouth, PA, USA), and protein bands were visualized
using enhanced chemiluminescence detection (Pierce). Images were
captured using Fujifilm LAS-3000 (Fujifilm, Sheffield, UK).
Flow cytometry and analysis of apoptosis
via Annexin V/propidium iodide staining
U251 and U87 cells were plated in 24-well plates
(15,000 cells/well) and were treated with bortezomib for 48 h.
Following the treatment, the monolayer cells were harvested with
trypsin-EDTA and washed with PBS. Cells were then incubated at room
temperature in binding buffer (10 mM HEPES, 135 mM NaCl, 5 mM
CaCl2) which contained Annexin V-FITC conjugate (1
μl/ml; BioVision, Mountain View, CA, USA) and propidium
iodide (PI; 1 μl/ml; BioVision) for 15 min. Excitation of
Annexin V-FITC was done with a 488 nm laser, and fluorescence
emission was collected in the FL1 channel through a 520-nm band
pass filter. PI was excited with a 488-nm laser and fluorescence
emission was collected in the FL3 channel through a 620-nm long
pass filter. Gated cells (1×104) were acquired for each
sample, and flow cytometric analysis was done on a FACSCalibur flow
cytometer (BD Biosciences, San Jose, CA, USA) using the CellQuest
software (BD Biosciences). Each analysis was performed using at
least 10,000 events.
Mcl-1 RNA interference assay
The sequence of the small interfering RNA (siRNA)
used to silence Mcl-1 gene (Mcl-1 siRNA) was
5′-GUAAUUAUUGACACAUUUCUU-3′. Both siRNA and negative control siRNA
were synthesized by Ambion (Life Technologies, Carlsbad, CA, USA).
U87 and U251 cells were electroporated using Amaxa Nucleofector
system (Lonza, Walkersville, MD, USA) according to the
manufacturer’s instructions. Briefly, 5×106 U87 and U251
cells were re-suspended in 100 μl of R nucleofector solution
containing 10 μM siRNA and electroporated using T01
nucleofector program. Cells were transferred to culture plates and
cultured at 5×105 cells/ml for 2 h before incubation
with bortezomib.
Statistical analysis
All data are given as the mean ± SD. N is the number
of the samples. Western blot data are expressed relative to the
control, assigning a value of 1 to the control group baseline mean.
Data were analyzed using Student’s t-test or 2-way ANOVA by SPSS
17.0 software as appropriate. P-value <0.05 was considered as a
significant difference between the data sets.
Results
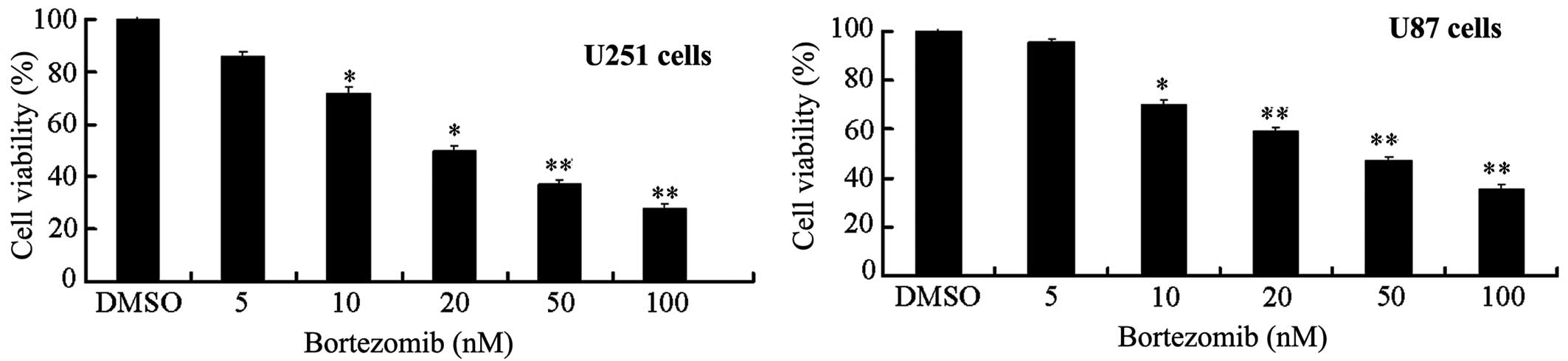
Bortezomib can reduce the viability of
U251 and U87 glioma cells in a dose-dependent manner
Previous research has shown that proteasome
inhibitor bortezomib can cause apoptosis in vitro in a
variety of tumor cells, such as prostate, breast and colon cancer
(4–7). Therefore, we first used the MTT method
to determine the effects of bortezomib on the glioma cell
viability. We chose two common glioma cell lines, U251 and U87, for
our experiments. Cells were cultured with or without various doses
of bortezomib for 24 h. Bortezomib significantly reduced the
survival rate of U251 and U87 cells in a dose-dependent manner
(Fig. 1). The median inhibitory
concentration (IC50) of bortezomib for U251 and U87
cells was 25.52 and 29.37 nM (n=6/cell line).
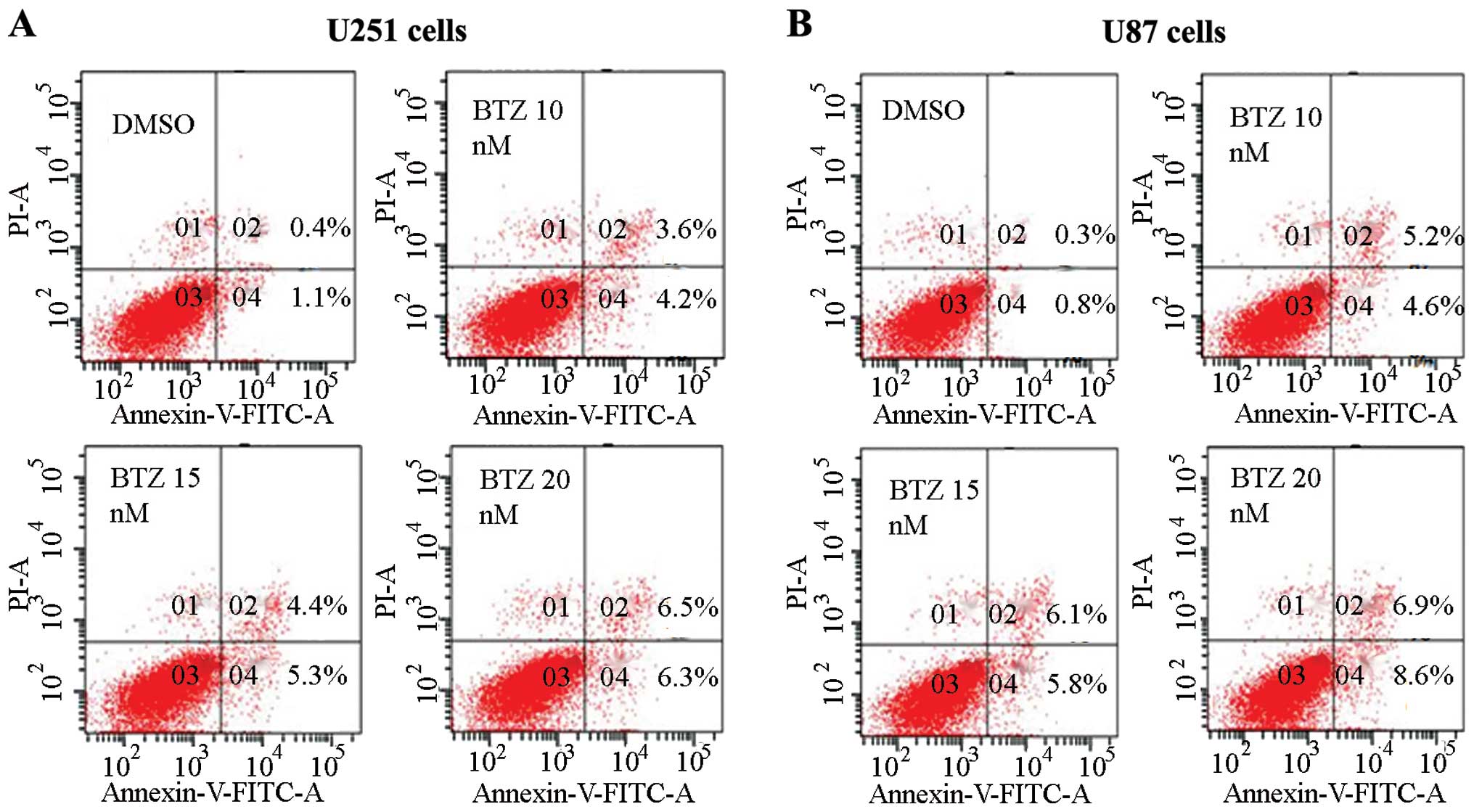
Bortezomib induces apoptosis in U251 and
U87 glioma cells
Since the MTT results showed that bortezomib can
reduce the U251 and U87 cell viability, we investigated the effect
of bortezomib on apoptosis in glioma cells. We recorded the
influence of different bortezomib concentration on the apoptosis by
Annexin V and PI double dye staining using flow cytometry.
Different concentrations of bortezomib can cause apoptosis of U251
and U87 cells (Fig. 2). Bortezomib
at concentration of 10–20 nM can induce apoptosis effectively in
U87 and U251 cells, as established mainly through the detection of
apoptosis-related proteins caspase-3 and PARP by western blot
analysis.
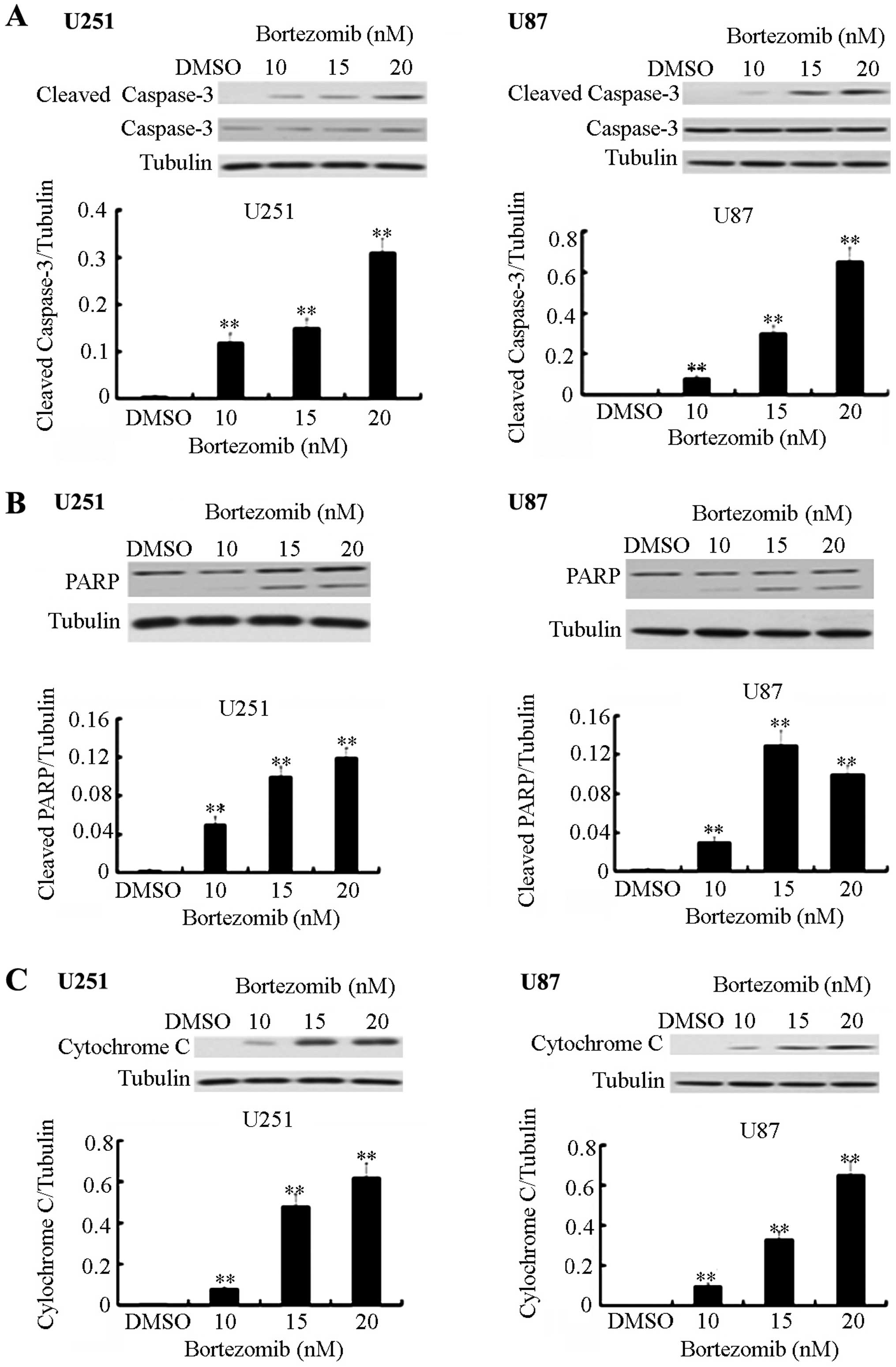
Bortezomib triggers dose-dependent
cleavage and activation of caspase-3, PARP activation and
upregulation of the cytochrome c expression in U251 and U87 glioma
cells
Several members of the Bcl-2 family are known to be
targets for bortezomib. In order to further determine the effect of
bortezomib on the apoptosis in the two glioma cell lines, we
checked the expression levels of apoptosis-related proteins by
using western blot analysis. Under normal circumstances, caspase-3
can exist in an inactive form. PARP, as a DNA repair enzyme, is
also the substrate of caspase. After being cleaved, the activated
form of caspase-3 mediates cell apoptosis. Cytochrome c is
an important component in the mitochondrial apoptosis signaling
pathways. Following the change of the mitochondrial membrane
permeability, cytochrome c which is located between the
mitochondrial membrane and the outer membrane can be released into
the cytoplasm and can trigger the apoptosis cascade. Bortezomib can
significantly increase the level of the cleaved form of caspase-3,
PARP and cytochrome c expression in a dose-dependent manner
(Fig. 3).
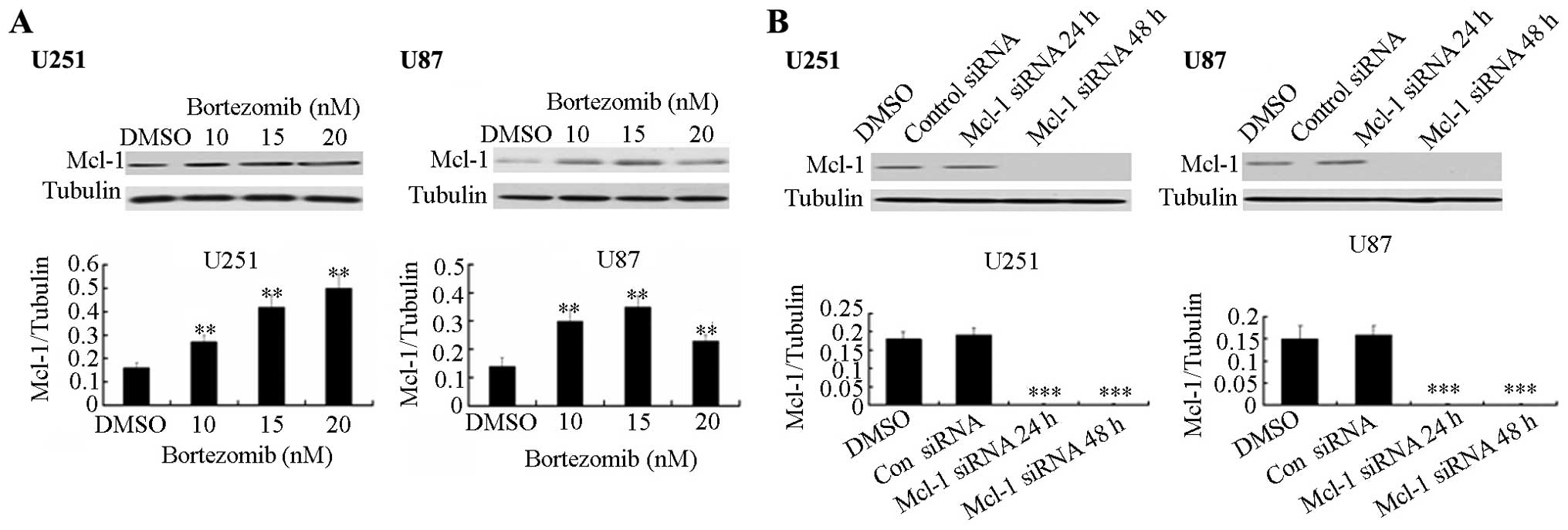
Bortezomib upregulates Mcl-1 while Mcl-1
siRNA down-regulates the Mcl-1 expression
Studies have shown that anti-apoptotic protein Mcl-1
is degraded by the ubiquitin-proteasome pathway. As a proteasome
inhibitor, bortezomib can induce apoptosis in glioma cells. Using
western blot analysis, we explored the effect of Mcl-1 on the
apoptosis induced by bortezomib in two types of glioma cells, U251
and U87. Bortezomib increases the protein expression level of Mcl-1
in both cell lines (Fig. 4A).
Furthermore, using siRNA technology, we checked the expression
levels of Mcl-1 after siRNA transfection. As shown in Fig. 4B, 24 and 48 h after transfection of
Mcl-1 siRNA plasmids, the Mcl-1 siRNA clearly inhibits the Mcl-1
expression in U251 and U87 cells.
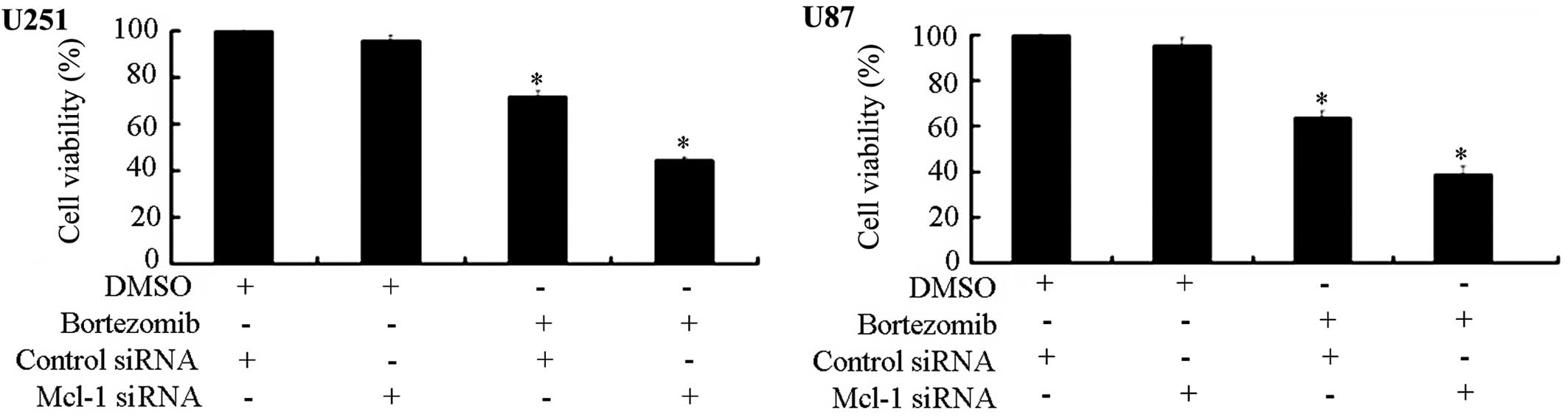
Knocking down Mcl-1 expression increases
glioma cell sensitivity to the inhibition of cell growth by
bortezomib
To definitely establish the role of Mcl-1 in the
bortezomib-induced apoptosis in glioma cells, we made use of RNA
interference (RNAi) technology. We transfected U87 and U251 cells
with Mcl-1 siRNA and control siRNA to evaluate the potential
effects on the cell viability due to Mcl-1 down-regulation. The MTT
assay demonstrated that the combined treatment with bortezomib and
Mcl-1 siRNA can significantly inhibit U251 and U87 cell viability.
The inhibition of Mcl-1 expression can increase the sensitivity of
both cell types to bortezomib (Fig.
5).
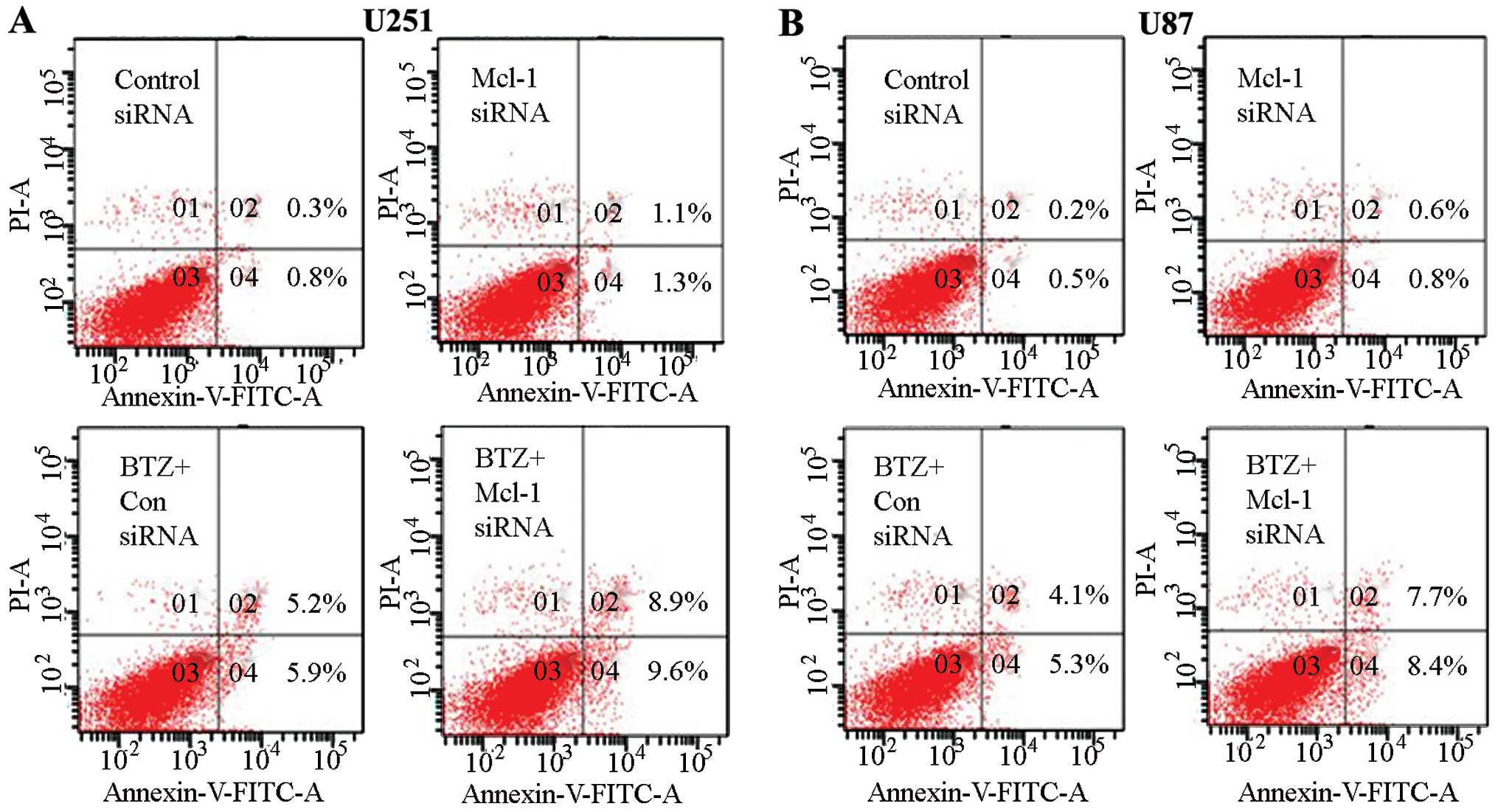
Knockdown of Mcl-1 augments apoptosis
induced by bortezomib in U251 and U87 glioma cells
Based on previous experimental results which
demonstrated that Mcl-1 siRNA can increase the inhibition of cell
growth, we used siRNA to knock down Mcl-1 expression in order to
determine whether Mcl-1 played an essential role in
bortezomib-induced apoptosis. Using flow cytometry, we observed
that Mcl-1 siRNA can significantly enhance bortezomib-induced
apoptosis compared with the control siRNA in U251 and U87 glioma
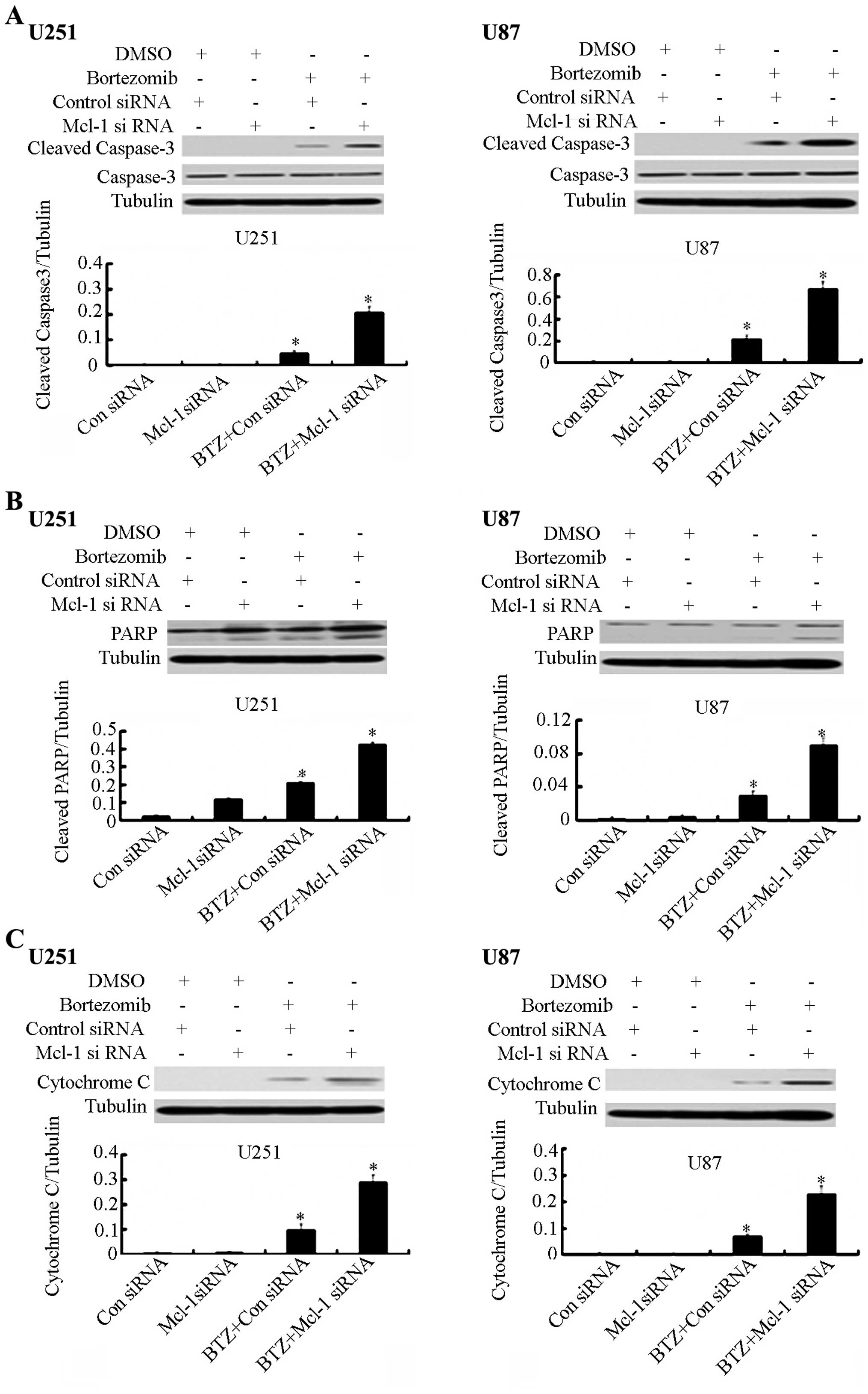
cells (Fig. 6). We also checked the
protein expression level of caspase-3 and PARP by western blot
analysis. We found that Mcl-1 siRNA in combination with bortezomib
can significantly increase the expression of caspase-3 and PARP
(Fig. 7A and B). Furthermore, the
expression level of cytochrome c was increased after the
Mcl-1 siRNA treatment (Fig.
7C).
Discussion
The ubiquitin-proteasome pathway represents the
major pathway for intracellular protein degradation (23,24).
The 26S proteasome is responsible for the degradation of ~80% of
all cellular proteins and plays an important role in the regulation
of cell growth and survival (25–27).
Inhibition of the proteasomal function in cancer cells would
promote apoptosis and has been shown to induce pro-apoptotic ER
stress in multiple myeloma (1),
pancreatic (21), prostate cancer
(18) and non-small cell lung
carcinoma (7). Therefore,
proteasome inhibitors represent a novel class of compounds with
promising antitumor activity and potential of becoming a new
important tool for the treatment of cancer. Bortezomib, the 26S
proteasome inhibitor, is the first proteasome inhibitor to enter
clinical trials (2), and has
demonstrated encouraging efficacy against multiple myeloma, where
it has become an approved therapy. In addition, bortezomib often
induces apoptosis on its own or when used together with other novel
therapeutic or chemotherapeutic agents (6–8). After
the major success achieved with bortezomib in hematological
malignancies, its effect on solid tumors was also investigated, but
the results reported so far were significantly less impressive
(28). In the present study, we
provided novel information on the cellular effects of bortezomib
and Mcl-1 in glioma cell lines.
Bortezomib has been observed to target many
molecules that are associated with tumor progression and treatment
resistance, such as nuclear factor-κB (NF-κB), pro-apoptotic and
anti-apoptotic Bcl-2 family proteins and p53 (1,3). One
major mechanism of bortezomib associated with its anticancer
activity has been attributed to the inhibition of IκB degradation,
which leads to the suppression of the NF-κB signaling pathway and
results in the downregulation of its anti-apoptotic target genes
(3,29). For example, bortezomib can decrease
the increment rate of multiple myeloma cells more than twice by
reducing the activity of NF-κB compared with chemotherapy-sensitive
cells. Bortezomib can enhance radiation sensitivity in colon cancer
cells when NF-κB activation is inhibited. Another important
mechanism of this drug involves upregulation of Noxa, a
pro-apoptotic protein which may interact with the anti-apoptotic
proteins of the Bcl-2 subfamily, Bcl-xL and Bcl-2 (1,30).
Bortezomib-mediated apoptosis is accompanied by induction of c-Jun
N-terminal kinase, generation of reactive oxygen species, release
of cytochrome c (the second mitochondria-derived activator
of caspases and apoptosis-inducing factor), and activation of the
intrinsic caspase-9 and extrinsic caspase-8 pathways. In non-small
cell lung cancer H460 cells, bortezomib can trigger cell arrest in
the G2-M phase and thus initiate apoptosis (31). Cell cycle arrest can increase the
phosphorylation and degradation of Bcl-2 and the accumulation of
cyclins A and B1 (32). It is very
important to further explore the different mechanisms of bortezomib
in the induction of apoptosis in different tumor cells.
The pivotal role Mcl-1 plays in protecting cancer
cells from apoptosis is well documented (33,34).
In glioma cells, the downregulation of Mcl-1 through the use of
siRNA has been proven to significantly decrease cell proliferation
and increase apoptosis (35). Since
upregulation of anti-apoptotic factors by bortezomib could
negatively affect its antitumor effect (4), we explored the implication of using
the RNAi technology on Mcl-1 level in glioma cells. Initially, we
observed Mcl-1 downregulation following Mcl-1 siRNA treatment.
However, this downregulation was not sufficient to induce a
significant level of apoptosis due, in part, to lack of caspase-3
activation in these cells. Compared with the limited effects
observed with the single agent therapy, addition of bortezomib
together with Mcl-1 inhibitor triggered the release of the
pro-apoptotic protein cytochrome c into the cytosolic
fraction and accumulation of increased levels of cleaved caspases
and PARP, hallmarks of apoptosis (36). We have previously shown that Mcl-1
downregulation is important to make multiple myeloma cells
susceptible to BH3-only proteins and therefore to mitochondrial
disruption (37,38). As a result of Mcl-1 downregulation
following siRNA treatment, BH3-only proteins may block the
anti-apoptotic effect of the members of the Bcl-2 superfamily
(Bcl-2, Bcl-xL or Mcl-1), and/or induce Bax/Bak conformational
change. Activated Bax and Bak form pores in the outer mitochondrial
membrane, resulting in the release of cytochrome c and
activators of caspases from the intermembrane space into the
cytosol (37–41). Noxa is capable of displacing the
activator BH3-only protein, Bim, and the pro-apoptotic protein,
Bak, from Mcl-1/Bim and Mcl-1/Bak complexes, respectively, thereby
triggering mitochondrial dysfunction, caspase activation and,
ultimately, apoptosis (42,43). Recently, Han et al (40) have demonstrated that Mcl-1 is a
direct substrate for caspase-8, and that loss of Mcl-1 generates
Mcl-1-free Bim that is involved in the apoptotic events. This
observation suggests the potential existence of novel cross-talk
mechanism between the extrinsic and the intrinsic apoptotic
pathways in the initiation of apoptosis.
Collectively, the present study demonstrates the
efficient induction of apoptosis by proteasome inhibitor bortezomib
in the glioma cell lines and provides novel evidence that a
combinational therapeutic treatment regime modulating the Mcl-1
expression levels may be an efficacious approach to sensitize
glioma to the apoptosis-inducing effects of bortezomib. Bortezomib
can inhibit the expression of Mcl-1 and increase the bortezomib
sensitivity in the glioma U251 and U87 cells. The combined use of
bortezomib and Mcl-1 inhibitor can effectively inhibit the
proliferation of glioma cells. Elucidation of the pathways of cell
death induced by Mcl-1 downregulation in combination with
bortezomib treatment may help to identify key molecular targets
that govern the viability of glioma cells. These targets could be
further manipulated to provide antitumor effects and warrant
further investigation.
References
|
1
|
Gomez-Bougie P, Wuillème-Toumi S, Ménoret
E, Trichet V, Robillard N, Philippe M, Bataille R and Amiot M: Noxa
up-regulation and Mcl-1 cleavage are associated to apoptosis
induction by bortezomib in multiple myeloma. Cancer Res.
67:5418–5424. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen D, Frezza M, Schmitt S, Kanwar J and
Dou QP: Bortezomib as the first proteasome inhibitor anticancer
drug: Current status and future perspectives. Curr Cancer Drug
Targets. 11:239–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lesinski GB, Raig ET, Guenterberg K, Brown
L, Go MR, Shah NN, Lewis A, Quimper M, Hade E, Young G, et al:
IFN-alpha and bortezomib overcome Bcl-2 and Mcl-1 overexpression in
melanoma cells by stimulating the extrinsic pathway of apoptosis.
Cancer Res. 68:8351–8360. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kane RC, Dagher R, Farrell A, Ko CW,
Sridhara R, Justice R and Pazdur R: Bortezomib for the treatment of
mantle cell lymphoma. Clin Cancer Res. 13:5291–5294. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Orlowski RZ, Eswara JR, Lafond-Walker A,
Grever MR, Orlowski M and Dang CV: Tumor growth inhibition induced
in a murine model of human Burkitt’s lymphoma by a proteasome
inhibitor. Cancer Res. 58:4342–4348. 1998.PubMed/NCBI
|
|
6
|
Fisher RI, Bernstein SH, Kahl BS,
Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard
JP, Lonial S, et al: Multicenter phase II study of bortezomib in
patients with relapsed or refractory mantle cell lymphoma. J Clin
Oncol. 24:4867–4874. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fanucchi MP, Fossella FV, Belt R, Natale
R, Fidias P, Carbone DP, Govindan R, Raez LE, Robert F, Ribeiro M,
et al: Randomized phase II study of bortezomib alone and bortezomib
in combination with docetaxel in previously treated advanced
non-small-cell lung cancer. J Clin Oncol. 24:5025–5033. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Berenson JR, Yang HH, Sadler K,
Jarutirasarn SG, Vescio RA, Mapes R, Purner M, Lee SP, Wilson J,
Morrison B, et al: Phase I/II trial assessing bortezomib and
melphalan combination therapy for the treatment of patients with
relapsed or refractory multiple myeloma. J Clin Oncol. 24:937–944.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Strauss SJ, Higginbottom K, Juliger S,
Maharaj L, Allen P, Schenkein D, Lister TA and Joel SP: The
proteasome inhibitor bortezomib acts independently of p53 and
induces cell death via apoptosis and mitotic catastrophe in B-cell
lymphoma cell lines. Cancer Res. 67:2783–2790. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fribley A and Wang CY: Proteasome
inhibitor induces apoptosis through induction of endoplasmic
reticulum stress. Cancer Biol Ther. 5:745–748. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lioni M, Noma K, Snyder A, Klein-Szanto A,
Diehl JA, Rustgi AK, Herlyn M and Smalley KS: Bortezomib induces
apoptosis in esophageal squamous cell carcinoma cells through
activation of the p38 mitogen-activated protein kinase pathway.
Cancer Ther. 7:2866–2875. 2008. View Article : Google Scholar
|
|
12
|
Zhang L, Lopez H, George NM, Liu X, Pang X
and Luo X: Selective involvement of BH3-only proteins and
differential targets of Noxa in diverse apoptotic pathways. Cell
Death Differ. 18:864–873. 2011. View Article : Google Scholar :
|
|
13
|
Kozopas KM, Yang T, Buchan HL, Zhou P and
Craig RW: MCL1, a gene expressed in programmed myeloid cell
differentiation, has sequence similarity to BCL2. Proc Natl Acad
Sci USA. 90:3516–3520. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang B, Gojo I and Fenton RG: Myeloid
cell factor-1 is a critical survival factor for multiple myeloma.
Blood. 99:1885–1893. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eramo A, Ricci-Vitiani L, Zeuner A,
Pallini R, Lotti F, Sette G, Pilozzi E, Larocca LM, Peschle C and
De Maria R: Chemotherapy resistance of glioblastoma stem cells.
Cell Death Differ. 13:1238–1241. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Strik H, Deininger M, Streffer J, Grote E,
Wickboldt J, Dichgans J, Weller M and Meyermann R: BCL-2 family
protein expression in initial and recurrent glioblastomas:
modulation by radiochemotherapy. J Neurol Neurosurg Psychiatry.
67:763–768. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stegh AH, Kim H, Bachoo RM, Forloney KL,
Zhang J, Schulze H, Park K, Hannon GJ, Yuan J, Louis DN, et al:
Bcl2L12 inhibits post-mitochondrial apoptosis signaling in
glioblastoma. Genes Dev. 2:98–111. 2007. View Article : Google Scholar
|
|
18
|
Papandreou CN, Daliani DD, Nix D, Yang H,
Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, et al:
Phase I trial of the proteasome inhibitor bortezomib in patients
with advanced solid tumors with observations in
androgen-independent prostate cancer. J Clin Oncol. 22:2108–2121.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Frankel A, Man S, Elliott P, Adams J and
Kerbel RS: Lack of multicellular drug resistance observed in human
ovarian and prostate carcinoma treated with the proteasome
inhibitor PS-341. Clin Cancer Res. 6:3719–3728. 2000.PubMed/NCBI
|
|
20
|
Sunwoo JB, Chen Z, Dong G, Yeh N, Crowl
Bancroft C, Sausville E, Adams J, Elliott P and Van Waes C: Novel
proteasome inhibitor PS-341 inhibits activation of nuclear
factor-kappa B, cell survival, tumor growth, and angiogenesis in
squamous cell carcinoma. Clin Cancer Res. 7:1419–1428.
2001.PubMed/NCBI
|
|
21
|
Nawrocki ST, Bruns CJ, Harbison MT, Bold
RJ, Gotsch BS, Abbruzzese JL, Elliott P, Adams J and McConkey DJ:
Effects of the proteasome inhibitor PS-341 on apoptosis and
angiogenesis in orthotopic human pancreatic tumor xenografts. Mol
Cancer Ther. 1:1243–1253. 2002.
|
|
22
|
Kukreja A, Hutchinson A, Mazumder A, et
al: Bortezomib disrupts tumour-dendritic cell interactions in
myeloma and lymphoma: therapeutic implications. Br J Haematol.
136:106–110. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Adams J: The proteasome: a suitable
antineoplastic target. Nat Rev Cancer. 4:349–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ciechanover A: The ubiquitin-proteasome
proteolytic pathway. Cell. 79:13–21. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hochstrasser M: Ubiquitin, proteasomes,
and the regulation of intracellular protein degradation. Curr Opin
Cell Biol. 7:215–223. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ciechanover A: The ubiquitin-proteasome
pathway: On protein death and cell life. EMBO J. 17:7151–7160.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peters JM, Cejka Z, Harris JR,
Kleinschmidt JA and Baumeister W: Structural features of the 26 S
proteasome complex. J Mol Biol. 234:932–937. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Engel RH, Brown JA, Von Roenn JH, O’Regan
RM, Bergan R, Badve S, Rademaker A and Gradishar WJ: A phase II
study of single agent bortezomib in patients with metastatic breast
cancer: a single institution experience. Cancer Invest. 25:733–737.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Berenson JR, Ma HM and Vescio R: The role
of nuclear factor-kappaB in the biology and treatment of multiple
myeloma. Semin Oncol. 28:626–633. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qin JZ, Ziffra J, Stennett L, Bodner B,
Bonish BK, Chaturvedi V, Bennett F, Pollock PM, Trent JM, Hendrix
MJ, et al: Proteasome inhibitors trigger NOXA-mediated apoptosis in
melanoma and myeloma cells. Cancer Res. 65:6282–6293. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Domina AM, Smith JH and Craig RW: Myeloid
cell leukemia 1 is phosphorylated through two distinct pathways,
one associated with extracellular signal-regulated kinase
activation and the other with G2/M accumulation or protein
phosphatase 1/2A inhibition. J Biol Chem. 275:21688–21694. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ma MH, Yang HH, Parker K, Manyak S,
Friedman JM, Altamirano C, Wu ZQ, Borad MJ, Frantzen M, Roussos E,
et al: The proteasome inhibitor PS-341 markedly enhances
sensitivity of multiple myeloma tumor cells to chemotherapeutic
agents. Clin Cancer Res. 9:1136–1144. 2003.PubMed/NCBI
|
|
33
|
Quinn BA, Dash R, Azab B, Sarkar S, Das
SK, Kumar S, Oyesanya RA, Dasgupta S, Dent P, Grant S, et al:
Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig
Drugs. 20:1397–1411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Le Gouill S, Podar K, Harousseau JL and
Anderson KC: Mcl-1 regulation and its role in multiple myeloma.
Cell Cycle. 3:1259–1262. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Derenne S, Monia B, Dean NM, Taylor JK,
Rapp MJ, Harousseau JL, Bataille R and Amiot M: Antisense strategy
shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential
survival protein of human myeloma cells. Blood. 100:194–199. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Clohessy JG, Zhuang J, de Boer J,
Gil-Gómez G and Brady HJ: Mcl-1 interacts with truncated Bid and
inhibits its induction of cytochrome c release and its role in
receptor-mediated apoptosis. J Biol Chem. 281:5750–5759. 2006.
View Article : Google Scholar
|
|
37
|
Craig RW: MCL1 provides a window on the
role of the BCL2 family in cell proliferation, differentiation and
tumorigenesis. Leukemia. 16:444–454. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Willis SN, Chen L, Dewson G, Wei A, Naik
E, Fletcher JI, Adams JM and Huang DC: Proapoptotic Bak is
sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by
BH3-only proteins. Genes Dev. 19:1294–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen L, Willis SN, Wei A, Smith BJ,
Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM and Huang DC:
Differential targeting of prosurvival Bcl-2 proteins by their
BH3-only ligands allows complementary apoptotic function. Mol Cell.
17:393–403. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Han J, Goldstein LA, Gastman BR and
Rabinowich H: Interrelated roles for Mcl-1 and BIM in regulation of
TRAIL-mediated mitochondrial apoptosis. J Biol Chem.
281:10153–10163. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gomez-Bougie P, Bataille R and Amiot M:
The imbalance between Bim and Mcl-1 expression controls the
survival of human myeloma cells. Eur J Immunol. 34:3156–3164. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Leu JI, Dumont P, Hafey M, Murphy ME and
George DL: Mitochondrial p53 activates Bak and causes disruption of
a Bak-Mcl1 complex. Nat Cell Biol. 6:443–450. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Weng C, Li Y, Xu D, Shi Y and Tang H:
Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis
in Jurkat leukemia T cells. J Biol Chem. 280:10491–10500. 2005.
View Article : Google Scholar : PubMed/NCBI
|