Introduction
Programmed cell death (PCD) is important in various
developmental pathways. Apoptosis, the best known type, is now
designated as type I PCD that eliminates damaged and infected
cells, as well as regulates tissue homeostasis. Two major apoptotic
pathways exist: the extrinsic pathway controlled by death receptors
and the intrinsic pathway mediated by mitochondria. These apoptotic
signaling pathways mediate an important cellular event, the
activation of cysteine proteases and caspases that cleave different
substrates eventually leading in cell death. However, there is
considerable evidence to suggest that PCD is not confined to
apoptosis but other mechanisms may also be involved. One of these
mechanisms is ‘autophagic PCD’, which is a process that involves
autophagosomes and autolysosomes (1,2).
Autophagosomes are double-membrane cytoplasmic vesicles that are
designed to engulf various cellular components, including
cytoplasmic organelles (3,4). Autophagosomes fuse to lysosomes to
become autolysosomes, which segregate cellular constituents for
digestion prolonging survival for a short time under starvation
conditions.
The molecular basis of autophagy has been
extensively studied. Mammalian target of rapamycin (mTOR), a
serine/threonine protein kinase, is one of the signaling proteins
that initiate autophagy. mTOR negatively regulates autophagy
related gene 1 (Atg1) or its mammalian homologs, Unc-151-like
kinase (ULK-1 and -2) in nutrient rich conditions, thus inhibiting
autophagy (5). During autophagosome
formation, Beclin-1, a mammalian homolog of the yeast Atg6 gene has
been shown to be involved in the nucleation step of the
autophagosome. When Bcl-2 is phosphorylated by JNK, Beclin-1 is
released from Bcl-2 at the level of the endoplasmic reticulum.
Beclin-1 subsequently combines with the class III
phosphatidylinositol 3-kinase Vps34, UVRAG and other partners
required for autophagy vesicle nucleation (6,7). The
next step in autophagophore elongation requires conjugation of
Atg5-Atg12 to localize at the outer membrane of the expending
membrane. Atg8 (also called LC3-I) is cleaved by Atg4, and is
subsequently conjugated with phophatidylethanolamine (PE) which
leads to the LC3-II isoform. LC3-II is recruited both at the inner
and the outer membranes of the growing vesicle. The completion of
the autophagosome is followed by its fusion with a lysosome.
While hepatocellular carcinoma (HCC) is the most
common malignancy worldwide, its incidence has been increasing in
east Asia and Western countries (8,9). Thus
far the therapies of liver cancer have included surgery, chemical
and target therapies, yet there is no perfect treatment (10). Therefore, it is necessary to
identify novel therapeutic strategies for the management of HCC.
Gartanin [1,3,5,8-tetrahydroxy-2,4-bis(3-methyl-2-butenyl)] is a
xanthone-type compound isolated from mangosteen, a tropical fruit
native to southeast Asia. Gartanin has been reported to possess
antioxidants, anticancer and antiproliferative properties (11,12).
Indeed, a recent study showed that gartanin sensitizes tumor
necrosis factor (TNF)-related apoptosis-inducing ligand
(TRAIL)-induced cell death through upregulation of DR5 expression
in TRAIL-resistant human gastric adenocarcinoma cells (13). However, the anticarcinoma effect of
gartanin on HCC and the molecular mechanism of its effect have not
yet been fully determined.
The purpose of the present study was to determine
whether gartanin induced PCD in HCC and to examine the relationship
between autophagy and apoptosis. In the present study, we
demonstrated for the first time that gartanin induced an autophagic
response in various cancer cell lines. Furthermore, genetic
inhibitors or inhibition of autophagy by specific chemical
inhibitors aggravated cell death in response to gartanin
stimulation. Furthermore, gartanin plus autophagy inhibitor (3-MA)
caused marked HCC growth inhibition compared with single agents
alone, suggesting that gartanin-induced autophagy may play a
self-protective role against its own cytotoxic effect. Our data
also showed that the autophagy induced by gartanin was independent
of mTOR and was mediated through activation of JNK and subsequent
phosphorylation of Bcl-2, ultimately leading to autophagy-dependent
interference with gartanin-induced apoptosis. Thus, gartanin may
has potential therapeutic use by targeting the autophagic
pathway.
Materials and methods
Cell culture
Human hepatoma cell lines Hep3B, HepG2 and Huh7 were
purchased from the American Type Culture Collection. Cells were
cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented
with 10% fetal bovine serum (FBS) and antibiotics (Life
Technologies) and maintained at 37°C with 5% CO2 in a
humidified atmosphere.
Chemicals
Gartanin isolated from mangosteen was kindly gifted
by Professor Y.-W.C. (Dongguk University). Acridine orange (AO) and
SP600125 were purchased from the Sigma Chemical Corporation.
MitoTracker was purchased from Invitrogen. Anti-p62, c-Jun,
Beclin-1 and LC3 antibodies were purchased from Santa Cruz
Biotechnology. Anti-Atg5, phosphorylated JNK (T183/Y185),
phospho-specific antibodies against c-Jun (S63), mTOR (S2448 and
S2481) antibodies were purchased from Cell Signaling. Other
antibodies were from Dakopatts.
Cell viability (MTT)
Cell viability assays were conducted as previously
described (14).
Observation of morphologic changes
Hep3B cells (5×105/well) were seeded into
6-well culture plates and incubated with gartanin for 24 h.
Cellular morphology was observed by a phase contrast microscope
(Leica, Nussloch, Germany).
Transmission electron microscopy
Electron microscopy was as previously described
(15). Briefly, cells were prefixed
in Karnovsky’s solution [1% paraformaldehyde, 2% glutaraldehyde, 2
mmol/l calcium chloride, 0.1 mol/l cacodylate buffer (pH 7.4)] for
2 h and washed with cacodylate buffer. Postfixing was carried out
in 1% osmium tetroxide and 1.5% potassium ferrocyanide for 1 h.
After dehydration with 50–100% alcohol, the cells were embedded in
Poly/Bed 812 resin (Pelco), polymerized and observed under an
electron microscope (EM 902A; Zeiss).
Acidic vesicular organelles with AO
staining
Gartanin-treated cells were stained with 1
µg/ml AO for 15 min and cells were observed under a
fluorescence microscope, to detect acidic vesicular organelles
(AVO). Gartanin-treated cells were stained with AO, detached by
trypsinization, and processed for the FACScan using CellQuest
software (Becton-Dickinson) to quantify the development of
AVOs.
Mitochondrial labelling within live
cells
Gartanin-treated cells were stained with 1
µg/ml MitoTracker for 15 min and observed under a
fluorescence microscope; samples were further analyzed by FACScan
using CellQuest software.
GFP-LC3 translocation
Hep3B cells were transfected with the plasmid
encoding GFP-LC3, and then treated with various dose of gartanin.
Translocation of GFP-LC3 from cytosol to autophagic vacuoles was
observed by fluorescence microscopy and the fluorescence intensity
of GFP of LC3 was assessed by FACScan using CellQuest software.
Western blotting
Western blotting experiments were conducted as
previously described in details (16).
Flow cytometric analysis
Cells were fixed with 1 U/ml of RNase A (DNase-free;
Sigma) and 10 µg/ml of propidium iodide (PI) (Sigma-Aldrich)
overnight at room temperature, in the dark. A flow cytometer was
used to analyze the level of apoptotic cells containing sub-G1 DNA
content. For Annexin V staining, live cells were incubated with
Annexin V (R&D Systems).
Statistical analysis
All data are presented as mean ± SE of at least 3
independent experiments. The statistical significance of
differences was assessed using ANOVA (GraphPad software) followed
by the Student-Newman-Keuls’ multiple comparison tests. P<0.05
was considered to indicate a statistically significant result.
Results
Gartanin inhibits proliferation of
HCC
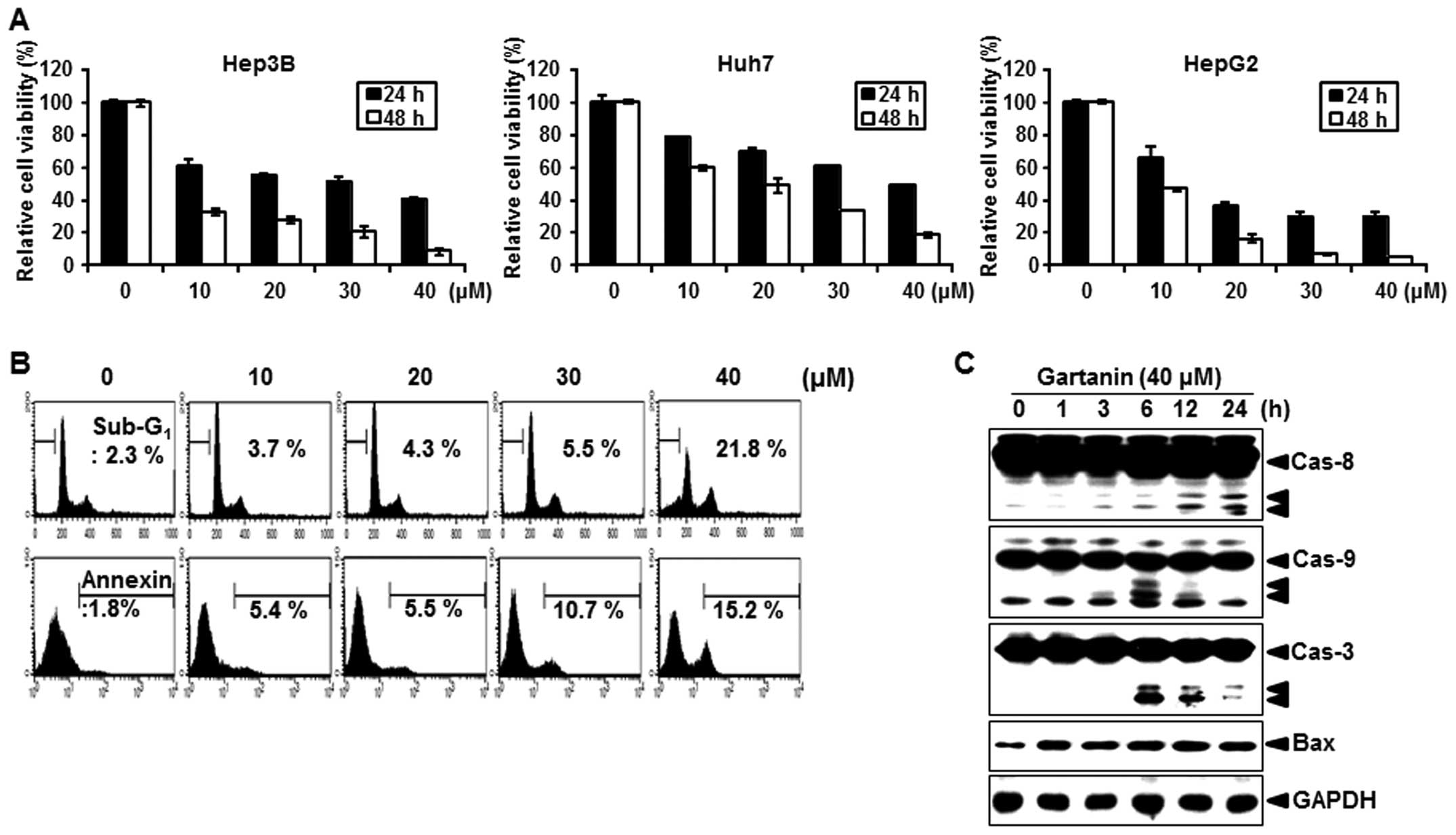
We first evaluated the effect of gartanin on the
proliferation of hepatoma cancer cells. Gartanin inhibited cell
proliferation in a time- and dose-dependent manner in Hep3B, HepG2
and Huh7 cells (Fig. 1A). Our
results revealed that 10–40 µmol/l gartanin decreased
viability in the tested HCC cell lines, suggestive of the
preferentially cytotoxicy of gartanin to malignant HCC cells. Next,
we investigated whether decreased cell viability by gartanin
treatment was due to the induction of apoptotic signaling.
Treatment with gartanin for 24 h increased the accumulation of
Hep3B cells in the sub-G1 phase (Fig.
1B, upper panel) and positive staining with Annexin V (Fig. 1B, lower panel). We also found that
the activity of caspase-8, -9 and -3 was significantly increased on
gartanin treatment (Fig. 1C).
Furthermore, gartanin enhanced the expression of pro-apoptotic
protein Bax (Fig. 1C). These
results indicated that the classic mitochondrial apoptosis pathways
were involved in gartanin-induced Hep3B cell apoptosis. Thus,
gartanin induced apoptotic cell death in HCC suggestive of its
potential as an effective therapeutic approach to malignant
HCC.
Gartanin induces autophagy of HCC
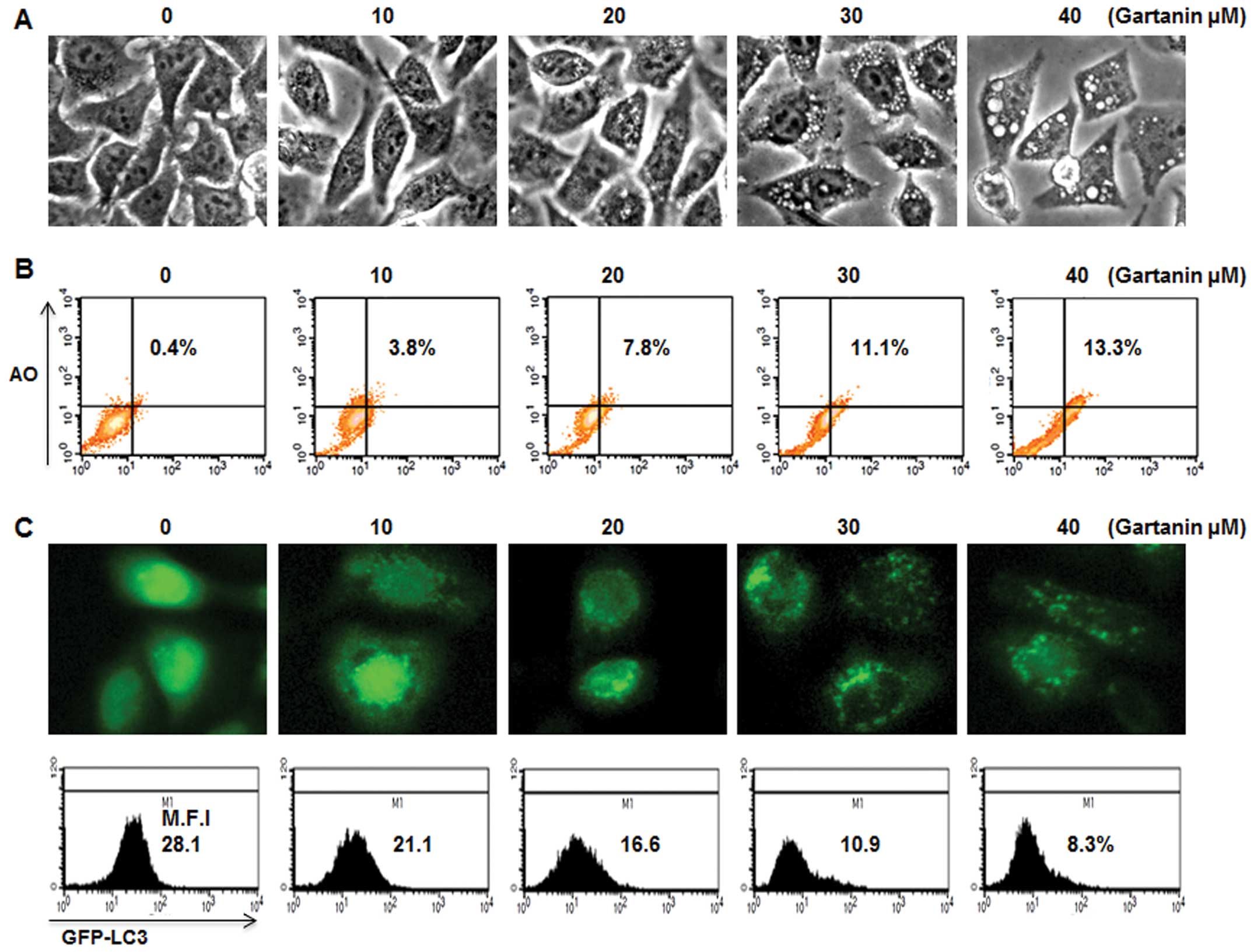
We next determined whether gartanin could induce
autophagy and apoptosis simultaneously. Gartanin-treated Hep3B
cells showed numerous small vesicles around their nuclei on
phase-contrast microscopy (Fig.
2A). We further investigated whether vacuolation was observed
in gartanin-treated Hep3B cells. AO, a lysosomotropic agent, is
able to stain the acidic vesicular organelle (AVOs) with cytoplasm
fluorescence changing from bright green to bright red, thereby
offering a rapid and quantitative measure of the induction of
autophagy. FACS analysis showed that the percentage of cells with
AVOs was dose-dependently higher in the gartanin-treated group vs.
the control group (Fig. 2B).
Vesicular accumulation of microtubule-associated protein 1 light
chain 3 (LC3) is a marker of autophagy, therefore, in order to
further evaluate autophagy, gartanin-treated cells were transfected
with GFP-LC3 plasmid. GFP-LC3 was diffusely expressed in untreated
cells, whereas gartanin-treated cells displayed a highly-intense
punctate expression (Fig. 2C, upper
panel). GFP-LC3 plasmid was also used in flow cytometry experiments
to measure the turnover of autophagic vesicles. We found that
gartanin significantly decreased the fluorescent intensity of
GFP-LC3 via rapid degradation by the autolysosome (Fig. 2C, lower panel). These results
suggested that fragmented intracellular granules may be engulfed by
autophagic vacuoles, on treatment with gartanin. We furthermore
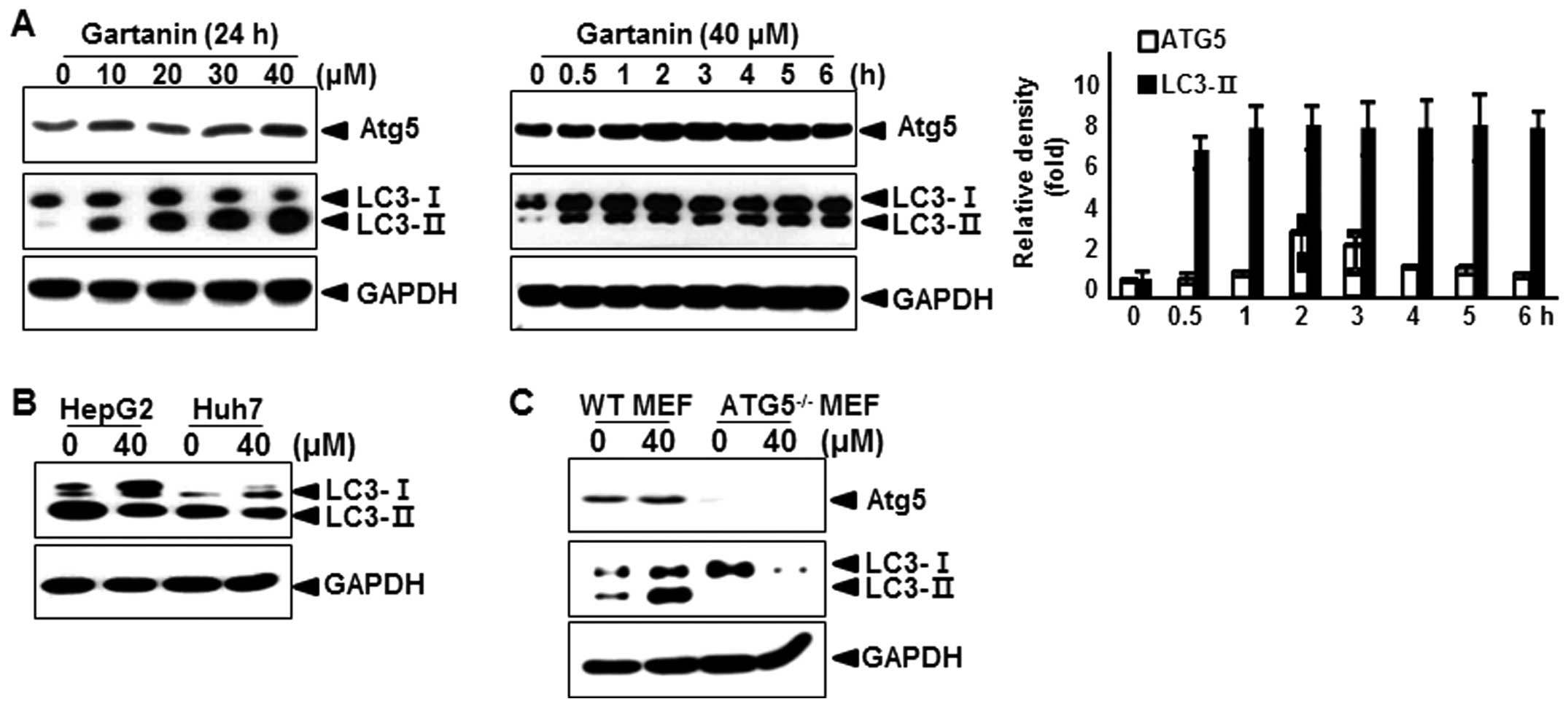
assessed the effect of gartanin on the levels of Atg5, an
autophagic marker (17), and the
conversion of the cytoplasmic form of the LC3-I protein (18 kDa) to
the pre-autophagosomal and autophagosomal membrane-bound form of
LC3-II (16 kDa). As shown in Fig.
3A, immunoblot analysis revealed that gartanin treatment dose-
and time-dependently led to an increase in the levels of Atg5 and
LC3-II proteins in Hep3B cells. Quantitative analysis also
indicated that gartanin treatment led to an increase in Atg5 and
LC3-II levels as early as 1 h after drug exposure and gradually
increased thereafter. Next, we examined whether gartanin induced
autophagy in other cell types. We found that gartanin increased
LC3-II proteins in HepG2 and Huh7 cell lines (Fig. 3B). Wild-type mouse embryonic
fibroblasts (WT MEF) and ATG5 knockout MEF (ATG5−/− MEF)
cells were incubated in the presence of gartanin, to determine the
role of ATG5 in gartanin-induced autophagy. Autophagy was measured
by immunoblot analysis of LC3. Gartanin failed to induce autophagy
in ATG5−/−MEF cells, while it dramatically induced
autophagy in WT MEF cells (Fig.
3C). These results indicated that ATG5 played an important role
in the induction of gartanin-mediated autophagy.
Gartanin-damaged mitochondria are
targeted by autophagy
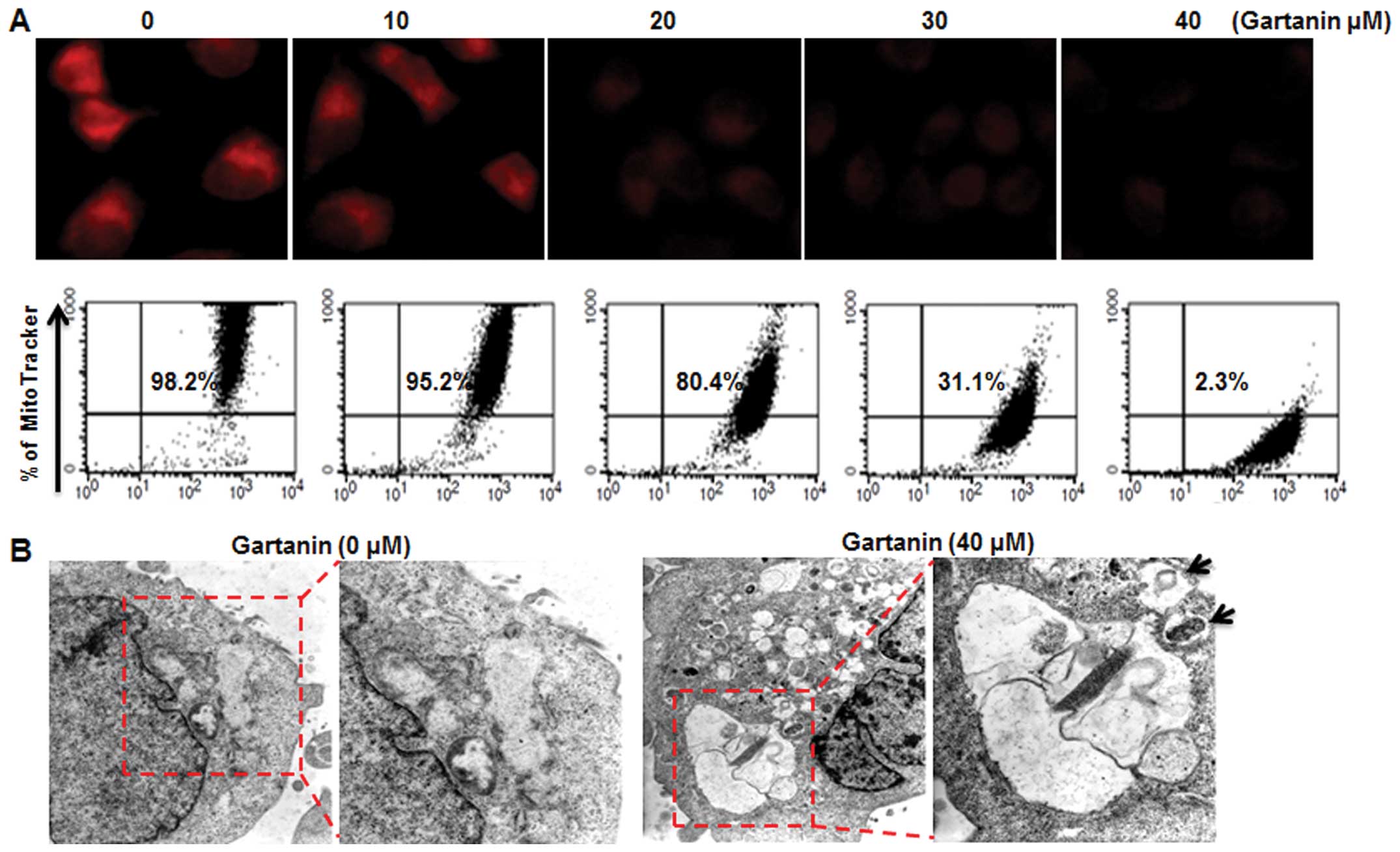
We next examined whether gartanin-induced autophagic
cell death in Hep3B cells was associated with alterations in
mitochondrial function and/or structure crucial to regulating cell
death. Gartanin induced a dose-dependent significant loss of
mitochondrial membrane potential (MMP) analyzed by MitoTracker on
fluorescence microscopy (Fig. 4A,
upper panel) and FACScan (Fig. 4A,
lower panel). Electron microscopy subsequently provided further
confirmation of gartanin-induced autophagy in Hep3B cells. Electron
micrographs of untreated cells showed normal morphology of all
organelles, with mitochondria scattered homogeneously throughout
the cells. Gartanin treatment for 24 h resulted in more advanced
mitochondrial disruption with frequent fusion between the vacuoles.
Vacuoles appeared in increasing sizes signifying ongoing fusion of
autophagosomes with lysosomes (Fig.
4B).
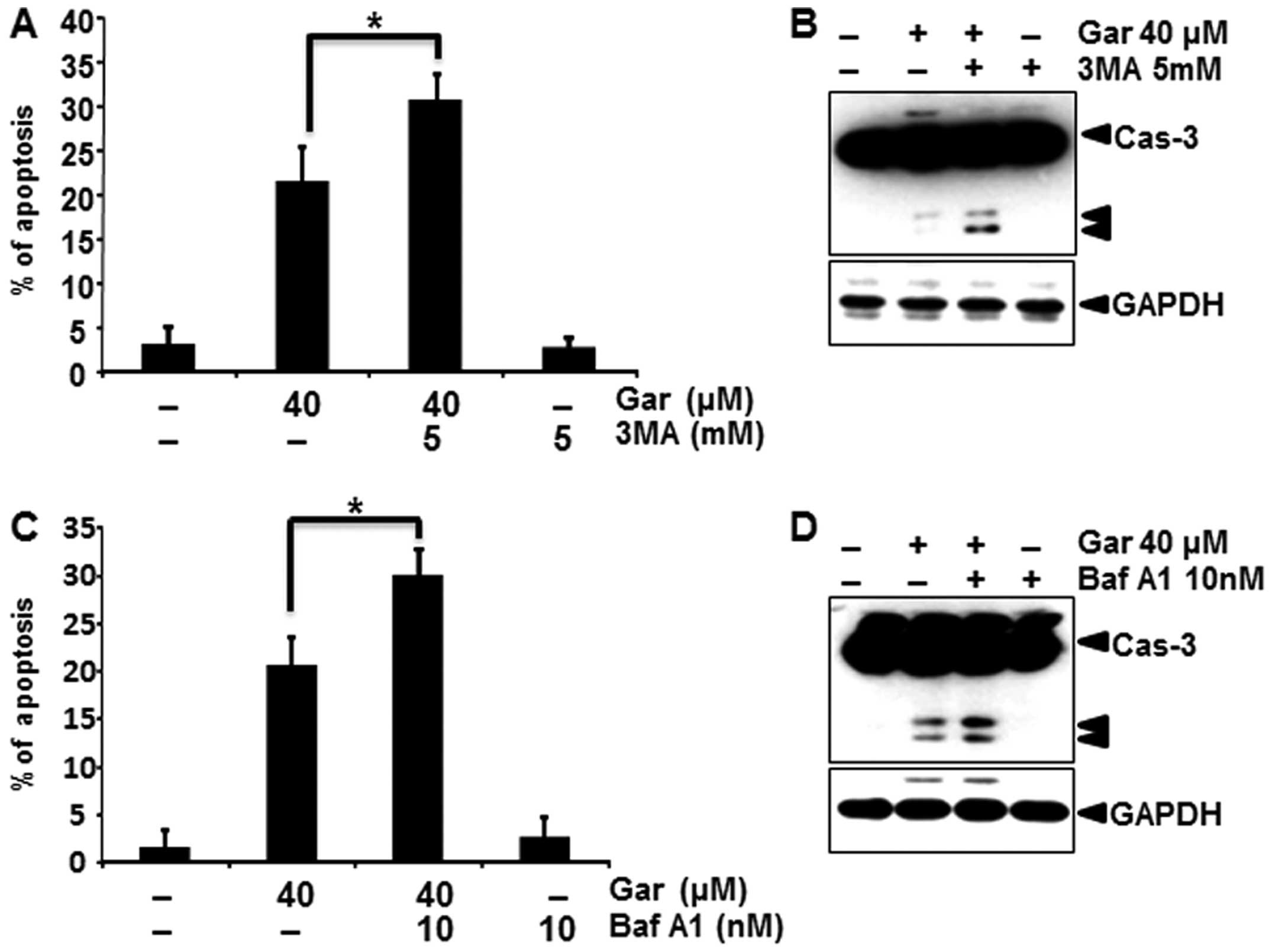
Autophagy inhibition enhances
gartanin-induced apoptosis
Recent research suggests that inhibition of
autophagy may enhance chemosensitization in human cancer cells.
Accordingly, we investigated whether pharmacological inhibition of
autophagy, enhanced gartanin-induced apoptosis. Firstly to inhibit
autophagy, we utilized the class III phosphatidylinositol 3-kinase
(PI3K) inhibitor 3-MA that was previously reported to sensitize
cancer cells to chemotherapy-induced apoptosis. As shown in
Fig. 5A, 3-MA plus gartanin treatment resulted in an
increased percentage of apoptotic cancer cells than gartanin alone
by a PI staining assay. Corroborating this finding, 3-MA enhanced
caspase-3 cleavage, a marker of apoptosis (Fig. 5B). Baf A1, a specific inhibitor of
vacuolar type H+-ATPase, prevents autophagy at the late
stage by inhibiting fusion between autophagosomes and lysosomes.
Baf A1 similarly enhanced gartanin-induced apoptotic signaling, as
shown by caspase-3 cleavage (Fig. 5C
and D). Consistent with the pharmacological approach,
suppression of autophagy by silencing Atg5 aggravated
gartanin-induced apoptosis in Hep3B cells as indicated by an
increase in sub-G1 population and cleaved caspase-3 and PARP (data
not shown). These results suggested that gartanin induced canonical
autophagy, and induction of autophagy had a protective role in
tumor cells subjected to the cytotoxicity of gartanin.
Gartanin-induced autophagy is mediated
through the JNK-Bcl-2 pathway
We analyzed the effect of gartanin on the main
cellular signaling pathways that are constitutively modulated in
Hep3B cells, to determine its mode of action. Since inhibition of
the mTOR signaling pathway is a well-characterized mechanism in the
initiation and maturation of autophagy, we examined whether
gartanin regulated the mTOR signaling pathway. Unexpectedly,
gartanin treatment up to 6 h induced a sharp increase in the
phosphorylation of mTOR, as well as its downstream targets p70
ribosomal protein S6 kinase (p70S6K), indicating that gartanin
induces autophagy through an Akt/mTOR-independent pathway (Fig. 6A). JNK has also been implicated in
the induction of autophagy by various stimuli, including starvation
(18), cytokine stimulation
(19) and ER stress (20); hence, we next investigated whether
JNK was involved in gartanin-induced autophagy. As shown in
Fig. 6B, gartanin triggered JNK
activation, within 0.5 h in Hep3B cells. This response was
sustained for at least 6 h (Fig.
6B). Indeed, JNK activation correlated with an increase in
c-Jun phosphorylation. One mechanism by which JNK contributes to
autophagy is via phosphorylation of the anti-apoptotic protein
Bcl-2 (21,22) and thereby, its dissociation from
Beclin-1 (23). Furthermore,
activated JNK mediates transcriptional activity of Bcl-2 (24), p62 (25) and Beclin-1 resulting in autophagy
(26). Western blotting was used to
determine whether JNK activation stimulated phosphorylation and/or
expression of these proteins. Gartanin unregulated the expression
as well as phosphorylation of Bcl-2, but had no effect on p62 and
Beclin-1 in our system (Fig. 6C).
In order to further evaluate the relationship between
gartanin-induced autophagy and the JNK signaling, cells were
treated with gartanin in the presence of SP600125 (a JNK
activity-specific inhibitor). Our data showed that pretreatment of
SP600125 significantly inhibited the 24 h gartanin-induced
phosphorylation of JNK, c-jun and Bcl-2 at 1 (Fig. 6D). Furthermore, gartanin-induced
AVOs were decreased by SP600125 pretreatment (Fig. 6E). SP600125 mediated JNK inhibition
stimulated gartanin-induced caspase-3 activation (Fig. 6F). Collectively, these results
validated the requirement of JNK activation in stimulation of
autophagy, and showed that JNK inhibition promoted gartanin-induced
cell death.
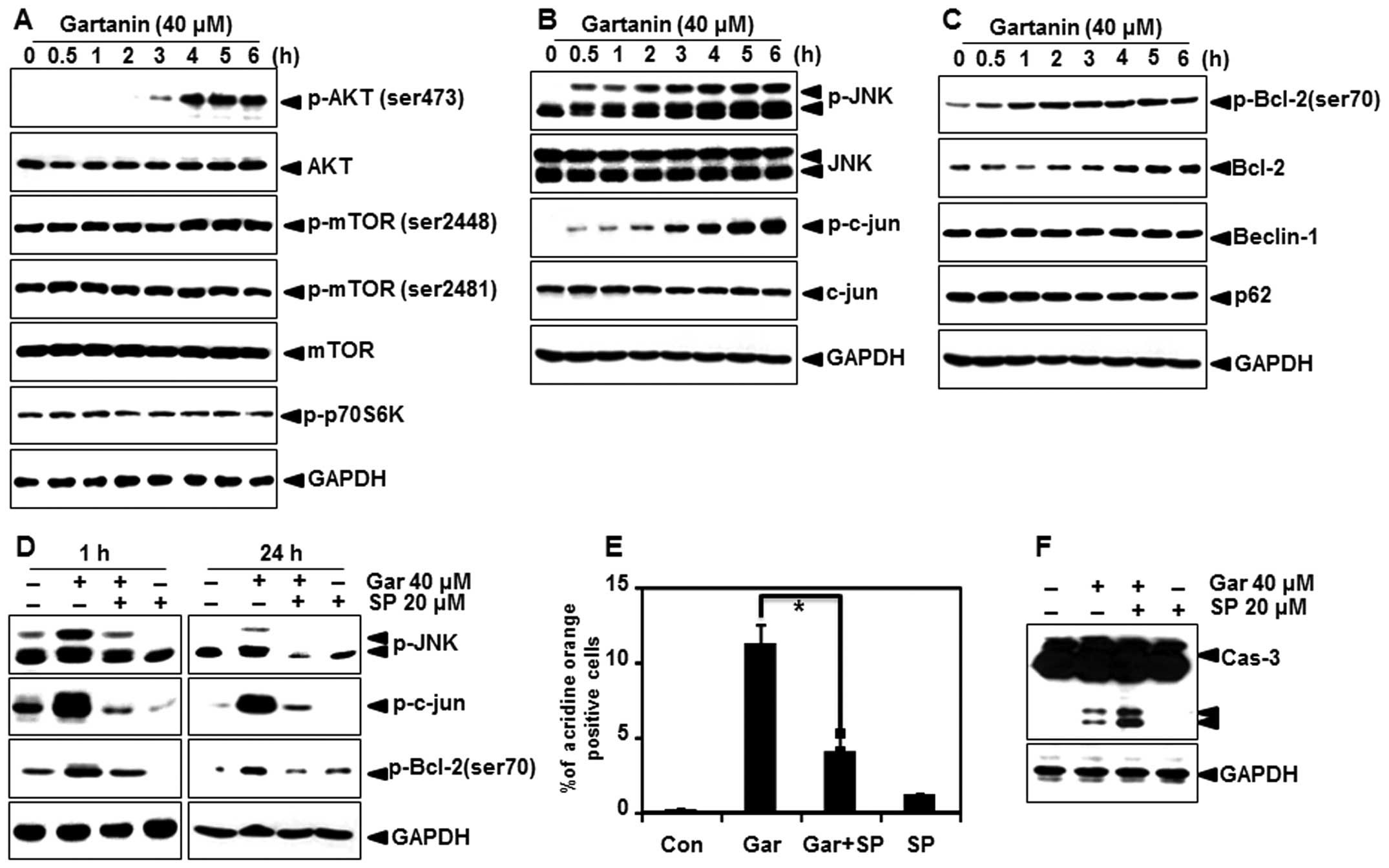 | Figure 6Autophagy-related signal transduction
in Hep3B cells with gartanin treatment. (A-C) Hep3B cells were
treated with indicated concentrations of gartanin for 6 h, after
which protein extracts were subjected to immunoblotting using the
specific antibodies, including p-Akt (ser473), p-mTOR (ser2448 and
ser2481), p-p70S6K, Beclin-1, p-JNK (Thr183/Tyr185), JNK, p-c-jun,
p-Bcl-2 (ser70) and Bcl-2. (D) Hep3B cells were treated with 20
µmol/l SP600125 for 0.5 h prior to gartanin treatment for
the indicated times. Total protein was subjected to 10% SDS-PAGE
followed by western blotting using anti-p-JNK, p-c-jun and p-Bcl-2.
(E) SP600125 (20 µmol/l) pretreated Hep3B cells were treated
40 µmol/l gartanin for 24 h. Following treatment, cells were
stained with 1 mg/ml acridine orange (AO) at 37°C for 15 min.
Staining of AOs was analyzed using flow cytometry. (F) Cell
extracts were used in western blot analyses. |
Discussion
Cancer remains one of the most aggressive and lethal
human malignancies worldwide. Although chemotherapy, radiotherapy
and surgery have been used for many years, these anticancer
therapies can only offer limited benefits to cancer patients due to
metastasis, acquired chemoresistance and toxicity. Evidence from
recent studies suggest that the consumption of vegetables and
fruits decreases the incidence of cancer (27). Natural products may serve as novel
adjunctive agents and chemoprevention of cancer. Mangosteen
(Garcinia mangostana Linn.), a well-known tropical fruit
contains considerable amounts of biologically active compounds,
such as xanthones. Among the xanthones, gartanin is most abundant
and frequently studied.
Autophagy is a degradative process in eukaryotic
cells that results in the breakdown of intracellular material
within lysosomes (28). It serves
as an alternative route of PCD called type-2 PCD or autophagic cell
death. Evidently autophagy is not always pro-death but instead can
be pro-survival under conditions of cellular stress induced by
nutrient deprivation or chemotherapy, thus allowing cells to evade
apoptosis. In tumor cells, the role of autophagy may depend on
tumor types, the stage of tumorigenesis, and the nature and extent
of the insult (29).
In the present study, we have firstly demonstrated
that gartanin induced apoptosis in Hep3B cells, through the classic
apoptotic pathways. We also identified the process of autophagy in
response to gartanin together with the underlying molecular
mechanisms. In the present study, gartanin-induced autophagy was
evidenced by autophagosomal marker LC3 conversion, accumulation of
AO-labeled acidic vesicles consistent with autophagolysosomes, and
punctate formation of GFP-LC3. We also showed that gartanin induced
autophagy in human HCC cell lines, Hep3B, HepG2 and Huh7. These
findings indicated that gartanin could generally induce autophagy
in various cancer cells, and not in a strict cell type-specific
manner. We used pharmacological and genetic approaches to inhibit
autophagy, in our attempt to define a pro-survival or pro-cell
death role for gartanin. 3-MA and Baf A1 were used as inhibitors of
autophagy. Our results showed that 3-MA or Baf A1 treatment
significantly enhanced gartanin-induced apoptosis in Hep3B cells,
indicating that gartanin-mediated autophagy was protective.
Furthermore, inhibition of autophagy-related genes (Atg5) resulted
in significant apoptotic cell death in Hep3B cells treated with
gartanin. Thus, we concluded that the gartanin-triggered autophagy
had protective effects in various cancer cells and inhibition of
autophagy could be a promising strategy to enhance the antitumor
efficiency of gartanin.
We next investigated the potential pathways involved
in gartanin-induced induction of autophagy, in an attempt to
identify means to enhance the efficacy of gartanin. We found an
important role for JNK signaling in gartanin-induced autophagy, in
our system. JNK has been reported to have critical functions
upstream of autophagy induced by growth factors, nutrients and
stress, including hypoxia and oxidative stress. It is well
established that the activation of JNK, also referred to as
stress-activated kinases, mediates Bcl-2 phosphorylation (23). Bcl-2 phosphorylation disrupts the
interactions between Beclin-1 and Bcl-2, releasing Beclin-1 to
promote autophagy. Our data showed that gartanin enhanced the
phosphorylation of JNK and Bcl-2 in Hep3B cells. Additionally,
SP600125-mediated inhibition of JNK abrogated gartanin-induced
autophagy via dephosphorylation of JNK and Bcl-2. Thus, it was
reasonable to hypothesize that gartanin treatment may induce JNK
activation, which results in Bcl-2 phosphorylation, thereby
disrupting Bcl-2 -Beclin-1 interaction, with net stimulation of
autophagy. JNK also mediates the late phase of autophagy through
regulation of microtubule stability via phosphorylation of MAP1B
(microtubule-associated protein1B) which mediates autophagosome
trafficking to the lysosome through interaction with both LC3-I and
LC3-II (30). Hence, further study
is required to investigate the role of JNK in MAP1B activation. We
also found that gartanin upregulated the gene expression of
anti-apoptotic Bcl-2. JNK is known to induce activation of
activator protein-1 (AP-1), hence an interesting possibility exists
for AP-1 activation in gartanin-mediated Bcl-2 regulation. More
prudent experiments will be needed for elucidating the role of AP-1
on gartanin-induced Bcl-2 expression. Summarily, we cautiously
concluded that gartanin-induced JNK activation may have an
anti-apoptotic action via upregulation and phosphorylation of
Bcl-2.
Additionally, the Akt/mTOR pathway is the classic
pathway often involved in regulating autophagy. Inhibition of the
Akt/mTOR pathway has consistently been associated with triggering
autophagy in cancer cells. Rapamycin is the mTORC1 inhibitor and
induces autophagy in different tumor cells. However, in the present
study, we found that the expression of phospho-mTOR (ser2448), as
well as the levels of phosphorylated p70S6 kinase was slightly
increased in Hep3B cells after exposure to gartanin. These results
suggested that gartanin induced autophagy via an mTOR-independent
pathway.
In conclusion, we demonstrated that autophagy
triggered by gartanin occurred in many types of cancer cells. In
addition, our data showed that activation of JNK signaling pathway
was responsible for gartanin-induced autophagy via increasing
phosphorylation of Bcl-2 which promoted its release from Beclin-1.
Thus, our data suggested that the JNK-Bcl-2 pathway was the
critical regulator of gartanin-induced autophagy and a potential
drug target for chemotherapeutic combination. These novel findings
improve our understanding of gartanin-mediated autophagy as well as
the mechanisms involved in its therapeutic role in hepatoma cancer
patients.
Acknowledgments
The present study was supported (in part) by the
Daegu University Research Grant, 2011.
References
|
1
|
Schweichel JU and Merker HJ: The
morphology of various types of cell death in prenatal tissues.
Teratology. 7:253–266. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Levine B and Yuan J: Autophagy in cell
death: An innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lieberman AP, Puertollano R, Raben N,
Slaugenhaupt S, Walkley SU and Ballabio A: Autophagy in lysosomal
storage disorders. Autophagy. 8:719–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sinha S and Levine B: The autophagy
effector Beclin 1: A novel BH3-only protein. Oncogene. 27(Suppl 1):
S137–S148. 2008. View Article : Google Scholar
|
|
7
|
Funderburk SF, Wang QJ and Yue Z: The
Beclin 1-VPS34 complex - at the crossroads of autophagy and beyond.
Trends Cell Biol. 20:355–362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
El-Serag HB and Mason AC: Rising incidence
of hepatocellular carcinoma in the United States. N Engl J Med.
340:745–750. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Parkin DM, Pisani P and Ferlay J: Global
cancer statistics. CA Cancer J Clin. 49:31–64. 1999. View Article : Google Scholar
|
|
10
|
Josephs DH and Ross PJ: Sorafenib in
hepatocellular carcinoma. Br J Hosp Med. 71:451–456. 2010.
View Article : Google Scholar
|
|
11
|
Suksamrarn S, Komutiban O, Ratananukul P,
Chimnoi N, Lartpornmatulee N and Suksamrarn A: Cytotoxic prenylated
xanthones from the young fruit of Garcinia mangostana. Chem Pharm
Bull. 54:301–305. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jung HA, Su BN, Keller WJ, Mehta RG and
Kinghorn AD: Antioxidant xanthones from the pericarp of Garcinia
mangostana (Mangosteen). J Agric Food Chem. 54:2077–2082. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kikuchi H, Ohtsuki T, Koyano T,
Kowithayakorn T, Sakai T and Ishibashi M: Activity of mangosteen
xanthones and teleocidin a-2 in death receptor expression
enhancement and tumor necrosis factor related apoptosis-inducing
ligand assays. J Nat Prod. 73:452–455. 2010. View Article : Google Scholar
|
|
14
|
Moon DO, Asami Y, Long H, Jang JH, Bae EY,
Kim BY, Choi YH, Kang CH, Ahn JS and Kim GY: Verrucarin A
sensi-tizes TRAIL-induced apoptosis via the upregulation of DR5 in
an eIF2α/CHOP-dependent manner. Toxicol In Vitro. 27:257–263. 2013.
View Article : Google Scholar
|
|
15
|
Colosetti P, Puissant A, Robert G, Luciano
F, Jacquel A, Gounon P, Cassuto JP and Auberger P: Autophagy is an
important event for megakaryocytic differentiation of the chronic
myelogenous leukemia K562 cell line. Autophagy. 5:1092–1098. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moon DO, Asami Y, Kim MO, Jang JH, Kim BY,
Ahn JS, Kim GY and Yun SG: Xestospongin C induces monocytic
differentiation of HL60 cells through activation of the ERK
pathway. Food Chem Toxicol. 55:505–512. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li C, Capan E, Zhao Y, Zhao J, Stolz D,
Watkins SC, Jin S and Lu B: Autophagy is induced in CD4+
T cells and important for the growth factor-withdrawal cell death.
J Immunol. 177:5163–5168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jia G, Cheng G, Gangahar DM and Agrawal
DK: Insulin-like growth factor-1 and TNF-alpha regulate autophagy
through c-jun N-terminal kinase and Akt pathways in human
atherosclerotic vascular smooth cells. Immunol Cell Biol.
84:448–454. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen JL, Lin HH, Kim KJ, Lin A, Forman HJ
and Ann DK: Novel roles for protein kinase Cdelta-dependent
signaling pathways in acute hypoxic stress-induced autophagy. J
Biol Chem. 283:34432–34444. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Geeraert C, Ratier A, Pfisterer SG, Perdiz
D, Cantaloube I, Rouault A, Pattingre S, Proikas-Cezanne T, Codogno
P and Poüs C: Starvation-induced hyperacetylation of tubulin is
required for the stimulation of autophagy by nutrient deprivation.
J Biol Chem. 285:24184–24194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wei Y, Pattingre S, Sinha S, Bassik M and
Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates
starvation-induced autophagy. Mol Cell. 30:678–688. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Puissant A, Robert G, Fenouille N, Luciano
F, Cassuto JP, Raynaud S and Auberger P: Resveratrol promotes
autophagic cell death in chronic myelogenous leukemia cells via
JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res.
70:1042–1052. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li DD, Wang LL, Deng R, Tang J, Shen Y,
Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al: The pivotal role of
c-Jun NH2-terminal kinase-mediated Beclin 1 expression during
anticancer agents-induced autophagy in cancer cells. Oncogene.
28:886–898. 2009. View Article : Google Scholar
|
|
27
|
Surh YJ: Cancer chemoprevention with
dietary phytochemicals. Nat Rev Cancer. 3:768–780. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shingu T, Fujiwara K, Bögler O, Akiyama Y,
Moritake K, Shinojima N, Tamada Y, Yokoyama T and Kondo S:
Stage-specific effect of inhibition of autophagy on
chemotherapy-induced cytotoxicity. Autophagy. 5:537–539. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang QJ, Ding Y, Kohtz DS, Mizushima N,
Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N and Yue Z:
Induction of autophagy in axonal dystrophy and degeneration. J
Neurosci. 26:8057–8068. 2006. View Article : Google Scholar : PubMed/NCBI
|