Introduction
Solid tumors are highly heterogeneous in terms of
oxygenation and contain hypoxic cell regions as a result of an
imbalance between cell proliferation and angiogenesis. As the
primary cellular and systemic response to hypoxic stress, tumor
cells in these regions induce the production of hypoxia-inducible
factors (HIFs), master transcription regulators that consist of two
subunits, the α subunit (HIF-1α, HIF-2α and HIF-3α) and the β
subunit (HIF-1β) (Arnt) (1,2). Of these three HIF-α family members,
HIF-1α is constitutively expressed, whereas HIF-2α is detected most
prominently in vascular endothelial cells during embryonic
development and HIF-3α is expressed in kidney and lung epithelial
cells (3–5). In addition, although HIF-1β is
unaffected by changes in the cellular oxygen concentrations, HIF-1α
is O2-dependently regulated (1,2,6). Under
normoxic conditions, HIF-1α is constantly degraded via the
hydroxylation of proline residues within the oxygen-dependent
degradation domain (ODD) of HIF-1α by prolyl hydroxylases (PHDs)
and the von Hippel-Lindau protein (VHL)-mediated
ubiquitin-proteasome pathway. However, under hypoxia, reactive
oxygen species (ROS) are generated by mitochondrial electron
transport chain and then released to the cytosol, thereby leading
to stabilization of HIF-1α through PHD inhibition due to oxidation
of Fe2+ within PHD (7).
The stabilized HIF-1α subunit is translocated into the nucleus,
where it forms a heterodimeric transcriptional complex with the
HIF-1β subunit and directly binds to the hypoxia-responsive element
(HRE, 5′-A/GCGTG-3′) to express a number of its target genes
involved in angiogenesis, metabolic adaptation, tolerance of
acidosis, cell survival and metastasis, such as vascular
endothelial growth factor (VEGF), SLC2A1, which encodes the glucose
transporter GLUT1, carbonic anhydrase (CA9), insulin-like growth
factor-2 (IGF2) and transforming growth factor-α (TGF-α),
respectively (1,8–11). As
a result, the expression of HIF-1 facilitates tumor cell survival
and growth in hypoxic regions.
Tempol is a member of the family of well-described
stable nitroxides that detoxify oxygen metabolites via redox
cycling with one-electron transfer reactions and is applied as a
standard substance in EPR spectroscopy. As for the biological
effects of tempol, its protective effects against radiation-induced
alopecia in guinea pigs have been confirmed (12). In addition, the administration of
tempol prior to fractionated whole brain radiotherapy resulted in
moderate protection against radiation-induced alopecia in a phase I
study (13).
In cancer therapy, anticancer drugs and ionizing
radiation enhance cell killing effects by targeting DNA in
proliferating tumor cells. On the other hand, hypoxic cells show
greater resistance to these treatments because these cells display
low levels of proliferation in the quiescent state (slow cycling or
G0) and exist in the microenvironment where the oxygen effect of
ionizing radiation is reduced (14,15).
Therefore, hypoxic cells demonstrate a higher possibility of
survival after these treatments than normoxic cells in the tumor
tissues, potentially resulting in metastasis or recurrence. In
other words, treatments targeting hypoxic cells appear to be a
promising strategy for eliminating cancer. To date, nitro-imidazole
compounds, such as misonidazole, pimonidazole and nimorazole, have
been synthesized as hypoxic cell radiosensitizers causing hypoxic
tumor cells to be more sensitive to radiation therapy, and these
agents have subsequently been tested in clinical trials. Although
these drugs exhibit significant radiosensitizing effects in
vitro, they show low levels of radiosensitization in
vivo and introduce harmful side effects, including peripheral
neuropathy (16). In recent years,
bioreductive prodrugs have also been actively developed as a novel
hypoxia-targeted drug. In particular, tirapazamine
(3-amino-1,2,4-benzotriazine 1,4-dioxide, TPZ), which produces
damage to hypoxic cells by ROS produced following one-electron
reduction by cytochrome P(450) reductase-enriched microsomes, is
currently undergoing evaluation in phase III clinical trials. In
addition to those mentioned above, new hypoxia-targeted treatments
combined with gene therapy have been developed. For example, Ido
et al and Harada et al showed significant tumor
regression and/or growth delay via the selective enhancement of
hypoxic cell killing induced by the hypoxia-regulated suicide gene
expression using hypoxia-targeted expression vectors harboring the
herpes simplex virus type 1 thymidine kinase (HSVtk) and caspase-3
genes under the control of HRE, respectively (17,18).
We found that tempol strongly induced the
accumulation of HIF-1α under a combination of hypoxic conditions.
This induction mechanism seems to enable us to enhance the hypoxic
cell killing by applying the vector bearing the suicide gene fused
downstream of HRE. The goal of this study was to evaluate the
enhancement of the cell killing effect in vitro applying the
plasmids that can regulate the suicide gene expression under a
combination of tempol and hypoxic conditions and to assess the
possibility of its application to gene therapy using tumor-bearing
mice boosted with tempol.
Materials and methods
Reagent and antibodies
Tempol (4-hydroxy-2,2,6,6-tetramethyl-piperidine
1-oxyl free radical) was purchased from Tokyo Chemical Industry
Co., Ltd. (Tokyo, Japan). Apigenin and echinomycin, HIF-1α
inhibitors, were purchased from Sigma-Aldrich, Inc. (St. Louis, MO,
USA). Anti-HIF-1α antibodies (cat# 3716S), anti-myc-tag antibodies
(cat# 2272S) and anti-β-actin antibodies (cat# 4970S) were
purchased from Cell Signaling Technology, K. K. (Japan).
Cell culture and bacteria
All cell lines used in this study, including MCF7
(human breast carcinoma), LNCap (human prostate carcinoma) and
Saos2 (human osteoblastic osteo-sarcoma), were grown in RPMI-1640
medium supplemented with 10% (v/v) heat inactivated fetal calf
serum, 100 U/ml of penicillin and 100 µg/ml of streptomycin.
The cells were incubated in a 5% CO2 incubator at 37°C.
For hypoxia treatment, the cells were incubated in a hypoxic
incubator containing 1.0% oxygen, 5% CO2 and 94%
nitrogen at 37°C.
The DH5α strain of Escherichia coli (E.
coli) (Takara Bio, Inc., Ohtsu, Japan) was used for the DNA
manipulation experiments. The E. coli cells were grown in LB
medium at 37°C. All medium compositions were purchased from BD
Diagnostics (Sparks, MD, USA) and all experiments with E.
coli were performed according to the methods described by
Sambrook and Russell (19).
Plasmid construction
In order to evaluate the rate of enhancement of the
HIF-1α expression induced by tempol treatment under hypoxic
conditions, we constructed a plasmid designated p4HRE-Luc-ODD
containing the luc gene to which the ODD fragment was added
under the control of four tandem copies of HRE fragments. To
complete the construction of the vector, a plasmid designated
p4HRE-Luc was constructed via self-ligation after EcoRI
digestion of PCR products amplified using pTA-Luc (Takara Bio,
Inc.) as a template with the following primers: a forward primer
with four copies of HRE consensus sequences,
5′-ATGAATTCTGCACGTACTGCACGTACTGCACGTACTGCACGTAGCGCGTGCTAGCCCGGGCTCG
AGA-3′ and reverse, 5′-ATGAATTCTATCGATAGAGAAATGTTCTGGCACCTGC-3′.
Underlining indicates HRE consensus sequences (20). A DNA fragment encoding the ODD of
HIF-1α (from the 557th to the 574th amino acid of HIF-1α) was
amplified using the pBC SK+ vector containing cDNA for
HIF-1α (cat# ORK01550; Kazusa DNA Research Institute, Kisarazu,
Japan) as a template with the following pair of primers,
5′-ATGGTACCGTTAGACTTGGAGATGTT AGCTCCC-3′ and 5′-ATAGAGCTCTAACTGGAA
GTCATC ATCCATTGG-3′, and then inserted in the frame at the
KpnI and SacI sites created at the 3′ end of the
luc gene in p4HRE-Luc following digestion of ODD fragments
amplified with KpnI and SacI to construct a plasmid
designated p4HRE-Luc-ODD. In addition, in order to express a
suicide gene under identical regulation to the luc gene in
p4HRE-Luc-ODD, a plasmid p4HRE-fcy::fur-ODD was constructed by
replacing the luc gene in the plasmid p4HRE-Luc-ODD with the
fcy::fur gene, which encodes cytosine deaminase (CD) and
uracil phosphoribosyl-transferase (UPRT), amplified using a plasmid
pORF5-fcyfur (InvivoGen, San Diego, CA, USA) as a template with the
following pair of primers: 5′-ATACTAGTATCACAGAGGAGACCATGGTCACA-3′
and 5′-ATGGTACCGCGACACAGTAGTATCTGTCCCCAAA-3′. The expression of the
suicide gene was confirmed by inserting a myc-tag sequence in the
frame at the end of the ODD sequence via self-ligation after
SphI digestion of PCR products amplified using
p4HRE-fcy::fur-ODD as a template with the following primers:
forward, 5′-ATGCATGCTAATTCTAGAGTCGGGGCGGCCGG-3′ and reverse,
5′-ATGCATGCTCACAGGTCCTCCTCTGAGATCAGCTTCTGCTCGAGCTCTAACTGGAAGTCATCATCCATT-3′.
The orientations and sequences of the constructed plasmids were
confirmed using a nucleotide sequencing analysis. Schematic
structures for each construct are shown in Figs. 2 and 3.
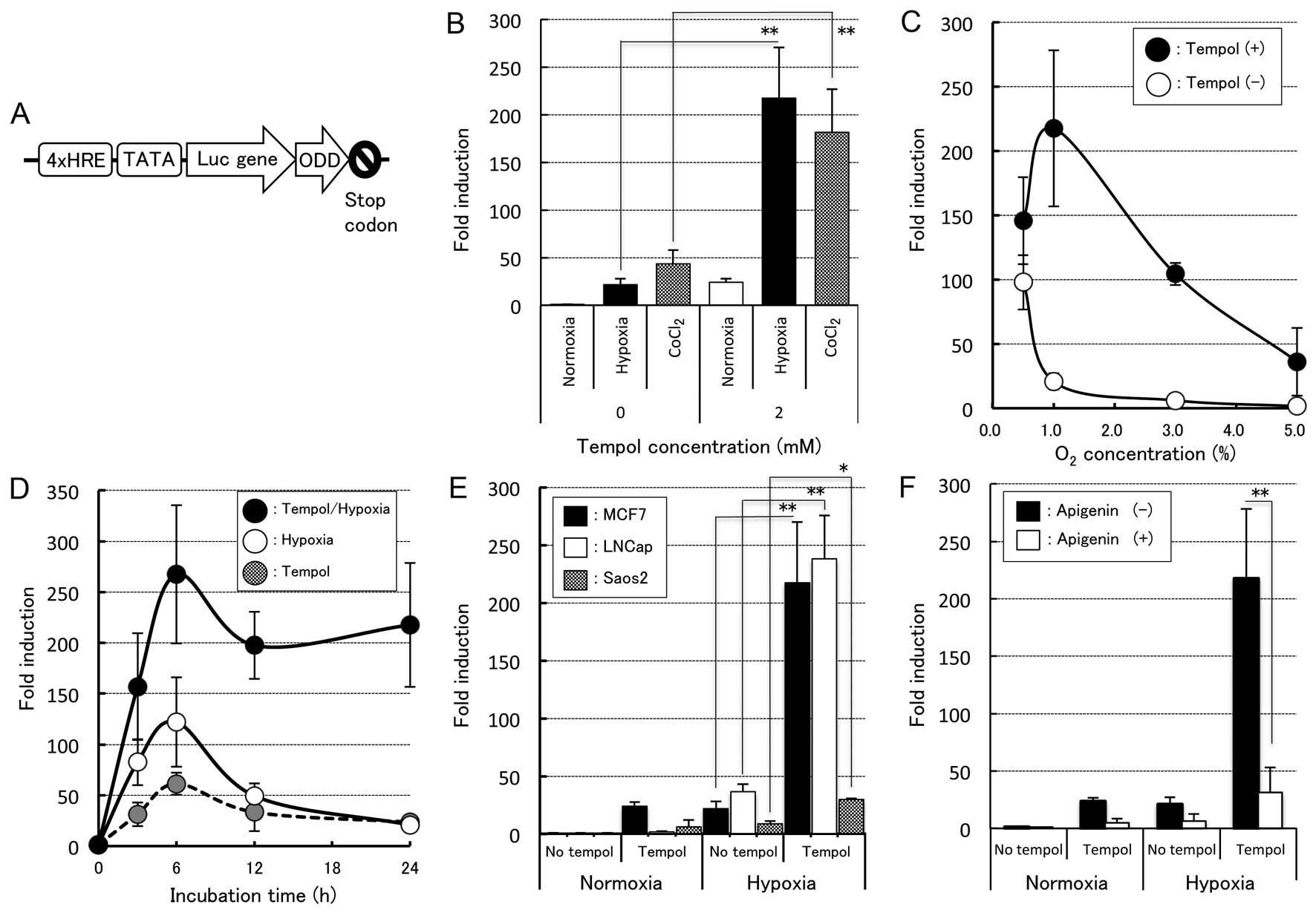 | Figure 2HIF1α-inducible properties induced by
tempol under various conditions. (A) Schematic structure of the
reporter plasmid p4HRE-Luc-ODD. The promoter sequence consisted of
four copies of HRE fragments and the TATA-box was fused upstream of
the luc gene. The ODD sequence amplified from HIF1α cDNA was
inserted downstream of this site. (B) Induction of the Luc activity
by tempol under hypoxia or CoCl2 treatment. MCF7 cells
transiently transfected with the plasmid p4HRE-Luc-ODD were allowed
to recover for 12 h after transfection under normoxia and exposed
to hypoxia (1.0% O2) or treated with 100 µM of
CoCl2 with or without 2 mM tempol for 24 h and then
assayed for Luc activity. The fold induction was calculated by
dividing the RLU of each stimulated cell by the RLU of the control
cells under normoxia without tempol. Each column presents the
average and standard deviation (n=5–7). **P<0.001,
significant increase vs. hypoxia and CoCl2 without
tempol (ANOVA with Bonferroni/Dunn). (C) Induction of the Luc
activity in the transiently transfected MCF7 as a function of the
oxygen concentration. The transfected cells were exposed to various
concentrations of O2 (0.5, 1.0, 3.0 and 5.0%
O2) with or without 2 mM tempol for 24 h. The fold
induction was calculated by dividing the RLU of the transfected
cells with or without tempol at various O2
concentrations by the RLU of the control cells under normoxia
without tempol. The error bars represent the standard deviation
(n=5). (D) Time courses of the Luc activity induced by tempol under
hypoxia and normoxia. The transfected MCF7 cells were exposed to
1.0% O2 or normoxia with or without 2 mM tempol, and the
fold induction was calculated by dividing the RLU of each
stimulated cell by the RLU of the control cells under normoxia
without tempol at the indicated time-points (3, 6, 12 and 24 h).
The error bars represent the standard deviation (n=5). (E)
Induction of the Luc activity in three cancer cell lines, MCF7,
LNCap and Saos2, by tempol under hypoxia and normoxia. Each cell
line was exposed to 1.0% O2 with 2 mM tempol for 24 h.
The fold induction was normalized to the RLU of the control cells
treated without tempol under normoxia. Each column presents the
average and standard deviation (n=3–7). *P<0.05 and
**P<0.01, significant increase vs. hypoxia (ANOVA
with Bonferroni/Dunn). (F) Suppression of the Luc activity by
HIF!1α, apigenin. The transfected cells were treated with 40
µM of apigenin or vehicle (0.1% DMSO) for 1 h prior to
exposure to 1.0% O2 with or without 2 mM tempol for 24
h. The fold induction was normal-ized to the RLU of the control
cells treated without tempol and apigenin under normoxia. Each
column presents the average and standard deviation (n=5–7).
**P<0.01, significant increase vs. without apigenin
(ANOVA with Bonferroni/Dunn). HIF, hypoxia-induced factor; ODD,
oxygen-dependent degradation domain; HRE, hypoxia-responsive
element; RLU, relative luciferase units. |
Transfection and establishment of stable
cell lines
For the transient transfection experiments,
3.0×105 cells were seeded prior to transfection into
60-mm glass Petri dishes and maintained for 12 h in the atmosphere
of a 5% CO2 incubator at 37°C. In order to determine the
rates of enhancement of the HIF-1α activity induced by tempol under
hypoxia, the p4HRE-Luc-ODD construct was co-transfected with
pGL-4.74, an internal control vector containing the Renilla
luc gene (Promega, Madison, WI, USA) at a ratio of 50:1
(p4HRE-Luc-ODD vs. pGL-4.70) using the Effectene transfection
reagent (Qiagen, Valencia, CA, USA), followed by incubation for 8
h. In addition, the p4HRE-fcy::fur-ODD construct was transfected
into MCF7 cells in accordance with the manufacturer’s instructions
in order to confirm the expression of the Fcy::Fur-ODD fused
protein. We established three stably transfected cell lines. For
this process, 1.0×106 MCF7 cells were seeded into 100-mm
culture dishes and subsequently transfected with the plasmids
p4HRE-Luc-ODD, pGL3-control and p4HRE-fcy::fur-ODD in addition to
the plasmid pIRES2-EGFP (Takara Bio, Inc.) containing the
neomycin/kanamycin resistance gene. The transfected cells were
cultured for ~14 days in culture medium containing 1 mg/ml of G418
(Nacalai Tesque, Kyoto, Japan), after which antibiotic-resistant
colonies were isolated and evaluated for the expression of the gene
of interest (Luc-ODD, Luc or Fcy::Fur-ODD) in order to select
successfully established stable transfectants. Representative
colonies were proliferated and designated the MCF7/HRE-Luc-ODD,
MCF7/SV40-Luc or MCF7/HRE-fcy::fur-ODD cell lines,
respectively.
Luciferase reporter assay
Following transfection, the cells were incubated for
12 h in fresh medium and then subjected to various combination
treatments with tempol and O2 at different durations of
incubation. As for the HIF-1α inhibition experiments with apigenin,
the transfected cells were treated with 40 µM of apigenin or
vehicle [0.1% dimethylsulfoxide (DMSO)] for 1 h prior to exposure
to the hypoxic conditions. Echinomycin was also used as an
inhibitor for HIF-1α in a similar manner. The treated cells were
then washed in phosphate-buffered saline (PBS) and lysed with 400
µl of passive lysis buffer (Promega) for 15 min at an
ambient temperature. A Luc assay was performed using
Dual-Luciferase assay reagent (Promega), in accordance with the
manufacturer’s instructions. The Luc activity of the p4HRE-Luc-ODD
construct was determined in relative luminescence units (RLU),
where the value of luminescence by the firefly Luc activity was
divided by that of the Renilla Luc activity in the same
lysate. The degree of enhancement of the Luc activity induced by
each treatment was expressed as the fold induction, where the RLU
value for a sample treated with a given treatment was divided by
that for an identically prepared sample treated without the
treatment.
Western blot analysis
Following the various treatments, 1.5×106
cells were washed in PBS twice and harvested via centrifugation.
The cells were then lysed in 150 µl of RIPA buffer [50 mM
Tris-HCl pH 7.4, 400 mM NaCl, 1% (v/v) Nonidet P-40 and 0.25% (w/v)
Na-deoxycholate] with protease inhibitor (Sigma-Aldrich) and stored
in a freezer at −20°C until use. The cell lysates (40
µg/lane) were subsequently separated using 4–20% gradient
polyacrylamide with SDS-polyacrylamide gel electrophoresis and
transferred onto nitrocellulose membranes (Schleicher &
Schuell, Keene, NH, USA). After blocking the membranes with 3% skim
milk in PBS for 1 h, the cells were incubated with anti-HIF-1α
antibodies (1:500), anti-Myc-tag antibodies (1:1,000) or
anti-β-actin antibodies (1:1,000) overnight at 4°C. After washing
the membranes three times with 0.1% Triton X-100 in PBS, the cells
were incubated with horseradish peroxidase-conjugated secondary
antibodies for 90 min, and the protein expression was visualized
with ECL and blotting detection reagents using an LAS-500
luminescence imaging analyzer (all from GE Healthcare, UK Ltd.,
Buckinghamshire, UK).
Colony formation assay
MCF7/HRE-fcy::fur-ODD cells were plated into culture
dishes and exposed to medium containing 2 mM tempol, 100 µM
of CoCl2 and 10 mM 5-fluorocytosine (5-FC) for 48 h.
After replacing the medium in the dishes with fresh medium, the
cells were incubated to form colonies for 14 days. For the
evaluation, the formed colonies were stained with methylene blue
and surviving colonies consisting of more than ~30 cells were
counted. The plating efficiency (PE) was defined as the number of
observed colonies divided by the number of plated cells treated
without any treatment. The surviving fraction (SF) was calculated
as the number of observed colonies divided by the number of plated
cells after treatment, with correction using the PE.
Luc activity in the tumor tissues
Suspensions of 3.5×106 MCF7/HRE-Luc-ODD
and MCF7/SV40-Luc cells in PBS were mixed with an equal volume of
Matrigel Basement Membrane matrix (BD Biosciences, Bedford, MA,
USA) and subcutaneously injected into the left and right flanks of
4-week-old KSN mice separately. When the tumors grew to 10 mm in
average diameter after inoculation, the tumor-bearing mice were
subjected to in vivo bioluminescence experiments. For the
control experiment, the mice were injected intra-peritoneally with
500 µl of tempol at 100 mM in PBS or PBS alone. For in
vivo bioluminescent imaging, the tumor-bearing mice were
injected with Luciferin EF (Promega) at a dose of 100 µg/g
body weight intraperitoneally 6 h after tempol or PBS
administration and then subjected to treatment with a
bioluminescence detection system (AEQUORIA-2D/c8600; Hamamatsu
Photonics K.K., Hamamatsu, Japan) 5 min after substrate
administration. Images of the Luc expression were captured with
integration time for 60 sec using the Wasabi application software
program (ver. 1.5; Hamamatsu Photonics K.K.). The Luc activity was
determined in RLU, where the luminescence value of the
MCF7/HRE-Luc-ODD tumor was divided by that of the MCF7/SV40-Luc
tumor in a given mouse. The degree of enhancement of the Luc
activity induced by tempol in each tumor was expressed as the ratio
of RLU, where the RLU value after tempol injection was divided by
that observed before tempol injection.
Statistical analysis
All values are expressed as the mean ± standard
deviation. Significant differences between groups were determined
using a one-way analysis of variance (ANOVA) or the Student’s
unpaired t-test. If the results of the ANOVA were significant, the
Bonferroni/Dunn procedure was used as a post hoc test. A P<0.05
was considered to be statistically significant.
Results
Tempol strongly enhances the expression
of HIF-1α in combination with hypoxia or CoCl2
treatment
Tempol is currently being assessed in a Phase II
clinical trial as a drug to prevent radiation-induced alopecia.
However, little is known about its detailed effects on the gene
expression profiles. We thus examined the influence of tempol on
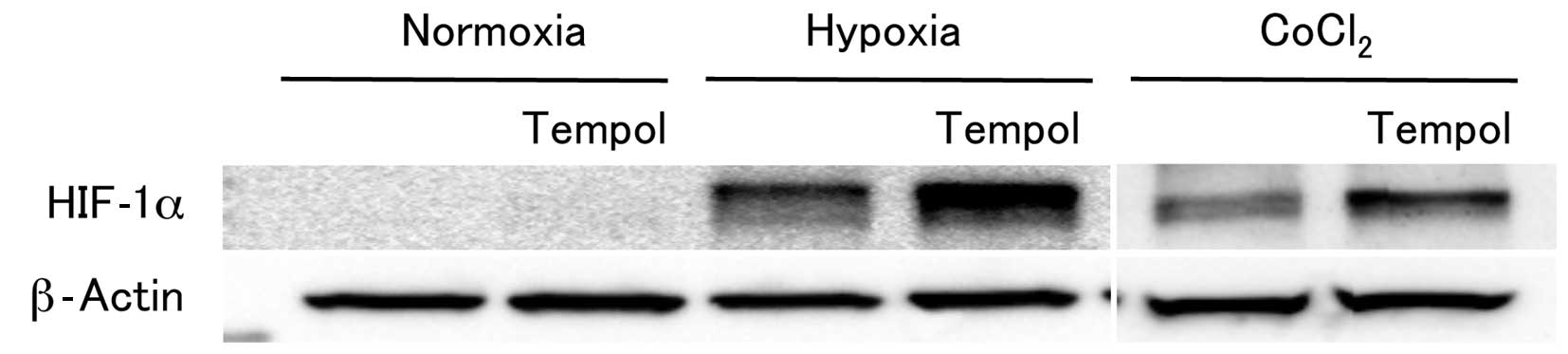
the accumulation of HIF-1α using a western blot analysis. Among the
genes tested, the expression of HIF-1α, which plays an essential
role in cellular responses to hypoxia, was markedly increased in
the MCF7 cells by treatment with 2 mM tempol and 1.0% O2
for 6 h (Fig. 1). The strongly
enhanced HIF-1α expression was observed 24 h after incubation with
tempol under conditions of hypoxia (data not shown). Similarly,
treatment with 100 µM CoCl2, a hypoxia-mimicking
agent, enhanced the expression of HIF-1α in combination with the
application of 2 mM tempol. Meanwhile, the HIF-1α expression was
detected at very low levels when the cells were treated with tempol
under normoxia (Fig. 1). These
results indicate that tempol selectively enhances the HIF-1α
expression under conditions of hypoxia.
We next carried out a Luc reporter assay to evaluate
the degree of enhancement of the activity of HIF-1α induced by
combination treatment with tempol and hypoxia. A plasmid,
p4HRE-Luc-ODD, containing the luc gene linked to four
tandemly repeated HRE/TATA boxes and a DNA fragment with the ODD
sequence was constructed and transiently transfected into MCF7
cells (Fig. 2A). Consequently, the
Luc activity in the cells transfected with p4HRE-Luc-ODD treated
under 1.0% O2 for 6 h was ~22-fold that observed in the
control cells under normoxia, while the Luc activity in the cells
transfected with p4HRE-Luc-ODD treated with 2 mM tempol under 1.0%
O2 increased up to ~217-fold compared to that noted in
the no tempol-treated cells under normoxia (~10-fold enhancement
compared to that in the no tempol-treated cells under 1.0%
O2; P<0.001, Fig.
2B). Similarly, the expression of HIF-1α in the cells treated
with both 2 mM tempol and 100 µM of CoCl2
demonstrated much higher enhancement (~181-fold) than that noted in
the no tempol-treated cells under normoxia (P<0.001, Fig. 2B). However, the transfected cells
treated with more than 2 mM tempol showed a lower Luc activity than
the no tempol-treated cells and high cytotoxicity associated with
alteration of their cell shape. In order to obtain more detailed
data regarding the enhancement of HIF-1α by tempol, we assessed the
O2 concentration- and time-dependent effects in
enhancing the HIF-1α expression. For the O2
concentration-dependent effects study, transient cells were exposed
to medium containing O2 at various concentrations (0.5,
1.0, 3.0 or 5.0%) with 2 mM tempol for 6 h. As shown in Fig. 2C, the degree of enhancement of the
HIF-1α expression in the no tempol-treated cells was highest at
0.5% O2 and decreased as the O2 concentration
increased. In contrast, the extent of enhancement of the HIF-1α
expression in the tempol-treated cells was highest at 1.0%
O2, indicating a different pattern from that observed in
the no tempol-treated cells. These results were also confirmed by
western blot analysis (data not shown). As for the time-dependent
effects study, enhancement of the HIF-1α expression in the cells
treated with tempol under 1.0% O2 was observed at 3 h
and peaked at 6 h, then gradually declined thereafter, as shown in
Fig. 2D. Moreover, the expression
of HIF-1α in these cells showed much greater enhancement (157- to
267-fold) at all incubation times than that observed in the cells
treated with either tempol alone or hypoxia alone (Fig. 2D).
We then confirmed whether the expression levels of
HIF-1α in the other cell lines were also affected by treatment with
tempol under conditions of hypoxia. Consequently, LNCaP cells and
Saos2 cells were subjected to similar assays. Following treatment
with tempol, the HIF-1α expression levels in the two cell lines
under hypoxia were significantly enhanced compared to that observed
in the two cell lines exposed to identical conditions with the
exception of tempol treatment (Fig.
2E). However, the degree of enhancement of the HIF-1α
expression induced by tempol treatment in the Saos2 cells at 1.0%
O2 was ~10 times less than that noted in the MCF7 or
LNCap cells, suggesting the presence of cell type-dependent
variation in the level of HIF-1α enhancement induced by tempol.
In order to verify that the increase in the Luc
activity of p4HRE-Luc-ODD after the combined treatment with tempol
and hypoxia was caused by the enhanced HIF-1α expression, we
performed experiments employing an inhibitor of the HIF-1
expression, apigenin. As a result, the addition of apigenin
significantly suppressed the induction of the Luc activity of
p4HRE-Luc-ODD in the MCF7 cells treated with 2 mM tempol under
hypoxia by up to 86%, confirming an HIF-1α-dependent increase in
Luc activity (Fig. 2F). We then
conducted similar experiments using echinomycin, an inhibitor of
the binding of HIF-1 to HRE. In the presence of this inhibitor, the
increase in the Luc activity induced by tempol in the cells
transfected with p4HRE-Luc-ODD under hypoxia was suppressed in a
concentration-dependent manner (data not shown). Based on these
results, we conclude that the enhancement of the Luc activity
caused by the enhancement of HIF-1α and tempol treatment strongly
enhances the expression of HIF-1α in combination with hypoxia or
CoCl2 treatment.
Selective enhancement of hypoxic cell
killing with the tempol-regulated suicide gene expression
The phenomenon in which the expression of HIF-1α is
enhanced by treatment with tempol under hypoxia may be applied to
selectively enhance cell killing under hypoxia. In order to test
this hypothesis, we constructed a plasmid, p4HRE-fcy::fur-ODD, by
replacing the luc gene of p4HRE-Luc-ODD with a suicide gene,
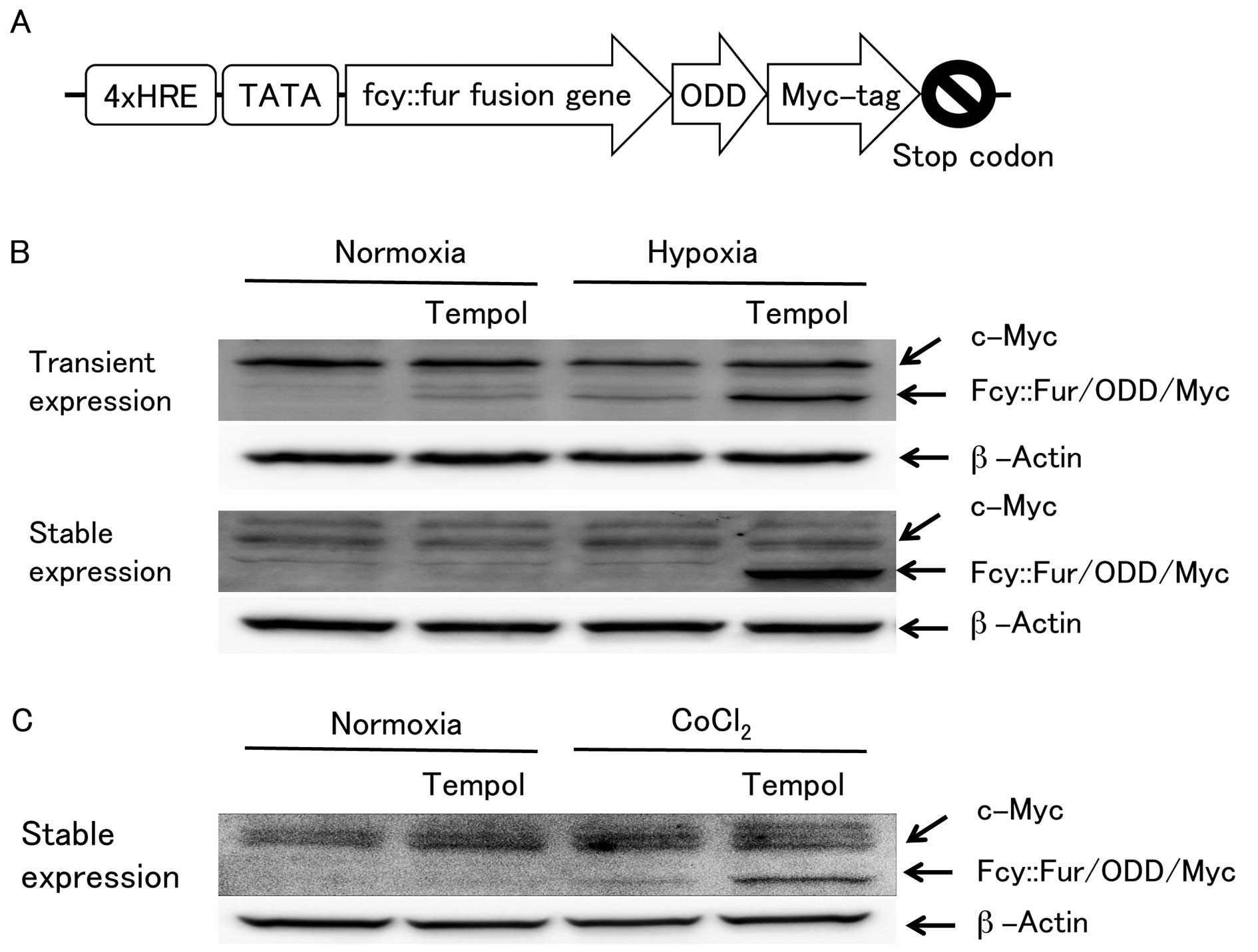
the fcy::fur gene (Fig. 3A).
The suicide gene was similarly modified in terms of the ODD
sequence with a myc-tag sequence attached at the end. The
fcy::fur fusion gene product exhibits a CD and UPRT
activity, which efficiently converts the less cytotoxic 5-FC to the
highly cytotoxic 5-FU and 5-FUMP. Therefore, cells expressing this
gene are killed in the presence of 5-FC. As shown in Fig. 3B and C, enhancement of the
fcy::fur fusion gene expression by tempol under hypoxia was
verified by western blot analysis using antibodies against myc-tag.
Moreover, strongly enhanced expression levels of the
fcy::fur fusion gene product were observed in both cells
transiently and stably transfected with MCF7/HRE-fcy::fur-ODD when
treated with tempol under hypoxia. A similar tendency was observed
among the stably transfected cells cultured with CoCl2
instead of under hypoxia. These results are consistent with those
obtained in the Luc reporter assay described above, indicating that
the fcy::fur fusion gene expression is also enhanced by
tempol, similar to the luc gene.
We then carried out a colony formation assay to
evaluate the degree of enhancement of selective cell killing
induced by tempol under hypoxic mimic conditions using
MCF7/HRE-fcy::fur-ODD cells. Stably transfected cells were cultured
in the presence of 2 mM tempol and 100 µM of
CoCl2 in addition to 10 mM 5-FC for 48 h, and the
cultured cells were washed in fresh medium and further incubated
without these chemicals for ~2 weeks to allow the cells to form
colonies. The number of colonies formed by the tempol-treated cells
was much lower than that formed by the control cells, even without
5-FC treatment, suggesting that these effects were possibly due to
the cytotoxicity of tempol. On the other hand, when stably
transfected cells were cultured in medium containing all of the
chemical reagents of tempol, CoCl2 and 5-FC, the number
of colonies was significantly reduced by 19-fold compared to that
formed by the cells treated similarly with the exception of 5-FC
(Fig. 4A and C). In addition,
alteration of the cellular morphology was observed only when the
cells were cultured in medium containing all of the chemical
reagents (Fig. 4B). These results
suggest that the application of this suicide gene expression
regulation system composed of an HRE promoter and the ODD sequence
in combination with tempol treatment may be useful for the
selectively killing of cells under hypoxia.
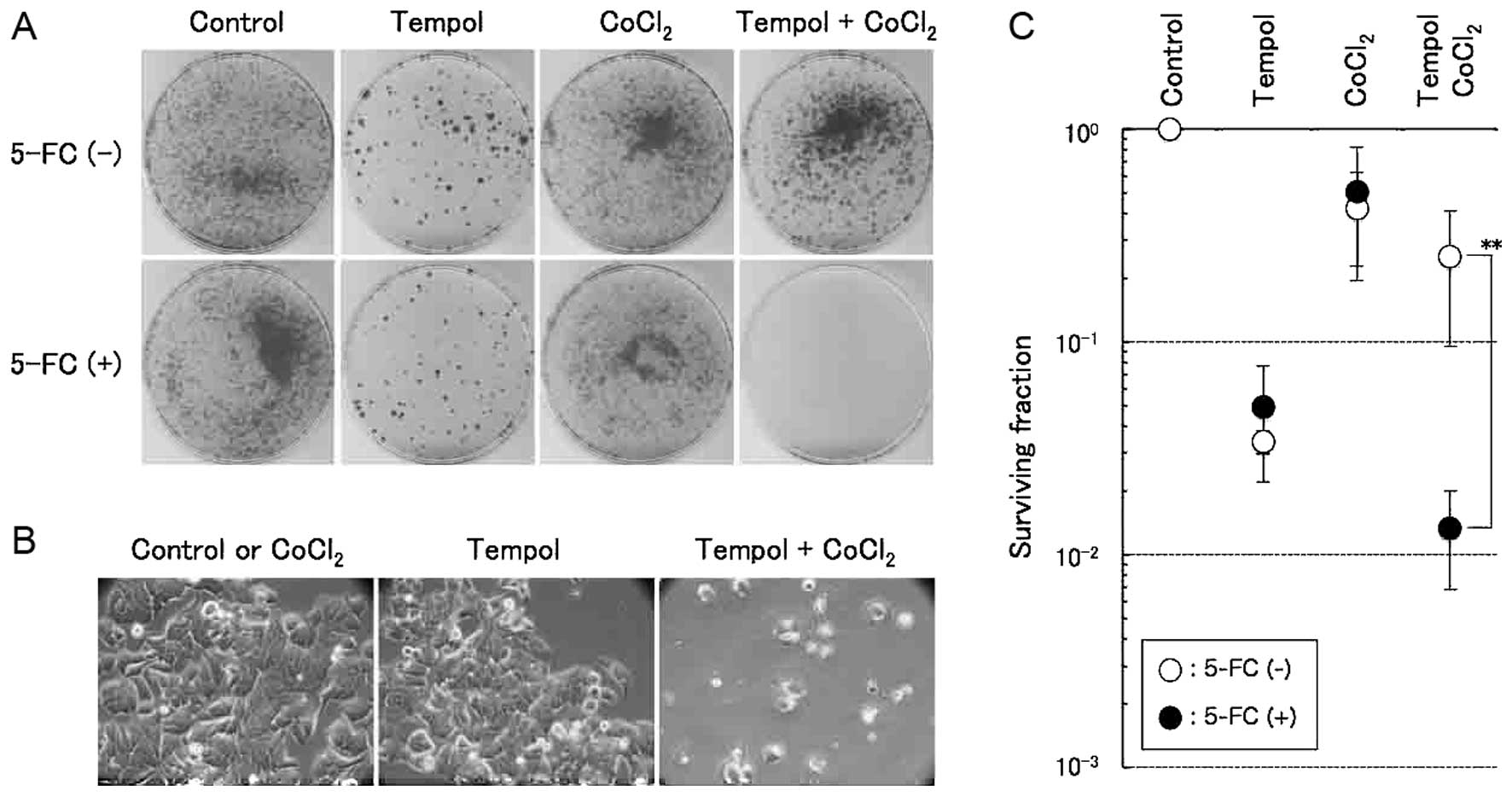 | Figure 4Selective enhancement of hypoxic cell
killing by Fcy::Fur fusion proteins induced by treatment with a
combination of tempol and CoCl2. (A) Selective hypoxic
cell killing was assessed using a colony formation assay. After the
stable expression clone, MCF7/HRE-fcy::fur-ODD cells, was treated
with 2 mM tempol and 100 µM of CoCl2 for 48 h in
10 mM 5-FC-containing medium, the cells were divided and plated for
a colony formation assay for 14 days. 5-FC (-), colony incubated
without 5-FC; 5-FC (+), colony incubated with 5-FC. (B) Alteration
of the cellular morphology of the stable cell lines treated with 2
mM tempol and/or 100 µM of CoCl2 in
5-FC-containing medium. (C) Surviving fraction of the stable
expression clone, MCF7/HRE-fcy::fur-ODD, after treatment with 2 mM
tempol and/or 100 µM of CoCl2. The error bars
represent the standard deviation (n=3). **P<0.01,
significance vs. without 5-FC (Student’s unpaired t-test). ODD,
oxygen-dependent degradation domain; HRE, hypoxia-responsive
element. |
Enhancement of the HIF-1α expression by
tempol in the solid tumors
Hypoxic regions have been reported to be found
inside solid tumor tissues. We thus investigated whether the gene
expression system developed in this report can be applied for gene
regulation in hypoxic regions of solid tumors in vivo. We
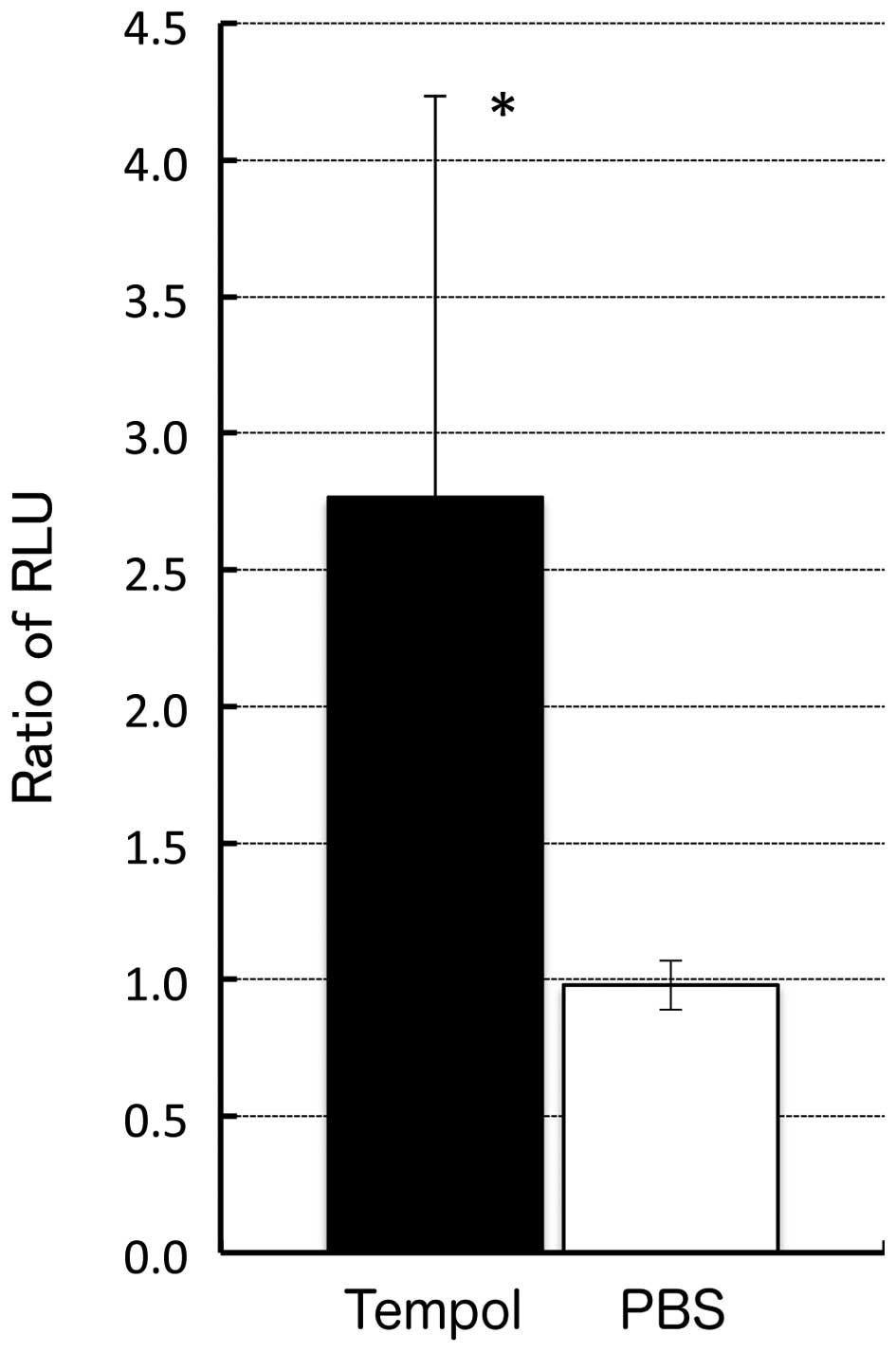
subsequently examined changes in the Luc activity in the xenograft
MCF7/HRE-Luc-ODD and MCF7/SV40-Luc tumor tissues in nude mice
following the injection of tempol by observing the extent of photon
generation due to the oxidation of a substrate administered to the
mice. As shown in Fig. 5, the ratio
of RLU in the mice injected with tempol was increased by more than
2.8±1.9 times compared with that observed in the control group
injected with PBS, suggesting the regulation of the gene expression
by our system following tempol injection both in vivo and
in vitro. However, the fold induction observed in
vivo was much lower than that obtained in vitro.
Discussion
In the present study, we demonstrated the effects of
tempol in upregulating the HIF-1α expression directly as well as
indirectly via an increase in the Luc expression of p4HRE-Luc-ODD.
Furthermore, under hypoxic conditions, it was confirmed that tempol
upregulated the luc gene expression of p4HRE-Luc-ODD in all
three transfected cell lines employed in this study (MCF7, LNCap
and Saos2), although to various degrees. Notably, all of these
episodes of upregulation were suppressed by treatment with either
apigenin, an inhibitor of the HIF-1 expression, or echinomycin, an
inhibitor of the binding of HIF-1 to HRE, thus confirming that
tempol upregulated the Luc expression via upregulation of the
HIF-1α expression. Considering the expression of the luc
gene under the control of four copies of HRE in the p4HRE-Luc-ODD
transfected MCF7 cells as an index of HIF-1α activity, the HIF-1α
expression was inversely regulated according to the oxygen
concentration. Namely, the rate of induction reached a maximum at
an oxygen concentration of 0.5%, the minimum applied in this study,
and subsequently declined in an exponential manner as the oxygen
concentration increased. On the other hand, when the cells were
treated with tempol, the rate of induction reached a peak at an
oxygen concentration of 1.0% rather than 0.5% (Fig. 2C). The p4HRE-Luc-ODD construct
carries a promoter composed of four copies of HRE and the TATA-box
to drive the luc gene. Therefore, the expression of this
gene is tightly controlled by the HIF-1α expression. In other
words, it inversely correlates with the oxygen concentration. In
addition, the Luc protein attaches with the ODD domain at its
C-terminal end, such that its accumulation is also inversely
regulated based on the oxygen concentration. Therefore, oxygen
suppresses the transcription of the luc gene and the
accumulation of Luc proteins in our gene regulation system applied
for p4HRE-Luc-ODD. However, the Luc expression was the highest at
1.0% oxygen concentration, rather than 0.5%, when MCF7 cells
transiently transfected with p4HRE-Luc-ODD were treated with
tempol, suggesting that the upregulation of HIF-1α is not
necessarily dependent on the degree of hypoxia. Although the reason
for this observation remains unknown, we consider that the
expression of the luc gene in our gene regulation system may be
determined by the balance between the increase in the HIF-1α
expression achieved with tempol treatment and the degradation of
HIF-1α and Luc via ODD induced by oxygen.
We then applied our gene regulation system for
cancer gene therapy targeting hypoxic regions using the
fcy::fur fusion gene that converts 5-FC, a pro-drug, to
5-FU, an anticancer drug as a suicide gene. Consequently, the rate
of cell survival in the MCF7/HRE-fcy::fur-ODD cells treated with
tempol under normoxia was not affected by the addition of 5-FC,
suggesting that the cell death observed under these conditions was
due to the cytotoxicity of tempol. In contrast, when these cells
were treated with 5-FC in the presence of tempol and
CoCl2, a hypoxic mimic compound, a 19-fold higher cell
death rate was observed compared with that noted in the same cells
treated identically with the exception of 5-FC. These results
indicate that our gene regulation system increased the suicide gene
expression in response to the addition of tempol only under
hypoxia, thereby facilitating cell killing specifically in hypoxic
regions. Interestingly, the rate of cell killing in the stably
transfected cells treated with tempol and CoCl2 only in
the absence of 5-FC was much lower than that observed in the same
cells treated identically with the exception of CoCl2.
Although the precise reasons for this phenomenon remain unclear, we
assume that the oxidized form of tempol is converted to its reduced
form via a reaction with cobalt, presumably reducing the
cytotoxicity of tempol.
Furthermore, we performed an in vivo
experiment to evaluate whether our gene regulation system works
in vitro in order to explore possible applications for
cancer gene therapy. We prepared two cell lines: MCF7/HRE-Luc-ODD
and MCF7/SV40-Luc. The former cell line was stably trans-fected
using our gene regulation system to express the luc gene in
response to tempol under hypoxia, while the latter was stably
transfected using a control gene expression system to express the
luc gene under the control of the strong SV40 promoter,
which is not affected by either tempol or hypoxic conditions. Mice
were subcutaneously administered the former cell line in the right
flank and the latter cell line in the left flank. Following tumor
formation, using an in vivo imaging apparatus, we evaluated
the increase in the Luc expression based on the degree of
luminescence emitted via the oxidation of a substrate injected
intraperitoneally before and after tempol stimulation. The amount
of luminescence emitted from the MCF7/HRE-Luc-ODD tumors in the
mice administered tempol was 2.8 times higher than that emitted
from the same tumors in the mice administered PBS. Although this
difference was statistically significant (P=0.0141), it was very
small compared to that observed in the in vitro experiment.
Tempol is a stable free radical of organic matter with an unpaired
electron. Based on the obtained results, the HIF-1α expression is
upregulated by the oxidized form of tempol. In the living body, the
oxidized form of tempol is converted to the reduced form by
antioxidants, including glutathione. Hyodo and colleagues reported
the period of the conversion from the oxidized forms to the reduced
forms in the living body to be 2–3 min (21). In addition, hypoxic regions in tumor
tissues are located 70–100 mm from blood vessels (22). Hence, in the living body, the access
of tempol to hypoxic regions is hampered by its short-lived
activity and the long distance from blood vessels. We consider one
reason for the smaller degree of HIF-1α enhancement observed in
this study to be that the number of molecules having an effect on
the HIF-1α expression in vivo is fewer than that seen in
vitro, thus reducing the rate of enhancement of the Luc
expression in the tumor xenograft model. Furthermore, the pressure
of oxygen in solid tumors is known to be highly heterogeneous, with
the overall hypoxic fraction (pO2 ≤2.5 mmHg) in tumors
estimated to be <25% according to clinical studies employing
computerized polarographic needle electrodes (23). Therefore, it is difficult to detect
the chemiluminescence of the Luc expression due to the
significantly reduced numbers of hypoxic cells in the tumor tissues
versus in vitro experiments, resulting in smaller levels of
enhancement of HIF-1α in the living body. Considering the
application of our system, which regulates the suicide gene
expression in hypoxic regions, for site-specific cancer gene
therapy, nitroxides with similar properties to tempol, including
high membrane permeability and a longer period of being in the
oxidized form in the living body, may be the ideal compound to
regulate suicide gene expression in hypoxic environments, if it is
less cytotoxic.
Hypoxic cells show resistance to radiation therapy
and chemotherapy due to the ability of a fraction of these cells to
survive after therapy, thus remaining a source for recurrence and
metastasis. Therefore, there have been many attempts to target
hypoxic cells. Among such approaches, gene therapy for only hypoxic
regions has been developed, in which a suicide gene is expressed
under the control of HRE to specifically target cells in hypoxic
regions. However, with current technology, it is very difficult to
transfer an adequate vector to eradicate all hypoxic cells.
Therefore, our method of enhancing the suicide gene expression
specifically in hypoxic regions using nitroxide is a promising
strategy for targeting hypoxic cells.
Acknowledgments
This research was supported in part by the
Intramural Research Program of the National Institutes of Health,
National Cancer Institute. In addition, this research was also
supported in part by Grants-in-Aid for Scientific Research (C)
(nos. 21591618 and 24591849) from the Japan Society for the
Promotion of Science and by a grant from the Kitasato University
School of Allied Health Sciences (Grants-in-Aid for Research
Project, no. 2012–1018).
Abbreviations:
|
HIF
|
hypoxia-inducible factor
|
|
Luc
|
luciferase
|
|
ODD
|
oxygen-dependent degradation
domain
|
|
HRE
|
hypoxia-responsive element
|
|
PHD
|
prolyl hydroxylase
|
|
VHL
|
von Hippel-Lindau protein
|
|
ROS
|
reactive oxygen species
|
|
VEGF
|
vascular endothelial growth factor
|
|
GLUT
|
glucose transporter
|
|
CA
|
carbonic anhydrase
|
|
IGF
|
insulin-like growth factor
|
|
TGF
|
transforming growth factor
|
|
TPZ
|
tirapazamine
|
|
HSVtk
|
herpes simplex virus type 1 thymidine
kinase
|
|
RLU
|
relative luciferase units
|
|
CD
|
cytosine deaminase
|
|
UPRT
|
uracil phosphoribosyltransferase
|
References
|
1
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar
|
|
2
|
Lee JW, Bae SH, Jeong JW, Kim SH and Kim
KW: Hypoxia-inducible factor (HIF-1)α: Its protein stability and
biological functions. Exp Mol Med. 36:1–12. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hu CJ, Wang LY, Chodosh LA, Keith B and
Simon MC: Differential roles of hypoxia-inducible factor 1α
(HIF-1α) and HIF-2α in hypoxic gene regulation. Mol Cell Biol.
23:9361–9374. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maynard MA, Evans AJ, Hosomi T, Hara S,
Jewett MA and Ohh M: Human HIF-3α4 is a dominant-negative regulator
of HIF-1 and is down-regulated in renal cell carcinoma. FASEB J.
19:1396–1406. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li QF, Wang XR, Yang YW and Lin H: Hypoxia
upregulates hypoxia inducible factor (HIF)-3α expression in lung
epithelial cells: Characterization and comparison with HIF-1α. Cell
Res. 16:548–558. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang BH, Semenza GL, Bauer C and Marti
HH: Hypoxia-inducible factor 1 levels vary exponentially over a
physiologically relevant range of O2 tension. Am J
Physiol. 271:C1172–C1180. 1996.PubMed/NCBI
|
|
7
|
Sabharwal SS and Schumacker PT:
Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’
heel? Nat Rev Cancer. 14:709–721. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Semenza GL: HIF-1: Upstream and downstream
of cancer metabolism. Curr Opin Genet Dev. 20:51–56. 2010.
View Article : Google Scholar :
|
|
9
|
Wykoff CC, Beasley NJP, Watson PH, Turner
KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell
PH, et al: Hypoxia-inducible expression of tumor-associated
carbonic anhydrases. Cancer Res. 60:7075–7083. 2000.
|
|
10
|
Feldser D, Agani F, Iyer NV, Pak B,
Ferreira G and Semenza GL: Reciprocal positive regulation of
hypoxia-inducible factor 1α and insulin-like growth factor 2.
Cancer Res. 59:3915–3918. 1999.PubMed/NCBI
|
|
11
|
Krishnamachary B, Berg-Dixon S, Kelly B,
Agani F, Feldser D, Ferreira G, Iyer N, LaRusch J, Pak B, Taghavi
P, et al: Regulation of colon carcinoma cell invasion by
hypoxia-inducible factor 1. Cancer Res. 63:1138–1143.
2003.PubMed/NCBI
|
|
12
|
Goffman T, Cuscela D, Glass J, Hahn S,
Krishna CM, Lupton G and Mitchell JB: Topical application of
nitroxide protects radiation-induced alopecia in guinea pigs. Int J
Radiat Oncol Biol Phys. 22:803–806. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Metz JM, Smith D, Mick R, Lustig R,
Mitchell J, Cherakuri M, Glatstein E and Hahn SM: A phase I study
of topical Tempol for the prevention of alopecia induced by whole
brain radiotherapy. Clin Cancer Res. 10:6411–6417. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dean M, Fojo T and Bates S: Tumour stem
cells and drug resistance. Nat Rev Cancer. 5:275–284. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rich JN: Cancer stem cells in radiation
resistance. Cancer Res. 67:8980–8984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Prasad KN: Handbook of Radiobiology. 2nd
edition. CRC Press; Boca Ranton, FL: 1995
|
|
17
|
Ido A, Uto H, Moriuchi A, Nagata K, Onaga
Y, Onaga M, Hori T, Hirono S, Hayashi K, Tamaoki T, et al: Gene
therapy targeting for hepatocellular carcinoma: Selective and
enhanced suicide gene expression regulated by a hypoxia-inducible
enhancer linked to a human α-fetoprotein promoter. Cancer Res.
61:3016–3021. 2001.PubMed/NCBI
|
|
18
|
Harada H, Hiraoka M and Kizaka-Kondoh S:
Antitumor effect of TAT-oxygen-dependent degradation-caspase-3
fusion protein specifically stabilized and activated in hypoxic
tumor cells. Cancer Res. 62:2013–2018. 2002.PubMed/NCBI
|
|
19
|
Sambrook J and Russell DW: Molecular
Cloning: A Laboratory Manual. 1. 3rd edition. Cold Spring Harbor
Laboratory Press; New York, NY: 2001
|
|
20
|
Kaluz S, Kaluzová M and Stanbridge EJ:
Rational design of minimal hypoxia-inducible enhancers. Biochem
Biophys Res Commun. 370:613–618. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hyodo F, Matsumoto K, Matsumoto A,
Mitchell JB and Krishna MC: Probing the intracellular redox status
of tumors with magnetic resonance imaging and redox-sensitive
contrast agents. Cancer Res. 66:9921–9928. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hall EJ: Radiobiology for the
Radiobiologist. 4th edition. J. B. Lippincott Company;
Philadelphia: 1994
|
|
23
|
Vaupel P, Höckel M and Mayer A: Detection
and characterization of tumor hypoxia using pO2
histography. Antioxid Redox Signal. 9:1221–1235. 2007. View Article : Google Scholar : PubMed/NCBI
|