Introduction
Slits are large secreted proteins (up to 200 kDa)
that regulate neuronal orientation and branching during nervous
system development by signaling through cognate Robo receptors that
consist of four known members (Robo1-4). In mammals, three
Slit genes, Slit1, Slit2 and Slit3,
have been identified. All the Slit proteins contain four
leucine-rich repeat domains, six EGF domains, a laminin G domain
and a cysteine-rich domain (1). All
three Slit proteins are proteolytically cleaved into a large
N-terminal fragment (140 kDa) and a shorter C-terminal fragment
(50–60 kDa) (2,3). The majority of the N-terminal fragment
and full-length Slits are membrane-associated, which mediates their
function. The C-terminal fragment is more diffuse and although its
function remains largely unknown, Condac et al recently
reported the pro-coagulation effects of the Slit3 C-terminal
fragment by neutralizing the anticoagulant activity of heparin
(4).
Slit1 expression is specific to neurons,
while Slit2 and Slit3 are detected in various
tissues, such as the lung, kidney and heart (5,6).
Compared with normal tissues, Slit genes are epigenetically
silenced in a wide variety of cancers, indicating that Slits are
potent tumor suppressors (7,8). To
date, most studies involving the effects of Slits on tumors have
focused on Slit2, including epigenetic modifications (9,10),
expression in various types of cancer and inhibition of tumor
growth, metastasis (11–17) and angiogenesis (18,19).
Although most results support the inhibitory effects of Slit2 on
tumor growth and metastasis, the role of Slit2 in growth and
metastasis is still controversial. Several studies have validated a
stimulative role of Slit2 on tumor growth and metastasis (20–22),
and this discrepancy reflects the complexity of Slit/Robo signaling
in different types of cancers.
Slit3 shares a high level of homology with Slit2,
both with the same proteolytic cleavage site, TSPCD, at the
beginning of the 6th EGF repeat (2). Slit3 is detected in various
normal tissues, with skin, brain cerebellum and lung having the
highest expression levels (8).
Genetic inactivation of Slit3 in mice revealed multiple
roles of Slit3 in organ development, particularly in the
diaphragm and kidney (23,24). In the vascular system, Slit3 is
secreted by endothelial cells and vascular smooth muscle cells to
stimulate angiogenesis by binding to the Robo4 receptor (25,26).
Similar to Slit2, frequent hypermethylation of promoters is the
main cause of Slit3 gene silencing in most cancers (8,27,28).
Lung carcinomas appear to be an exception. The frequency of
Slit3 methylation in lung cancers is far less than
Slit2 (8,29), indicating that an alternative
mechanism apart from methylation is responsible for the
downregulation of Slit3. Recombinant Slit3 was reported to
inhibit the migration of melanoma cells (30); the second leucine-rich repeat domain
is sufficient to induce such repressive effects (31). Reduction of miR-218, which resides
in the introns of the Slit2 and Slit3 genes, causes
downregulation of Slit3, resulting in promotion of
metastasis in thyroid cancer (32).
Slit3 overexpression in human breast carcinoma cells
resulted in tumor growth by suppressing CXCR4 expression
(17). These results suggest the
negative regulation of Slit3 on tumor growth and metastasis,
similar to the most reported functions of Slit2. However, the
functions of Slit3 in lung carcinoma have not been widely
reported.
In the present study, we investigated the effects of
Slit3 on lung carcinoma cells by Slit3 gene silencing in
A549 cells, which have a high level of Slit3 expression
compared to other cancer cell lines. We validated the inhibitory
effects of Slit3 on proliferation, migration and invasion in the
cells, confirming a potential tumor-suppressor role of slit3 in
lung carcinoma.
Materials and methods
Cell culture
Human glioblastoma U87MG cells, and human normal
lung fibroblasts (HFL-1) were purchased from the National Platform
of Experimental Cell Resources for Sci-Tech (Beijing, China). Human
lung adenocarcinoma A549, Spc-a-1 and Ltep-a-2 cells were purchased
from the Cell Resource Center at the Institute of Life Sciences,
Chinese Academy of Science (Shanghai, China). All cells were
cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM;
HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum
(FBS; Gibco-BRL, Carlsbad, CA, USA).
Lentiviral infection
shRNA Slit3 oligos were annealed and cloned
into the lentiviral vector pLentiLox 3.7 (a gift from Professor
Jiahuai Han, State Key Laboratory of Cellular Stress Biology,
School of Life Sciences, Xiamen university, China). The
Slit3 N-terminal (33aa–1120aa, ~140 kDa) was amplified from
pSecTag2B-hSlit3 (gift from the Institut National de la Santé et de
la Recherche Médicale, Paris, France) and subcloned into the
lentiviral expression vector pBobi (gift from Professor Jiahuai
Han) with a myc tag at the carboxyl terminus, and the same vector
encoding GFP was used as a control. Virus stocks were prepared by
co-transfecting pll3.7 or pBobi with two packaging plasmids (pHR
and pVSVG) into 293T cells. Viral supernatants were harvested after
48 h, filtered and centrifuged (90 min at 75,000 × g). A549 cells
were infected with pll3.7-Slit3-shRNA in the presence of 8
μg/ml Polybrene (Sigma, St. Louis, MO, USA) for two days.
Infected A549 cells were sorted by clone picking.
Real-time quantitative PCR
Total RNA was extracted with TRIzol reagent (Life
Technologies, Carlsbad, CA, USA). cDNA for the PCR template was
generated using the ReverTra Ace qPCR RT kit (FSQ-101; Toyobo,
Osaka, Japan) as recommended by the manufacturer’s protocols.
Primer sequences are listed in Table
I. Real-time PCR was performed in an MJ Mini System (Bio-Rad)
using Thunderbird SYBR qPCR Mix (QPS-201; Toyobo). Amplification
was performed under the following conditions: 95°C for 1 min
followed by 40 cycles at 95°C for 15 sec, and 62°C for 45 sec. To
normalize the real-time PCR results, GAPDH was used as an
internal control. Relative genes expression in various tumor cell
lines was calculated using the Livak and Schmittgen (33) method and compared with the U87MG
cells.
 | Table IPrimers for real-time quantitative
PCR. |
Table I
Primers for real-time quantitative
PCR.
|
hSLIT1-F |
GCAATTCCACACCGTTGAGC |
|
hSLIT1-R |
AGCTGAGTTGACATCCACGG |
|
hSLIT2-F |
TGAAGGTCTTGCCGAAAGGTAT |
|
hSLIT2-R |
AAAGCGTGCTTATTCTGTTGTT |
|
hSLIT3-F |
GCGATTTGGAGATCCTTACCCT |
|
hSLIT3-R |
GCAGTACAGGTGGTTGGAGTGG |
|
hROBO1-F |
GCATCGCTGGAAGTAGCCATACT |
|
hROBO1-R |
CATGAAATGGTGGGCTCAGGAT |
|
hROBO2-F |
GGGTTACTACATCTGCCAGGCTT |
|
hROBO2-R |
AGGTGGAGGTCTATCTGTCAAAACAT |
|
hROBO3-F |
CAGTGTCCGATGGAAGAAGG |
|
hROBO3-R |
GTCCATCTCCTGCACATTGG |
|
hROBO4-F |
GACACTTGGCGTTCCACCTC |
|
hROBO4-R |
AGAGCAAGGAGCGACGACAG |
| hMMP2-F |
CTTCTTCCCTCGCAAGCC |
| hMMP2-R |
ATGGATTCGAGAAAACCG |
| hMMP9-F |
ACGCAGACATCGTCATCC |
| hMMP9-R |
CCACAACTCGTCATCGTC |
|
hGAPDH-F |
GGCTGAGAACGGGAAGCTTGTCAT |
|
hGAPDH-R |
CAGCCTTCTCCATGGTGGTGAAGA |
Immunofluorescence
Cells were fixed with 4%
paraformal-dehyde/phosphate-buffered saline (PBS) for 15 min at
room temperature. Fixed cells were washed twice with PBS and
permeabilized with 0.25% Triton X-100/PBS for 15 min. Cells were
blocked with 1.0% FBS/PBS for 1 h and labeled with rabbit
polyclonal primary antibody for tubulin (T3526; Sigma) at a 1:80
dilution in 0.1% BSA/PBS buffer with 0.05% Tween-20 (PBST) at 37°C
for 1 h. After incubation, the cells were washed 3 times with PBST
and incubated with Alexa Fluor 594-conjugated goat anti-rabbit
secondary antibody (A11037; Invitrogen) at a 1:1,000 dilution at
room temperature for 1 h in the dark. After washing with PBST, the
cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5
min. After extensive washing, the cells were examined and recorded
with an inverted fluorescence microscope (IX71; Olympus, Japan)
equipped with a SPOT Insight QE CCD camera.
Cell proliferation assay
A total of 500 cells were seeded/well in 96-well
microplates (Sunub, Shanghai, China) and cultured for 4 consecutive
days. The medium was changed every other day. On each day, one
group of culture media was replaced with 100 μl serum-free
fresh medium containing 10% MTT (Sigma) and maintained at 37°C for
4 h. After discarding the medium, 150 μl of
dimethylsulfoxide (DMSO) was added to each well to dissolve MTT
formazan. Optical densities were measured at 560 nm with a
microplate reader (Multiskan MK3; Thermo Fisher Scientific,
USA).
Cell migration and invasion assays
Cell migration was assessed by a wound healing
recovery assay. Cells that were 90% confluent in a 24-well plate
(Sunub) were scratched with a 200-μl pipette tip to form
wound gaps. After washing out cell debris, the wound gaps were
photographed under a microscope at a magnification of ×40 to
acquire a baseline image. The cells were then cultured in DMEM for
the indicated time periods and photographed to obtain the second
set of images. Gap width was measured using Image-Pro Plus 6.0
software. Cell migration was quantitatively analyzed by subtracting
the gap width of the second image from the baseline image.
Cell invasion was assessed by the Matrigel invasion
assay. Chambers (8 μm; Millipore, Billerica, MA, USA) were
coated with 20 μl diluted Matrigel (0.1 mg protein/ml; BD)
for 30 min at 37°C and inserted into a 24-well plate. A total of
1×104 cells/chamber in 100 μl serum-free DMEM
were plated on the upper Matrigel-coated chambers. Medium in the
lower chambers contained 10% FBS as the source of chemoattractant.
The plates were incubated for 24 h at 37°C. Cells that had invaded
the lower surface at 37°C were fixed with methanol and stained with
crystal violet. Three random fields were counted under a light
microscope.
Gelatin zymography
A total of 2 million cells were cultured in a 6-cm
dish (Sunub) with 5 ml DMEM (10% FBS) for 8 h. Media were replaced
with 2 ml serum-free DMEM and incubated for an additional 12 h. The
conditioned media was collected and filtered with a 0.22-μm
filter (Millipore). A total of 10 μl of conditioned media
was mixed with 10 μl sample buffer [0.25 M Tris-HCl, pH 6.0,
8.5% glycerol, 4% sodium dodecyl sulfate (SDS) and 0.01%
bromophenol blue] and electrophoresed on a 7.5% SDS-polyacrylamide
gel containing 2 mg/ml gelatin (Sigma). After electrophoresis, the
gel was washed 3 times for 10 min in 2.5% Triton X-100 and placed
in incubation buffer (50 mM Tris-HCl, pH 7.6, 10 mM
CaCl2, 50 mM NaCl and 0.05% Brij-35) over night at 37°C.
After incubation, the gel was stained with a solution of 0.25%
Coomassie blue R250, 40% methanol and 10% acetic acid for 1 h at
room temperature and destained with 40% methanol and 10% acetic
acid until the protein bands were apparent.
Immunoblotting
To detect E-cadherin and vimentin, the cells were
washed with cold PBS (pH 7.4), incubated with cold RIPA lysis
buffer (P0013; Beyotime, China) on ice for 10 min, and collected
with a scraper. Lysates were then centrifuged at 12,000 × g for 10
min. To detect membrane-associated Slit3 protein, a Qproteome Cell
Compartment kit (Qiagen, Hilden, Germany) was used to isolate the
protein. Protein samples were boiled at a ratio of 3:1 with sample
buffer (250 mM Tris-HCl, pH 6.8, 40% glycerol, 20% mercaptoethanol,
8% SDS and 0.04% bromophenol blue) and separated by SDS-PAGE.
Following electrophoresis, proteins on the gel were transferred to
an Immobilon-P membrane (PVDF; 0.45 μm; Merck Millipore,
Molsheim, France), which was blocked with 5% skim milk powder
dissolved in TBS buffer with 0.05% Tween-20 (TBST). After washing,
the membranes were incubated with rabbit polyclonal primary
antibody for Slit3 (AB5703P; Merck Millipore) at a 1:500 dilution,
rabbit monoclonal primary antibody for E-cadherin (#3195; Cell
Signaling) at a 1:1,000 dilution, rabbit polyclonal primary
antibody for vimentin (sc-5565; Santa Cruz) at a 1:1,000 dilution,
mouse monoclonal antibody for α-tubulin (T6074; Sigma) at a 1:4,000
dilution and appropriate horseradish peroxidase-linked secondary
antibodies at a 1:2,000 dilution. After washing 3 times with TBST,
the bound antibody was developed using the ECL Plus Western
Blotting Detection System (Thermo, USA).
Statistical analysis
GraphPad Prism software was used for all statistical
analysis. Quantitative data are expressed as the mean ± SD and were
compared using the unpaired Student’s t-test.
Results
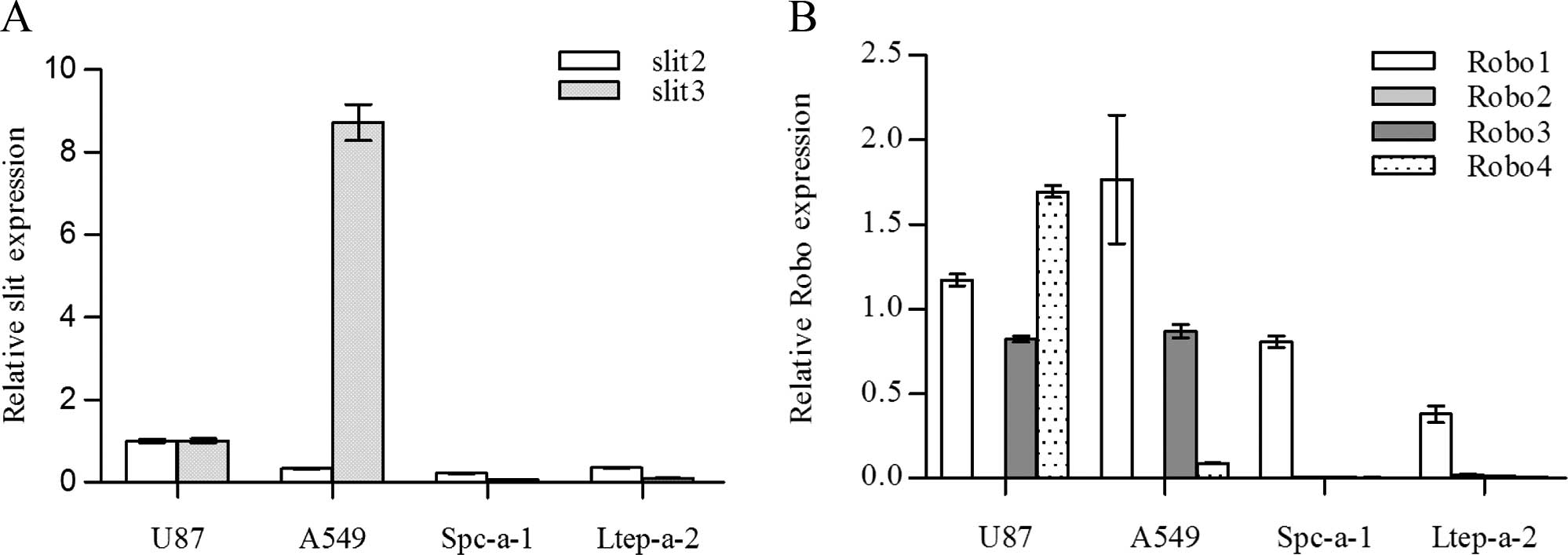
Slit and Robo expression in the lung
carcinoma cell lines
The Oncomine database indicates that Slit3
expression is downregulated in lung carcinoma compared to normal
tissues (34,35). To verify the results in the tumor
cell lines, we examined Slit and Robo expression in three lung
adenocarcinoma cell lines (A549, Spc-a-1 and Ltep-a-2). Since
Slit3 has been reported to be overexpressed in gliomas and
is considered a biomarker for gliomas (36), we used the glioblastoma cell line
u87MG as a positive control for Slit3.
Slit1 expression was not detected in any of
the cell lines analyzed. Slit2 was detected in all of the
cell lines and in general maintained a relatively lower expression
when compared with that in the U87MG cells (Fig. 1A). Slit3 was only detected in
the A549 and U87MG cells. A549 cells showed high Slit3
expression levels that were nearly 9-fold more than that of the
U87MG cells. Of the three lung adenocarcinoma cell lines, high
Slit3 expression was only detected in the A549 cells,
indicating cell-specific Slit3 expression. However, high
Slit3 expression in the A549 cells was not evident at the
protein level. Although Slit3 is a secreted protein, both ELISA of
the cell supernatant and immunoblotting of the cell lysates (up to
80 μg of total loading protein) did not detect Slit3 (data
not shown). We theorized that the membrane-associated
characteristics of Slit and the relatively low amount of Slit3 may
account for the lack of Slit3 detection in the supernatant and cell
lysates. Unexpectedly, immunofluorescence only detected a very weak
signal on the A549 cell surface (data not shown).
Robo receptors are heterogeneously expressed by
tumor cell lines. While Robo2 was not detected in any of the cell
lines assayed, Robo1 was detected in all of the cell lines. A549
cells maintained a relatively high level of Robo1 expression
compared with the other cell lines. Robo3 and Robo4 were only
detected in the U87MG and A549 cells, yet not in the Spc-a-1 and
Ltep-a-2 cells (Fig. 1B).
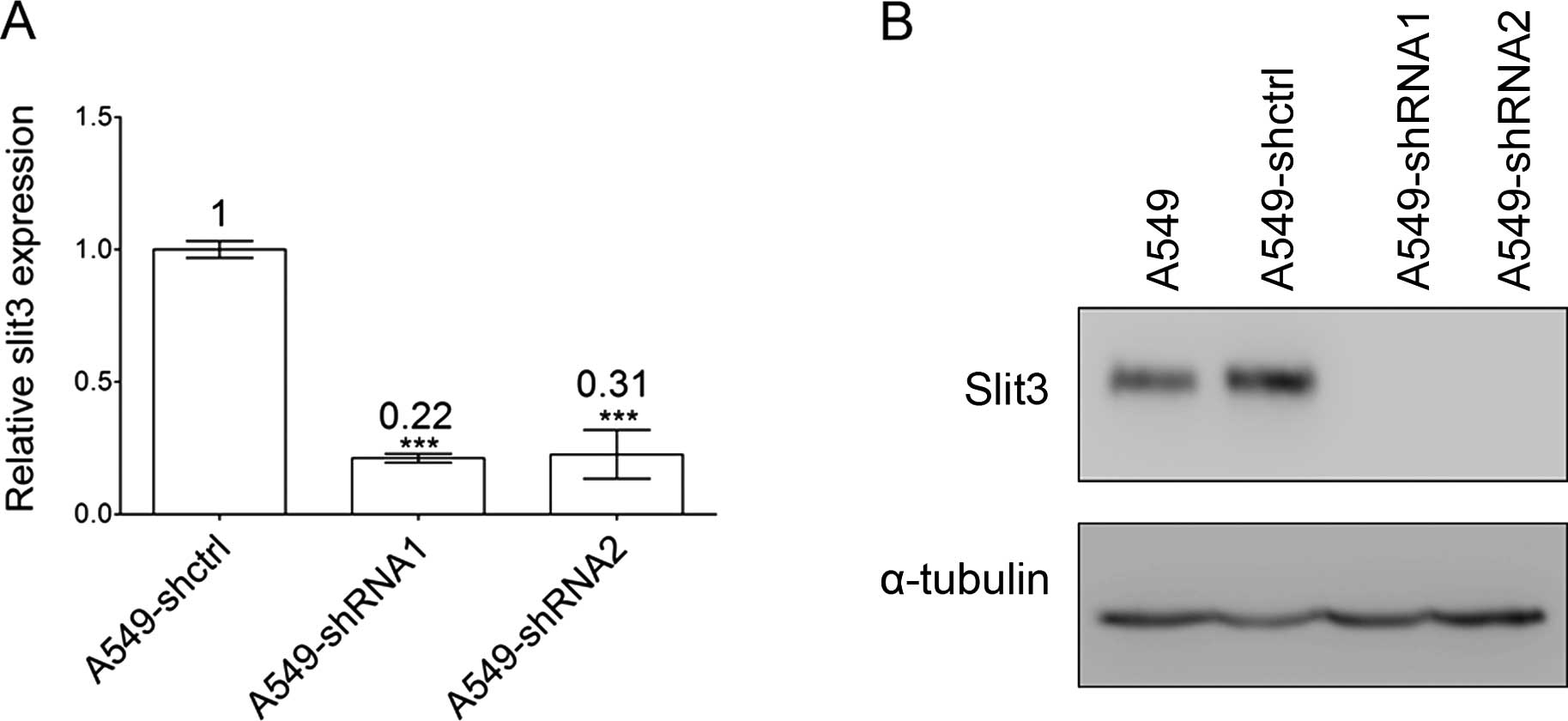
Slit3 knockdown in A549 cells
To evaluate the role of Slit3, A549 cells were
transduced with the VSVG pseudotyped lentivirus plasmid (pLL3.7)
for stable Slit3 silencing (A549-shRNA) or an empty control
plasmid without Slit3-shRNA insertion (A549-shctrl).
Silencing of Slit3 mRNA in the A549 cells was first verified
by real-time quantitative PCR. Slit3 mRNA expression in the
A549 cells was significantly decreased, with a knockdown efficiency
of 78 and 69%, respectively (Fig.
2A). Given that Slit3 is predominantly membrane-associated, we
isolated the membrane-associated proteins in the A549 cells using a
Qproteome Cell Compartment kit to detect the changes in Slit3
protein levels. We did not detect Slit3 protein bands when using
60–120 μg of total loading protein, yet detected it when
using 180 μg of total sample protein (Fig. 2B). The discordance between high
Slit3 mRNA expression and protein abundance suggests that
post-translational mechanisms influenced Slit3 abundance in the
A549 cells. The molecular size of the Slit3 protein band detected
in the A549 cells was slightly larger than the theoretically
calculated value of N-terminal Slit3 (~140 kDa), which reflects
cellular glycosylation of the slit3 protein. The results are
consistent with the findings that the majority of Slits are
secreted in a membrane-associated manner. Notably, full-length
Slit3 (~200 kDa) was not detected, indicating that proteolytically
processed N-terminal Slit3 is the primary form of secreted Slit3 in
the A549 cells.
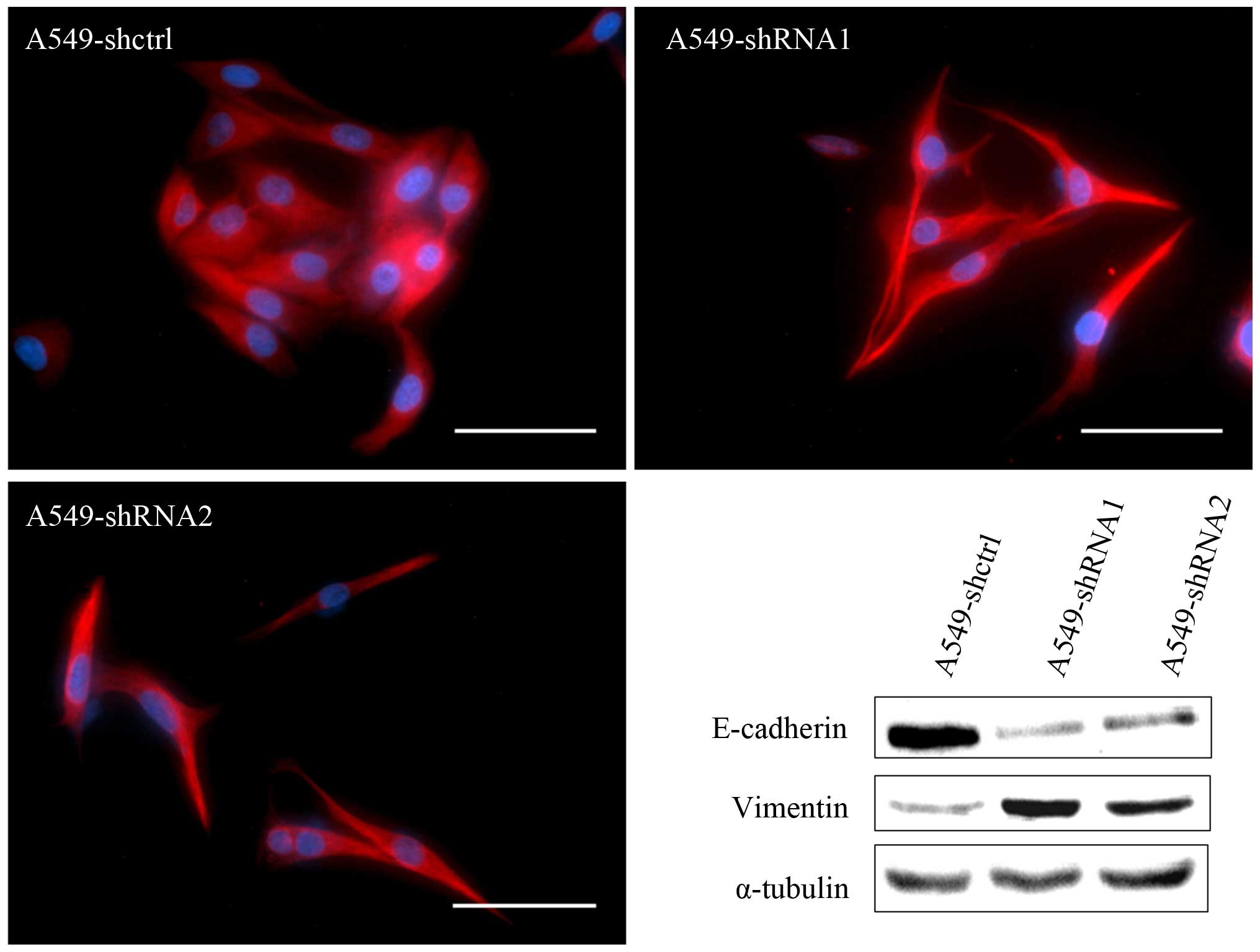
Slit3 silencing induces EMT in A549
cells
Slit3 silencing induced significant
morphologic changes in the A549 cells as demonstrated by an
elongated fibroblast-like morphology, a phenomenon reminiscent of
EMT (Fig. 3). We further examined
the effects of Slit3 on the expression of molecular hallmarks of
EMT, including the epithelial marker E-cadherin and the mesenchymal
marker vimentin. Immunoblotting analysis confirmed that E-cadherin
protein expression was obviously reduced in the A549-shRNA cells
when compared with the controls. Corresponding to reduced
E-cadherin expression, vimentin expression was significantly
increased.
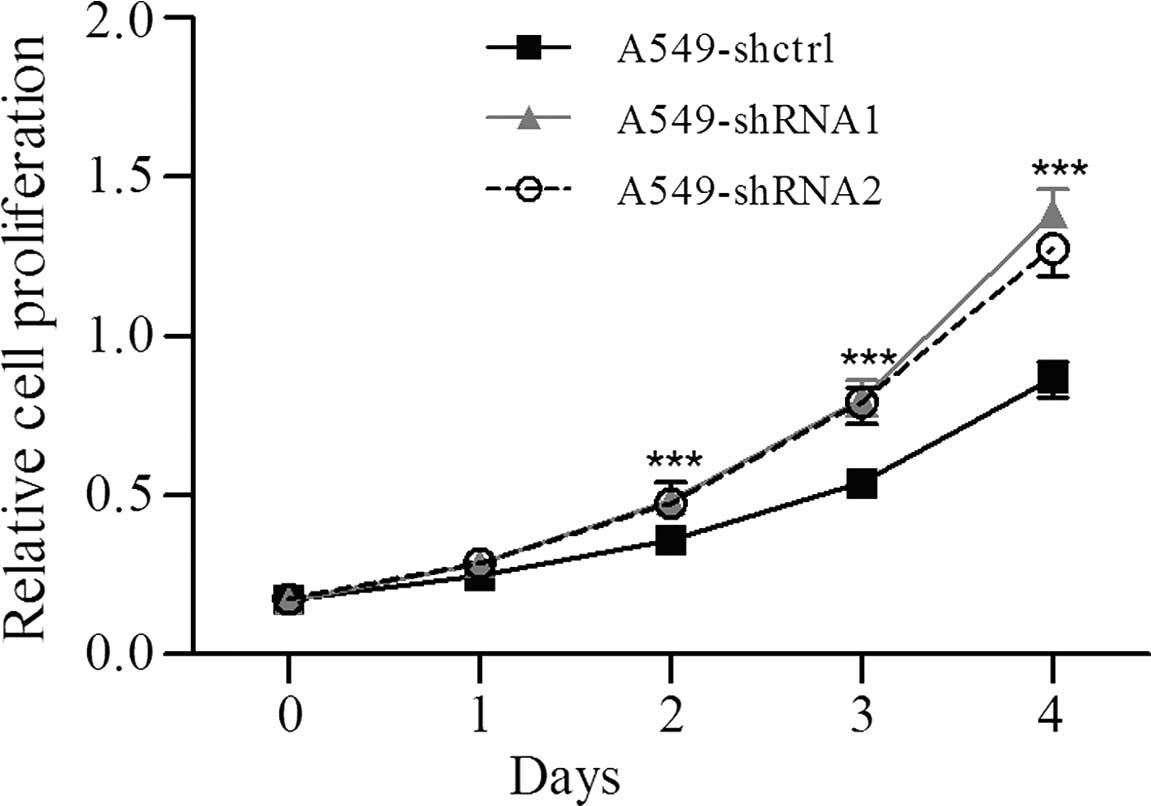
Slit3 silencing enhances the
proliferation, migration and invasion of A549 cells
To evaluate the effects of Slit3 on the
proliferation of A549 cells with Slit3 knockdown, the MTT
assay was performed. Compared with the controls, the Slit3
knockdown significantly stimulated the proliferation of the A549
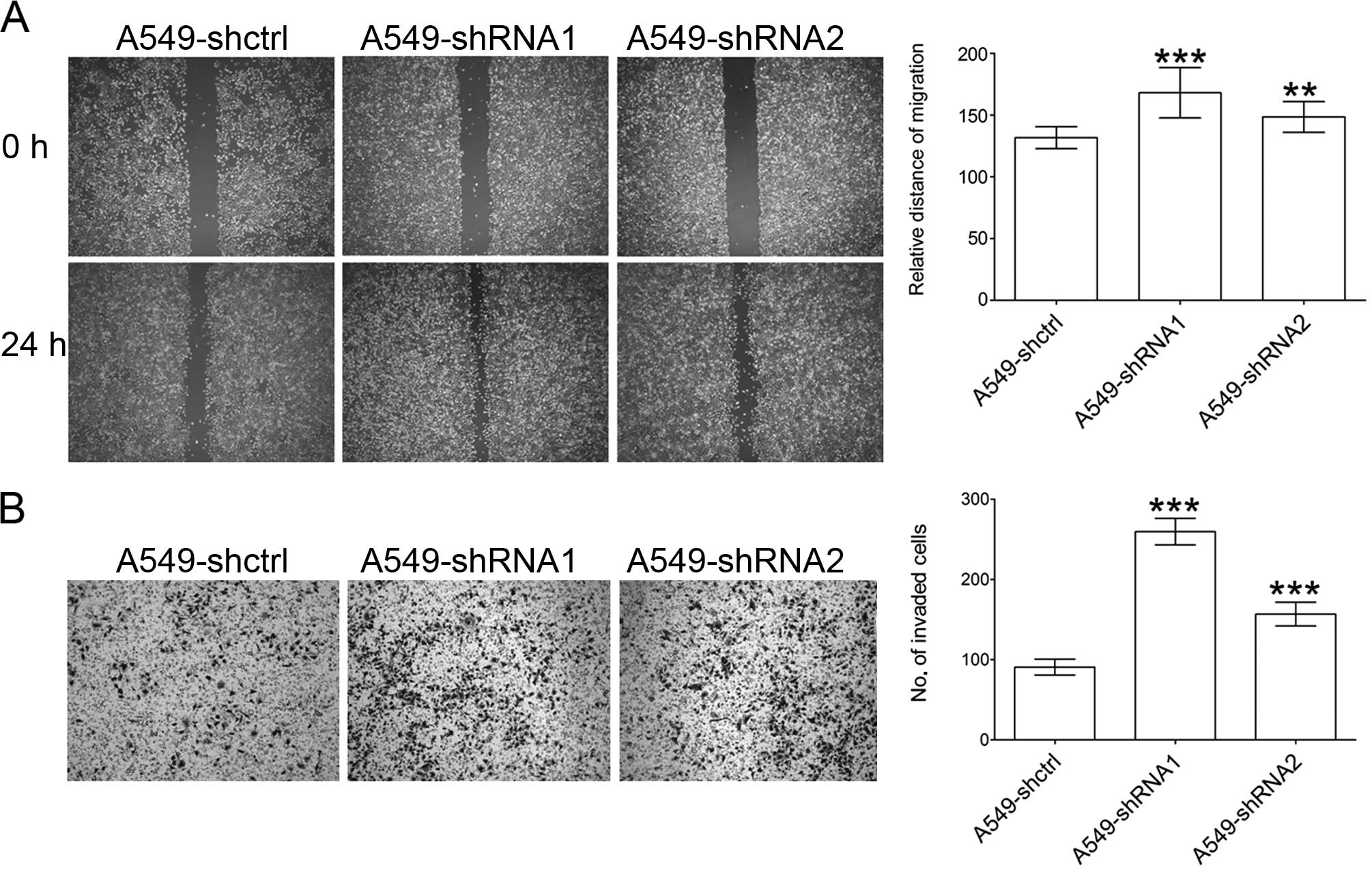
cells (Fig. 4). Cell migration was
measured by a wound healing assay in which the Slit3 knockdown
significantly promoted the migration of the A549 cells when
compared to that of the controls (Fig.
5A). Similar results were also observed in the Matrigel
invasion assay. Slit3 knockdown enhanced the ability of A549
cells to traverse the Matrigel-coated membrane (Fig. 5B). These results suggest that Slit3
exerts negative regulation on the proliferation and invasive
behavior of A549 cells, thereby confirming the role of slit3 as a
tumor suppressor.
Slit3 silencing upregulates MMP2 and MMP9
expression
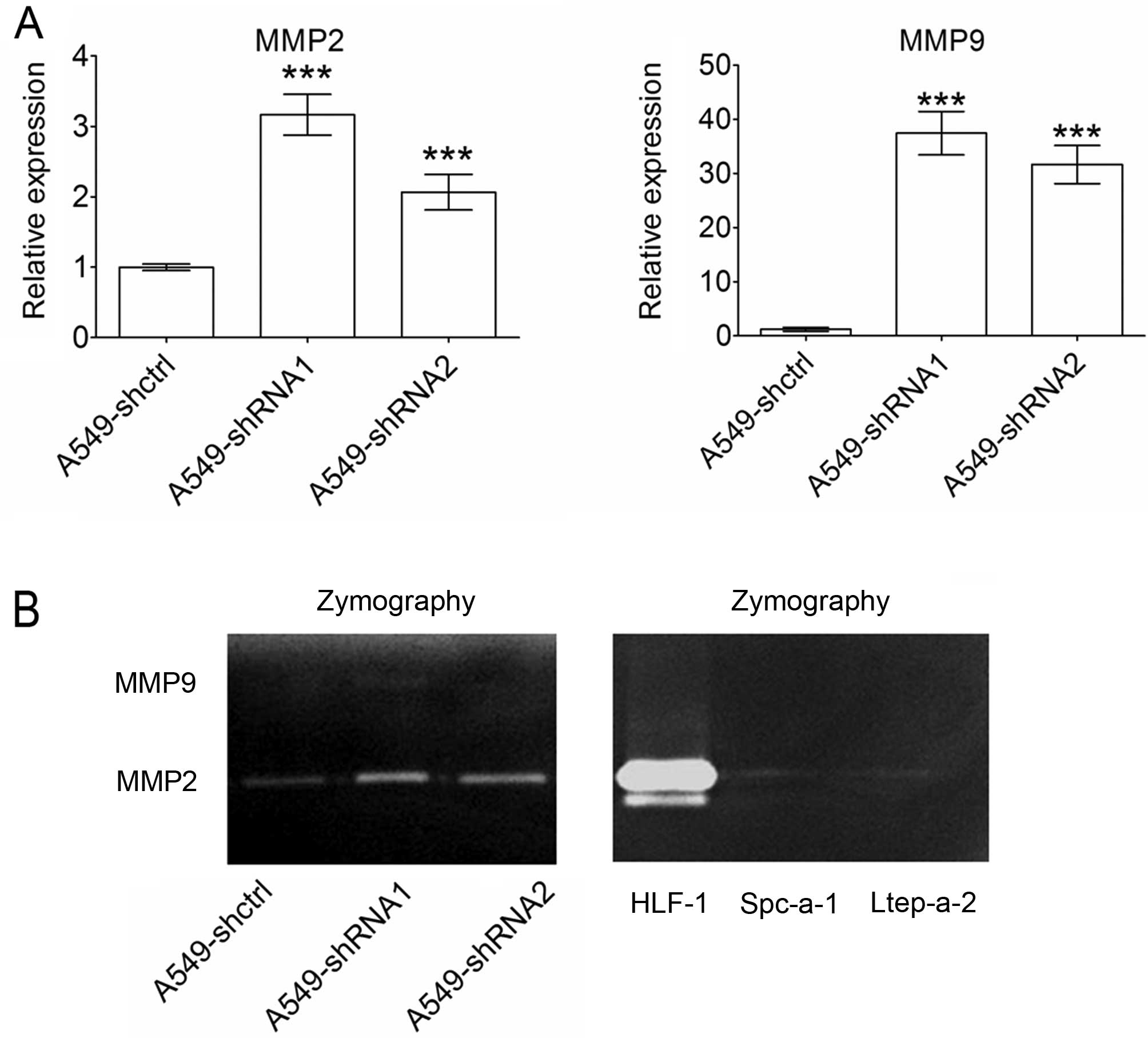
To explore the mechanisms underlying the stimulative
effects of Slit3 silencing on the invasion of A549 cells, we
detected MMP2 and MMP9 expression, which are key
enzymes for degradation of type IV collagen, a major component of
the basement membrane. Quantitative PCR results showed that
silencing of Slit3 in the A549 cells caused significant
upregulation of both MMP2 and MMP9 (Fig. 6A). To verify the effects of Slit3
silencing on MMP2 and MMP9 secretion, gelatin zymography was
performed. Supernatants were prepared by incubating the cells with
serum-free DMEM for 12 h to decrease the effects of cell
proliferation on MMP production. As shown in Fig. 6B, A549 control cells maintained very
weak MMP2 secretion, whereas MMP9 production was not detected.
Slit3 silencing in the A549 cells induced an increase in
MMP2 production. Although MMP9 was not detected (Fig. 6B, left), Slit3 silencing in
the A549 cells caused significant upregulation of MMP9 at
the mRNA level (Fig. 6A). Lack of
MMP9 detection could be due to low MMP9 expression in A549
cells. Quantitative PCR confirmed that MMP9 expression was
significantly lower than MMP2 in the A549 cells (data not
shown). To determine whether weak MMP2 and MMP9
expression is common in lung carcinoma cell lines, culture
supernatants from the Spc-a-1 and Ltep-a-2 cell lines were also
assayed using gelatin zymography. Human normal lung fibroblasts
(HFL-1) were used as a positive control. HFL-1 maintained high
levels of MMP2, while the two lung carcinoma cell lines showed weak
MMP2 production. MMP9 was not detected in any of the cell lines
(Fig. 6B, right). These results
suggest that relatively weak MMP2 and MMP9 expression
may be a common characteristic in the lung carcinoma cell lines
analyzed.
Discussion
Although a number of studies have explored the roles
of Slit2 in tumor progression, relatively little progress has been
made in defining the effects of Slit3 on tumors. In contrast to
Slit2, which was reported to be upregulated in various
tumors (18,20), few results concerning Slit3
upregulation in tumor tissues have been reported. Although
microarray analysis revealed Slit3 upregulation in separated
invasive hepatocellular carcinoma cells, no evidence of protein
level upregulation has been provided (37). Moreover, Slit3 upregulation
at the mRNA level does not necessarily lead to an increase in Slit3
at the protein level. Chen et al examined the correlation
between mRNA expression and protein levels in lung adenocarcinomas
and found a significant correlation in only a small subset of the
proteins studied (38). Towner
et al recently reported high Slit3 expression on the
surface of high-grade human gliomas compared to low-grade gliomas
and normal brain tissues using immunohistochemistry and suggested
Slit3 as a potent biomarker for human high-grade gliomas in
clinical diagnosis (36). These
results are in contrast to reports that Slit3 is frequently
methylated in gliomas (8),
indicating heterogeneous Slit3 expression in gliomas. The
present study confirmed Slit3 expression in the u87MG
glioblastoma cell line and detected higher Slit3 expression
in lung carcinoma A549 cells. These results are consistent with
reports that the frequency of promoter methylation is lower in lung
carcinomas (8,29) and partially explained high
Slit3 expression in A549 cells. However, the high
slit3 expression in A549 cells was not confirmed in two
other lung adenocarcinoma cell lines (Spc-a-1 and Ltep-a-2),
indicating cell-specific Slit3 expression that was not
apparent at the protein level.
As in the u87MG cells, Slit3 proteins were not
detected in the supernatant using ELISA. However,
immunofluorescence detected a very weak signal on the cell surface,
indicating that Slit3 in A549 cells remains mostly
membrane-associated. Immunoblotting further validated the trace
amounts of Slit3 associated with the cell membrane. These results
suggest that post-translational mechanisms influence Slit3 protein
abundance in cells despite mRNA expression. Given that Slit3
functions as a latent tumor suppressor, we suggest that
post-translational regulation of Slit3 confers a
self-protective advantage against the tumor-suppressor effects of
Slit3.
In contrast to the findings that both full length
and N-terminal fragments of Slit are present in neuronal cells
(39) or tissues (40), only the N-terminal of the Slit3
protein (~140 kDa) was detected at the A549 cell surface,
indicating that the extent of proteolytic cleavage of the Slit3
protein differs by tissue type. The cleaved Slit fragment also
exerts different functions. Nguyen et al demonstrated that
full length Slit2 neutralized the branching of dorsal root ganglia
axons induced by N-terminal Slit2 (41), suggesting that the function of Slit
was partly associated with its structure. In the present study,
lack of full length Slit3 in A549 cells may imply the loss of an
unknown regulatory mechanism that balances the effects of the
N-terminal Slit3 fragment.
Recombinant Slit3 was reported to inhibit the
migration of malignant melanoma cells through downregulation of
AP-1 (30). Slit3
overexpression in human breast carcinoma cells inhibits tumor
growth by suppressing Sdf1 and Cxcr4 expression
(17). The present study indicated
that Slit3 silencing in A549 cells promoted proliferation,
migration and invasion of the cells even though only trace amounts
of Slit3 protein were detected. These results are evidence that
Slit3 exerts negative regulatory effects on tumor proliferation and
migration. Results are in accordance with reported suppressive
effects of Slit2 on growth and migration in various types of tumor.
In nervous system development, Slits function as repellent factors
that prevent axon growth into particular regions (42). Increased migration due to Slit3
silencing supports the idea that repellent effects may also
influence tumor cell migration.
EMT plays a critical role in tumor metastasis by
which tumor cells weaken E-cadherin-dependent intercellular
adhesion and enhance motility, facilitating tumor cells to invade
surrounding tissues. Chen et al reported an inhibitory
effect of Slit2 on EMT wherein Slit2 overexpression in
colorectal cancer cells inhibited cell migration as a result of
increasing E-cadherin levels, while Slit2 knockdown led to opposite
results (11). However, Slit2
promotion of EMT was also reported in colorectal cancer cells
(21), the different cell lines
analyzed accounted for conflicting results. The results of the
present study confirmed the inhibitory effects of Slit3 on EMT in
lung carcinoma A549 cells given that Slit3 silencing induced an
elongated fibroblast-like morphology, downregulation of E-cadherin
and upregulation of vimentin.
MMPs play a critical role in tumor metastasis. The
prometastatic or antimetastatic effects of Slit are likely
associated with MMP expression or protein activities. Qi et
al recently reported upregulation of MMP2, yet not MMP9 in skin
tissue of Slit2-transgenic mice and proposed that MMP2 upregulation
accounted for loss of the basement membrane in Slit2-transgenic
mice, indicating a positive correlation between Slit2 and MMP2
expression (20). However, Prasad
et al reported that Slit2 inhibits the CXCL12-induced
activities of MMP2 and MMP9 in breast cancer cells (43). Our results showed that MMP2 and MMP9
expression was negatively associated with alteration of
Slit3 expression. Slit3 silencing promoted MMP2 and
MMP9 expression in A549 cells, whereas Slit3 overexpression
reduced expression, supporting the negative correlation between
Slit3 and MMP2 and MMP9 expression, even though only MMP2 secretion
was detected. Conflicting results, however, were recently reported
in primary amnion and myometrial cells wherein Slit3
knockdown with siRNA decreased MMP9 gene expression and secretion
(44). Mechanisms underlying these
results may be associated with heterogeneous expression of Robo
receptors in various tissues. Of the four Robo receptors, Robo4 is
structurally divergent from the other three members both in
extracellular and intracellular domains and may stimulate signaling
pathways that differ from other receptors, resulting in a
functional difference. For example, Slit2/Robo4 and Slit2/Robo1
signaling pathways induce opposite effects on the migration of
endothelial cells (45). In the
present study, Robo1 and Robo4 were detected in A549 cells and
Robo1 maintained a relatively high level of expression. Therefore,
we suggest that Slit3 most likely exerts its effects in A549 cells
by binding to Robo1.
In summary, these results demonstrated that Slit3
functions as a tumor suppressor in lung carcinoma. Due to promoter
hypermethylation, overall expression of Slit3 is
downregulated in most tumors. Even if hypomethylation of the
promoter occurs in some cancer tissues, such as lung carcinoma, and
results in high slit3 expression, transcription of the slit3
protein is still strictly controlled. Slit3 represses migration and
invasion of A549 cells by blocking EMT and suppressing both MMP2
and MMP9 expression and secretion. Further studies are needed to
decipher the mechanisms underlying the post-translational
regulation of Slit3 and suppression of MMP2 and MMP9 expression and
secretion.
Acknowledgments
This study was supported by grants of National
Natural Science Foundation of China (grant no. 30971210), the Key
Project of Chinese Ministry of Education (grant no. 109093) and the
Fujian Provincial Department of Science and Technology (grant no.
2007J0113).
References
|
1
|
Katoh Y and Katoh M: Comparative genomics
on SLIT1, SLIT2, and SLIT3 orthologs. Oncol Rep. 14:1351–1355.
2005.PubMed/NCBI
|
|
2
|
Brose K, Bland KS, Wang KH, Arnott D,
Henzel W, Goodman CS, Tessier-Lavigne M and Kidd T: Slit proteins
bind Robo receptors and have an evolutionarily conserved role in
repulsive axon guidance. Cell. 96:795–806. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Patel K, Nash JA, Itoh A, Liu Z,
Sundaresan V and Pini A: Slit proteins are not dominant
chemorepellents for olfactory tract and spinal motor axons.
Development. 128:5031–5037. 2001.PubMed/NCBI
|
|
4
|
Condac E, Strachan H, Gutierrez-Sanchez G,
Brainard B, Giese C, Heiss C, Johnson D, Azadi P, Bergmann C,
Orlando R, et al: The C-terminal fragment of axon guidance molecule
Slit3 binds heparin and neutralizes heparin’s anticoagulant
activity. Glycobiology. 22:1183–1192. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu JY, Feng L, Park HT, Havlioglu N, Wen
L, Tang H, Bacon KB, Jiang Zh, Zhang Xc and Rao Y: The neuronal
repellent Slit inhibits leukocyte chemotaxis induced by chemotactic
factors. Nature. 410:948–952. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shyamsundar R, Kim YH, Higgins JP,
Montgomery K, Jorden M, Sethuraman A, van de Rijn M, Botstein D,
Brown PO and Pollack JR: A DNA microarray survey of gene expression
in normal human tissues. Genome Biol. 6:R222005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dallol A, Da Silva NF, Viacava P, Minna
JD, Bieche I, Maher ER and Latif F: SLIT2, a human homologue of the
Drosophila Slit2 gene, has tumor suppressor activity and is
frequently inactivated in lung and breast cancers. Cancer Res.
62:5874–5880. 2002.PubMed/NCBI
|
|
8
|
Dickinson RE, Dallol A, Bieche I, Krex D,
Morton D, Maher ER and Latif F: Epigenetic inactivation of SLIT3
and SLIT1 genes in human cancers. Br J Cancer. 91:2071–2078. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dallol A, Krex D, Hesson L, Eng C, Maher
ER and Latif F: Frequent epigenetic inactivation of the SLIT2 gene
in gliomas. Oncogene. 22:4611–4616. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dunwell TL, Dickinson RE, Stankovic T,
Dallol A, Weston V, Austen B, Catchpoole D, Maher ER and Latif F:
Frequent epigenetic inactivation of the SLIT2 gene in chronic and
acute lymphocytic leukemia. Epigenetics. 4:265–269. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen WF, Gao WD, Li QL, Zhou PH, Xu MD and
Yao LQ: SLIT2 inhibits cell migration in colorectal cancer through
the AKT-GSK3β signaling pathway. Int J Colorectal Dis. 28:933–940.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim HK, Zhang H, Li H, Wu TT, Swisher S,
He D, Wu L, Xu J, Elmets CA, Athar M, et al: Slit2 inhibits growth
and metastasis of fibrosarcoma and squamous cell carcinoma.
Neoplasia. 10:1411–1420. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Göhrig A, Detjen KM, Hilfenhaus G, Körner
JL, Welzel M, Arsenic R, Schmuck R, Bahra M, Wu JY, Wiedenmann B,
et al: Axon guidance factor SLIT2 inhibits neural invasion and
metastasis in pancreatic cancer. Cancer Res. 74:1529–1540. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Werbowetski-Ogilvie TE, Seyed Sadr M,
Jabado N, Angers-Loustau A, Agar NY, Wu J, Bjerkvig R, Antel JP,
Faury D, Rao Y, et al: Inhibition of medulloblastoma cell invasion
by Slit. Oncogene. 25:5103–5112. 2006.PubMed/NCBI
|
|
15
|
Dallol A, Morton D, Maher ER and Latif F:
SLIT2 axon guidance molecule is frequently inactivated in
colorectal cancer and suppresses growth of colorectal carcinoma
cells. Cancer Res. 63:1054–1058. 2003.PubMed/NCBI
|
|
16
|
Qiu H, Zhu J, Yu J, Pu H and Dong R: SLIT2
is epigenetically silenced in ovarian cancers and suppresses growth
when activated. Asian Pac J Cancer Prev. 12:791–795.
2011.PubMed/NCBI
|
|
17
|
Marlow R, Strickland P, Lee JS, Wu X,
Pebenito M, Binnewies M, Le EK, Moran A, Macias H, Cardiff RD, et
al: SLITs suppress tumor growth in vivo by silencing Sdf1/Cxcr4
within breast epithelium. Cancer Res. 68:7819–7827. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang B, Xiao Y, Ding BB, Zhang N, Yuan X,
Gui L, Qian KX, Duan S, Chen Z, Rao Y, et al: Induction of tumor
angiogenesis by Slit-Robo signaling and inhibition of cancer growth
by blocking Robo activity. Cancer Cell. 4:19–29. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang LJ, Zhao Y, Han B, Ma YG, Zhang J,
Yang DM, Mao JW, Tang FT, Li WD, Yang Y, et al: Targeting
Slit-Roundabout signaling inhibits tumor angiogenesis in
chemical-induced squamous cell carcinogenesis. Cancer Sci.
99:510–517. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qi C, Lan H, Ye J, Li W, Wei P, Yang Y,
Guo S, Lan T, Li J, Zhang Q, et al: Slit2 promotes tumor growth and
invasion in chemically induced skin carcinogenesis. Lab Invest.
94:766–776. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou WJ, Geng ZH, Chi S, Zhang W, Niu XF,
Lan SJ, Ma L, Yang X, Wang LJ, Ding YQ, et al: Slit-Robo signaling
induces malignant transformation through Hakai-mediated E-cadherin
degradation during colorectal epithelial cell carcinogenesis. Cell
Res. 21:609–626. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang XM, Han HX, Sui F, Dai YM, Chen M and
Geng JG: Slit-Robo signaling mediates lymphangiogenesis and
promotes tumor lymphatic metastasis. Biochem Biophys Res Commun.
396:571–577. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu J, Zhang L, Wang D, Shen H, Jiang M,
Mei P, Hayden PS, Sedor JR and Hu H: Congenital diaphragmatic
hernia, kidney agenesis and cardiac defects associated with
Slit3-deficiency in mice. Mech Dev. 120:1059–1070. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yuan W, Rao Y, Babiuk RP, Greer JJ, Wu JY
and Ornitz DM: A genetic model for a central (septum transversum)
congenital diaphragmatic hernia in mice lacking Slit3. Proc Natl
Acad Sci USA. 100:5217–5222. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang B, Dietrich UM, Geng JG, Bicknell R,
Esko JD and Wang L: Repulsive axon guidance molecule Slit3 is a
novel angiogenic factor. Blood. 114:4300–4309. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paul JD, Coulombe KL, Toth PT, Zhang Y,
Marsboom G, Bindokas VP, Smith DW, Murry CE and Rehman J:
SLIT3-ROBO4 activation promotes vascular network formation in human
engineered tissue and angiogenesis in vivo. J Mol Cell Cardiol.
64:124–131. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Narayan G, Goparaju C, Arias-Pulido H,
Kaufmann AM, Schneider A, Dürst M, Mansukhani M, Pothuri B and
Murty VV: Promoter hypermethylation-mediated inactivation of
multiple Slit-Robo pathway genes in cervical cancer progression.
Mol Cancer. 5:162006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nones K, Waddell N, Song S, Patch AM,
Miller D, Johns A, Wu J, Kassahn KS, Wood D, Bailey P, et al:
Genome-wide DNA methylation patterns in pancreatic ductal
adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2
and MET signaling. Int J Cancer. 135:1110–1118. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dammann R, Strunnikova M, Schagdarsurengin
U, Rastetter M, Papritz M, Hattenhorst UE, Hofmann HS, Silber RE,
Burdach S and Hansen G: CpG island methylation and expression of
tumour-associated genes in lung carcinoma. Eur J Cancer.
41:1223–1236. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Denk AE, Braig S, Schubert T and
Bosserhoff AK: Slit3 inhibits activator protein 1-mediated
migration of malignant melanoma cells. Int J Mol Med. 28:721–726.
2011.PubMed/NCBI
|
|
31
|
Schubert T, Denk AE, Ruedel A, Kaufmann S,
Hustert E, Bastone P and Bosserhoff AK: Fragments of SLIT3 inhibit
cellular migration. Int J Mol Med. 30:1133–1137. 2012.PubMed/NCBI
|
|
32
|
Guan H, Wei G, Wu J, Fang D, Liao Z, Xiao
H, Li M and Li Y: Down-regulation of miR-218-2 and its host gene
SLIT3 cooperate to promote invasion and progression of thyroid
cancer. J Clin Endocrinol Metab. 98:E1334–E1344. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
34
|
Bhattacharjee A, Richards WG, Staunton J,
Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, et
al: Classification of human lung carcinomas by mRNA expression
profiling reveals distinct adenocarcinoma subclasses. Proc Natl
Acad Sci USA. 98:13790–13795. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hou J, Aerts J, den Hamer B, van Ijcken W,
den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens
JA, Hoogsteden HC, et al: Gene expression-based classification of
non-small cell lung carcinomas and survival prediction. PLoS One.
5:e103122010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Towner RA, Jensen RL, Vaillant B, Colman
H, Saunders D, Giles CB and Wren JD: Experimental validation of 5
in-silico predicted glioma biomarkers. Neuro Oncol. 15:1625–1634.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lin ZY and Chuang WL: Genes responsible
for the characteristics of primary cultured invasive phenotype
hepatocellular carcinoma cells. Biomed Pharmacother. 66:454–458.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen G, Gharib TG, Huang CC, Taylor JM,
Misek DE, Kardia SL, Giordano TJ, Iannettoni MD, Orringer MB,
Hanash SM, et al: Discordant protein and mRNA expression in lung
adenocarcinomas. Mol Cell Proteomics. 1:304–313. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hu H: Chemorepulsion of neuronal migration
by Slit2 in the developing mammalian forebrain. Neuron. 23:703–711.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Niclou SP, Jia L and Raper JA: Slit2 is a
repellent for retinal ganglion cell axons. J Neurosci.
20:4962–4974. 2000.PubMed/NCBI
|
|
41
|
Nguyen Ba-Charvet KT, Brose K, Ma L, Wang
KH, Marillat V, Sotelo C, Tessier-Lavigne M and Chédotal A:
Diversity and specificity of actions of Slit2 proteolytic fragments
in axon guidance. J Neurosci. 21:4281–4289. 2001.PubMed/NCBI
|
|
42
|
Bagri A, Marín O, Plump AS, Mak J,
Pleasure SJ, Rubenstein JL and Tessier-Lavigne M: Slit proteins
prevent midline crossing and determine the dorsoventral position of
major axonal pathways in the mammalian forebrain. Neuron.
33:233–248. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Prasad A, Fernandis AZ, Rao Y and Ganju
RK: Slit proteinmediated inhibition of CXCR4-induced chemotactic
and chemoinvasive signaling pathways in breast cancer cells. J Biol
Chem. 279:9115–9124. 2004. View Article : Google Scholar
|
|
44
|
Lim R, Barker G and Lappas M: SLIT3 is
increased in supracervical human foetal membranes and in labouring
myometrium and regulates pro-inflammatory mediators. Am J Reprod
Immunol. 71:297–311. 2014. View Article : Google Scholar
|
|
45
|
Autiero M, De Smet F, Claes F and
Carmeliet P: Role of neural guidance signals in blood vessel
navigation. Cardiovasc Res. 65:629–638. 2005. View Article : Google Scholar : PubMed/NCBI
|