Introduction
Prostate cancer has a high cancer incidence and
mortality worldwide (1). Metastasis
occurs in over one-third of patients leading to a poor prognosis
despite the use of surgery, chemotherapy and radiotherapy (2–4). Thus,
it is essential to develop efficient anticancer agents to inhibit
metastasis. Invasion, as a key step of metastasis, contributes to
cancer progression. The development of anti-invasion drugs is
therefore an important strategy to combat prostate cancer.
Dietary intake of cruciferous vegetables was
demonstrated to reduce the risks of various types of cancer
(5–7). Previous findings have shown that the
potential anti-carcinogenic properties of cruciferous vegetables
result from sulforaphane (SFN), which is a strong anticancer
component (8–11). SFN may suppress cancer progression
via a few anti-proliferative mechanisms (12–15).
In a previous study, we found that SFN inhibited invasion in human
glioblastoma cells through activation of the ERK1/2 signaling
pathway (16). However, whether SFN
inhibits invasion in human prostate cancer cells remains to be
determined. Thus, these effects and the signaling pathways
underlying invasion inhibition should be investigated.
Extracellular signal-regulated kinases (ERK1/2) have
been shown to have pivotal roles in cell proliferation,
differentiation, apoptosis and invasion (17–21).
The ERK1/2 pathway triggered oncogene and tumor-suppressor gene
expression depending on the time and strength of ERK1/2 activation
(17,21,22).
It has been indicated that transient ERK1/2 phosphorylation (5–15
min stimulation) resulted in cell proliferation, migration and
invasion. By contrast, sustained (>15 min stimulation)
activation of ERK1/2 exhibited the opposite effects (17,21–25).
Previous results demonstrated that human chorionic gonadotropin β
(hCGβ) promoted cell migration and invasion in human glioblastoma
and prostate cancer cells by increasing the expression and activity
of MMP-2, resulting from the sustained activation of ERK1/2
(26,27). It was also found that the sustained
phosphorylation of ERK1/2 by SFN contributed to the suppression of
migration and invasion in human glioblastoma cells (16). Thus, in the present study, we
investigated whether SFN inhibited invasion in human prostate
cancer cells via sustained activation of ERK1/2 and the downstream
signaling.
Cadherins, a superfamily of transmembrane
glycoproteins, mediate calcium-dependent cell adhesion (28). E-cadherin, a hallmark of
epithelial-mesenchymal transition (EMT), belongs to the classic
cadherin subfamily and is thought to maintain cell polarity and
strengthen intercellular adhesion and acts as a suppressor of
invasion. Downregulation of E-cadherin is frequently observed in
various metastatic human epithelial cancers (29). It was reported that E-cadherin
expression was linked to ERK1/2 activation in tumor cell migration
and invasion (30–32). In the present study, we investigated
whether E-cadherin was regulated through SFN-induced ERK1/2
phosphorylation in human prostate cancer cells.
The cellular adhesion molecule CD44 is a
transmembrane glycoprotein and an EMT marker correlated with
breast, prostate, colon, head and pancreatic cancers (33). CD44 isoforms (CD44v) are produced
through alternative splicing of the CD44v precursor mRNA (33). Altered expression or dysfunction of
CD44 variant 6 (CD44v6) is involved in the aggressive behavior of
various types of cancer and correlates with poor prognosis in human
malignancies such as breast, leukemia, gastric and colorectal
cancer (34–37). Additionally, SFN-induced ERK1/2
phosphorylation contributed to CD44v6 expression in human
glioblastoma cells (16).
Therefore, we hypothesized that SFN regulates CD44v6 via the
activation of ERK1/2 in human prostate cancer cells.
The extracellular matrix (ECM) and basement
membranes are crucial barriers against the invasion of malignant
cells (38). Matrix
metalloproteinases (MMPs) are undoubtedly associated with tumor
invasion (39). MMP-2, which is
involved in the degradation of type IV collagen and gelatin, is
expressed at elevated levels in ovarian, uterine, breast, prostate
cancer and melanoma (40–42). Previous results showed that ERK1/2
phosphorylation decreased the expression and activity of MMP-2 in
glioblastoma cells (16). Thus, SFN
may regulate MMP-2 through the ERK1/2 signaling pathway and
contribute to the suppression of invasion in human prostate cancer
cells.
In summary, in the present study we investigated the
potential of SFN to suppress invasion and identified the underlying
molecular mechanisms. Particularly, we aimed to determine key
target proteins and establish more powerful strategies to prevent
and treat prostate cancer.
Materials and methods
Reagents
D, L-SFN was purchased from Sigma (St. Louis, Mo,
USA). RPMI-1640 culture medium was purchased from Hyclone (Logan,
UT, USA). Fetal bovine serum (FBS) and penicillin-streptomycin were
purchased from Invitrogen-life Technologies (Carlsbad, Ca, USA).
Dimethyl sulfoxide (DMSO) was purchased from AppliChem GmbH
(Darmstadt, Germany). An MTS assay kit was purchased from Promega
(Madison, WI, USA). Transwell plates used for the invasion assay
were purchased from Costar (Corning, Cambridge, MA, USA). Matrigel
basement membrane matrix for cell invasion assay was purchased from
BD Biosciences (Bedford, MA, USA). PD98059 was purchased from Cell
Signaling Technology, Inc. (Shanghai, China). The antibodies,
anti-ERK1/2, anti-phospho-ERK1/2 and anti-E-cadherin were purchased
from Cell Signaling Technology. Anti-MMP-2 antibody and anti-CD44v6
were purchased from Abcam (Shanghai, China).
Cell culture
The human DU145 prostate carcinoma cell line was
purchased from the Cell Resource Center, Peking Union Medical
College (CRC/PUMC). The cells were cultured in RPMI-1640 medium
supplemented with 10% FBS, 100 U/ml penicillin and 100 U/ml
streptomycin at 37°C with 5% CO2 in a standard
humidified incubator. The cells were treated with SFN and ERK1/2
inhibitor PD98059 (25 µM) during the logarithmic growth
phase for 24 h.
Cell viability assay
DU145 cells were seeded in 96-well plates at
2×103 cells/well. After 12 h, the medium in each well
was replaced with the medium containing different concentrations of
SFN and the plate was incubated for 24 h. Subsequently, the cell
viability was detected using an MTS assay kit according to the
manufacturer's instructions. The absorbance values were measured by
a BioTek® microplate reader (Synergy™ HT, Winooski, VT,
USA) at a wavelength of 490 nm.
Morphological observation
DU145 cells were plated and grown in a 6-well
culture plate until they reached 70% confluence. The cells were
then washed with phosphate-buffered saline (PBS) and exposed to
different concentrations of SFN (0, 5, 10, 15, 20, 25, 30, 35 and
40 µM) for 24 h. Cell morphology was photographed using a
digital camera (Olympus, Tokyo, Japan) connected to a
phase-contrast microscope (Leica DMIRB, Wetzlar, Germany).
Invasion assay
The Transwell system (24 wells, 8 µm pore
size with polycarbonate membrane) coated with Matrigel was used for
the in vitro invasion assays. Matrigel was diluted with
serum-free RPMI-1640 to a final concentration of 2 mg/ml. The
Transwell inserts were rehydrated with pre-warmed serum-free medium
at 37°C for 30 min prior to seeding cells. A total of
2×105 cells were suspended in 100 µl serum-free
medium and were added to the upper chamber. Medium containing 10%
FBS and different concentrations of SFN was then added to the lower
chamber, respectively. After 24 h, the cells in the upper chamber
were carefully wiped with a cotton swab. The cells invading the
lower surface of the filter were fixed with 100% methanol and
stained with crystal violet. The invaded cells on the lower surface
of the membrane filter were counted in five randomly fields under
microscope, and the cell number was analyzed statistically.
Western blot analysis
DU145 cells from different treatment groups were
collected and lysed with RIPA (Thermo Scientific, Waltham, MA, USA)
containing protease inhibitors (Roche, Mannheim, Germany). The cell
lysate was centrifuged at 12,000 rpm for 20 min. Proteins were
separated by SDS-PAGE and transferred onto a nitrocellulose
membranes via wet transfer. The membranes were blocked in 1.5% BSA
in TBS Tween-20 (TBS-T) buffer and then incubated with primary
antibody at 4°C overnight. Subsequently, the membranes were
incubated with fluorescence-labeled secondary antibody (LI-COR
Bioscience, Lincoln, NE, USA). After being washed with TBS-T for
3×5 min each, the membranes were scanned using the Odyssey Infrared
Imaging System (LI-COR Bioscience).
Gelatin zymography
Following treatment, the medium with MMP-2 activity
was collected from an equal number of cells and was centrifuged at
3,000 rpm for 5 min to remove cell debris. The samples were mixed
with non-reduced loading buffer and subjected to a 10%
SDS-polyacrylamide gel containing 0.1% gelatin as a substrate at
4°C. After electrophoresis, the gel was rinsed four times with
denaturation buffer (2.5% Triton X-100) on a shaker to remove SDS
at room temperature and then incubated in developing buffer (50 mM
Tris-HCl, pH 7.5, 0.2 M NaCl, 5 mM
CaCl2·2H2O, and 0.02% Brij-35, ph 7.6) for 42
h at 37°C. After incubation, the gel was stained using 0.5%
Coomassie Brilliant Blue R-250 for 30 min and then destained with
destaining solution (50% methanol, 10% acetic acid, 40%
ddH2O). Enzyme activity was identified as bright bands
against the dark blue background of the substrate.
Statistical analysis
Data are presented as means ± SD. Data were analyzed
by ANOVA and subjected to analysis of variance with post-tests for
comparison among specific groups. Bonferroni corrections for
multiple comparisons against a single group were used. P<0.05
was considered statistically significant. Each experiment was
repeated a minimum of three times.
Results
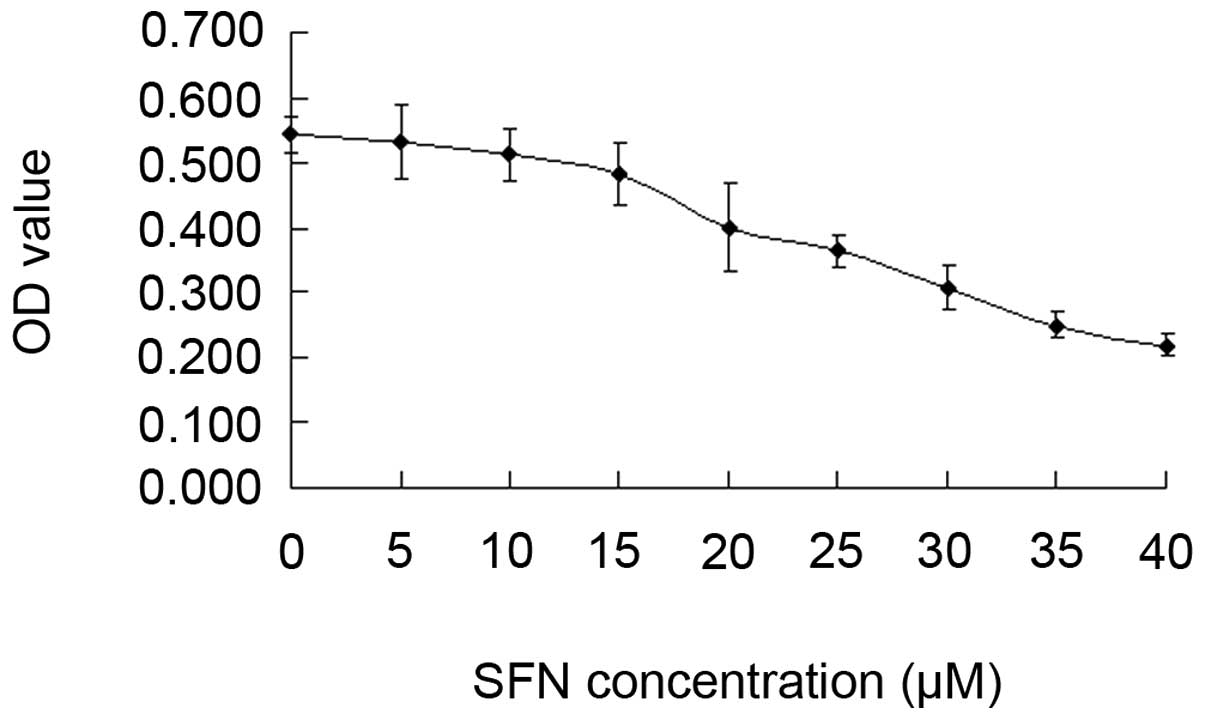
SFN inhibits cell viability in a
dose-dependent manner
DU145 cells were incubated with increasing
concentrations of SFN (0, 5, 10, 15, 20, 25, 30, 35 and 40
µM) for 24 h. SFN was dissolved in DMSO. The final
concentration of DMSO in the medium was <0.01%. The medium
containing DMSO without SFN was used in the control group. The cell
viability was detected using the CellTiter 96® AQueous
One Solution Cell Proliferation Assay kit (Promega). The results
showed that SFN significantly inhibited cell viability in a
dose-dependent manner at concentrations of 15–40 µM for 24 h
(P<0.05, Fig. 1). Thus, a
non-toxic concentration (<15 µM) was used as the optimal
concentration of SFN for the invasion study.
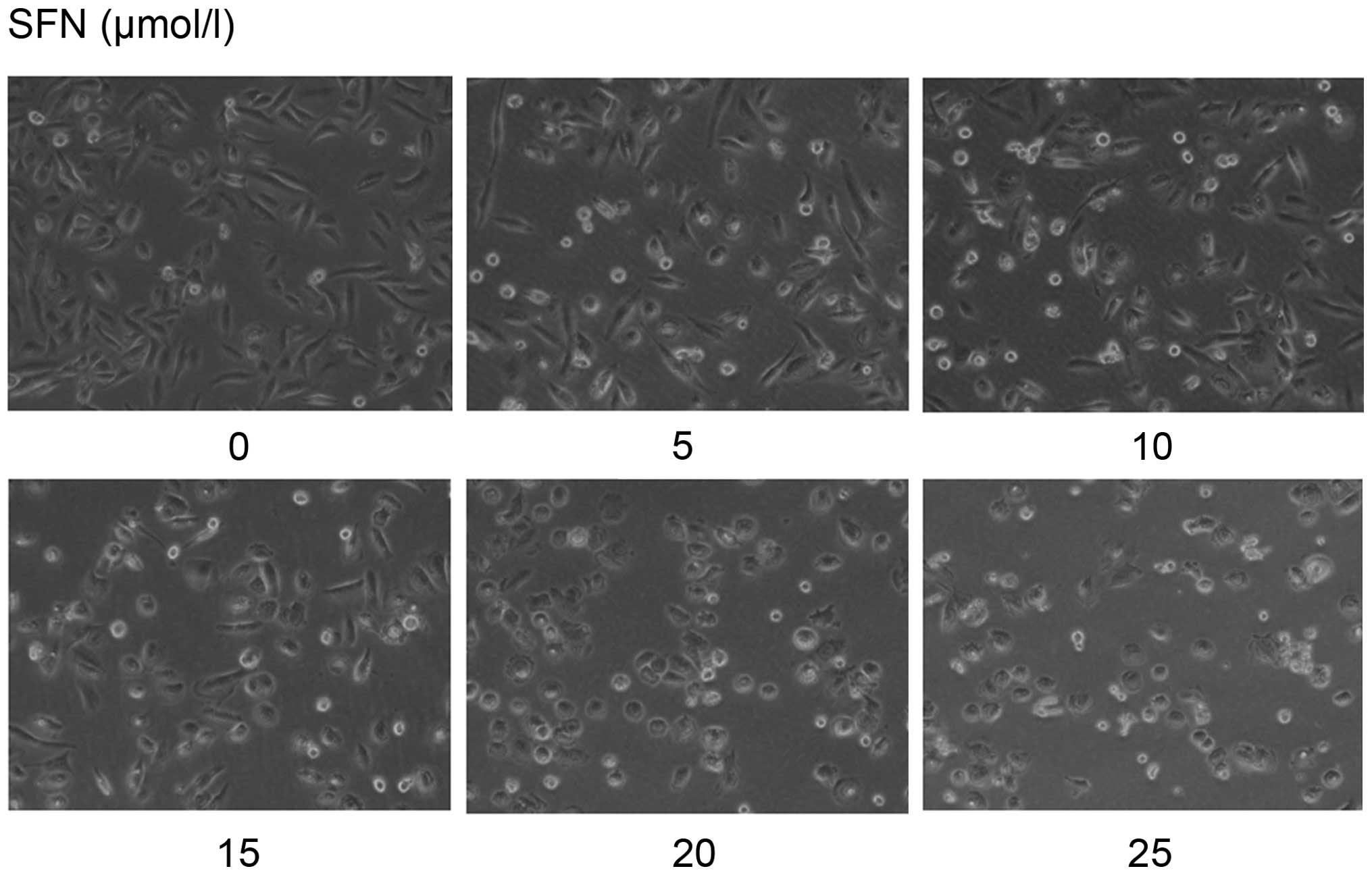
SFN alters cell morphology in a dose- and
time-dependent manner
Untreated DU145 cells presented a morphological
change with some cell pseudopodia. We found that SFN-treated cells
exhibited less pseudopodia and became rounded with the doses and
times (Fig. 2). Pseudopodia are
required in the migration and invasion of tumor cells. Therefore,
we suggested that SFN may inhibit invasion in DU145 cells.
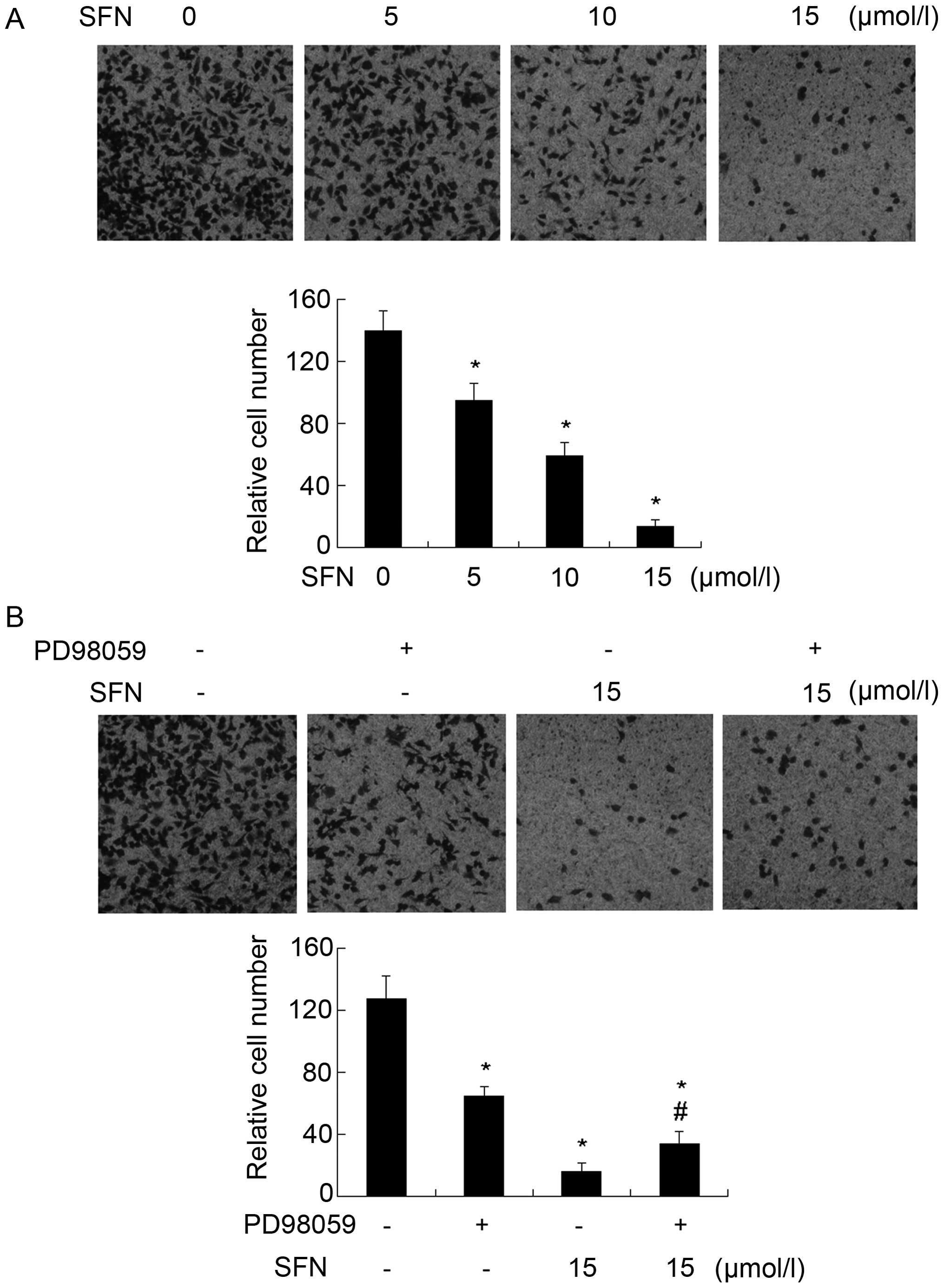
SFN inhibits cell invasion in a
dose-dependent manner
Matrigel-coated Transwells were used to determine
the effect of SFN on cell invasion. DU145 cells were treated with
different concentrations of SFN (0, 5, 10 and 15 µM). The
results showed that SFN significantly reduced cell invasion vs. the
control group in DU145 cells (Fig.
3a). In order to determine whether SFN inhibited cell invasion
by activating ERK1/2, we continued to treat cells with or without
PD98059, the blocker of ERK1/2. The invasive ability of the cells
treated with PD98059 and SFN was improved vs. the SFN-only
treatment cells (Fig. 3B).
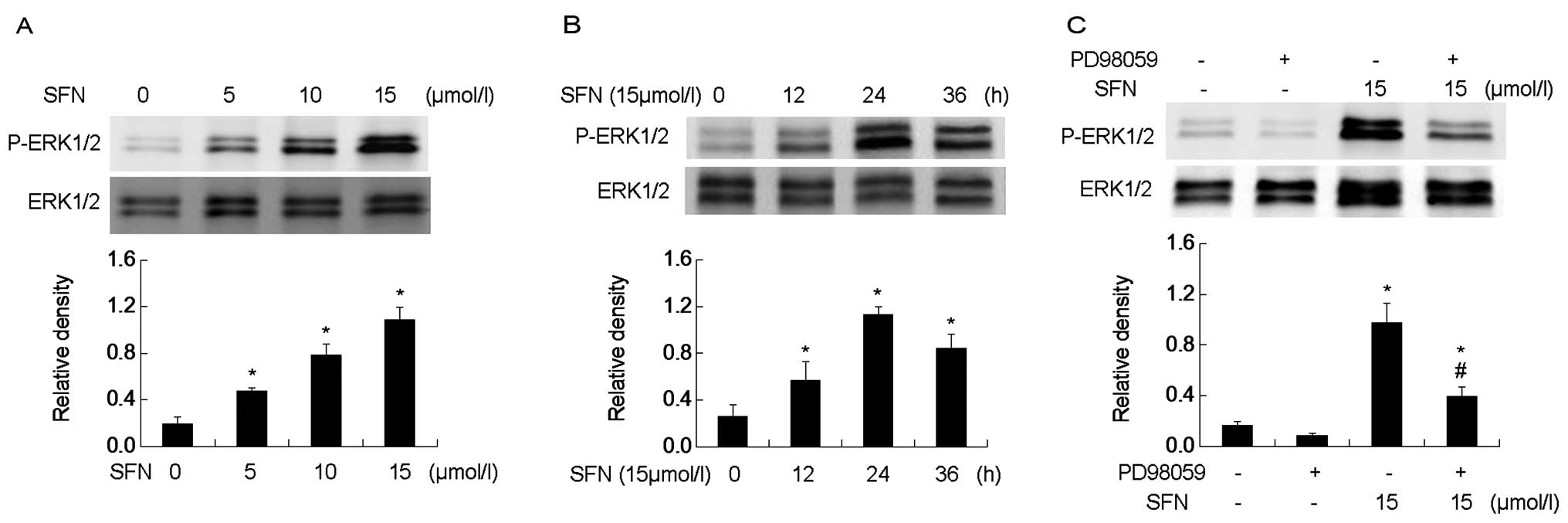
SFN activates ERK1/2 in a dose- and
time-dependent manner
The ERK1/2 phosphorylation was tested in SFN-treated
DU145 cells. SFN was added to the medium at different
concentrations (0, 5, 10 and 15 µM) for 24 h. Western blot
analysis showed that SFN induced ERK1/2 phosphorylation in a
dose-dependent manner (Fig. 4A). We
treated the cells with 15 µM SFN at different time-points
(0, 12, 24 and 36 h). Western blot analysis revealed that ERK1/2
phosphorylation followed a time-dependent manner and increased to
the peak at 24 h (Fig. 4B). PD98059
significantly diminished ERK1/2 phosphorylation and the level of
activation was higher than that of the control (0 µM of SFN)
(Fig. 4C).
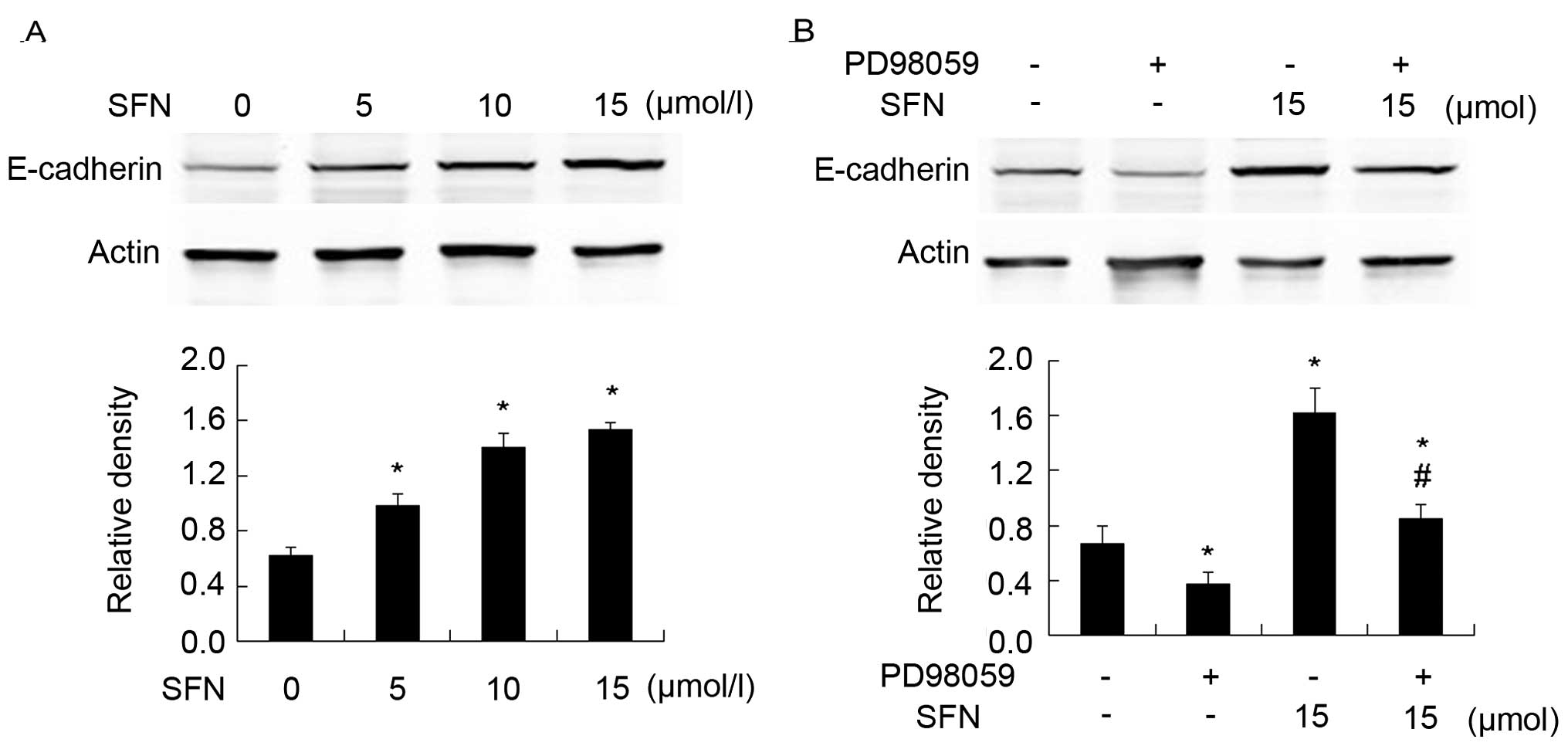
SFN upregulates E-cadherin by activating
ERK1/2
In the present study, we investigated whether SFN
regulated E-cadherin in DU145 cells. Western blot analysis showed
that SFN upregulated E-cadherin significantly in a dose-dependent
manner (Fig. 5A). To clarify
whether the increased level of E-cadherin by SFN treatment was
caused by phosphorylation of ERK1/2, we pretreated cells with
PD98059. Western blot analysis showed that PD98059 significantly
abolished the upregulation of E-cadherin triggered by SFN (Fig. 5B). This result indicated that SFN
upregulated E-cadherin via sustained ERK1/2 phosphorylation.
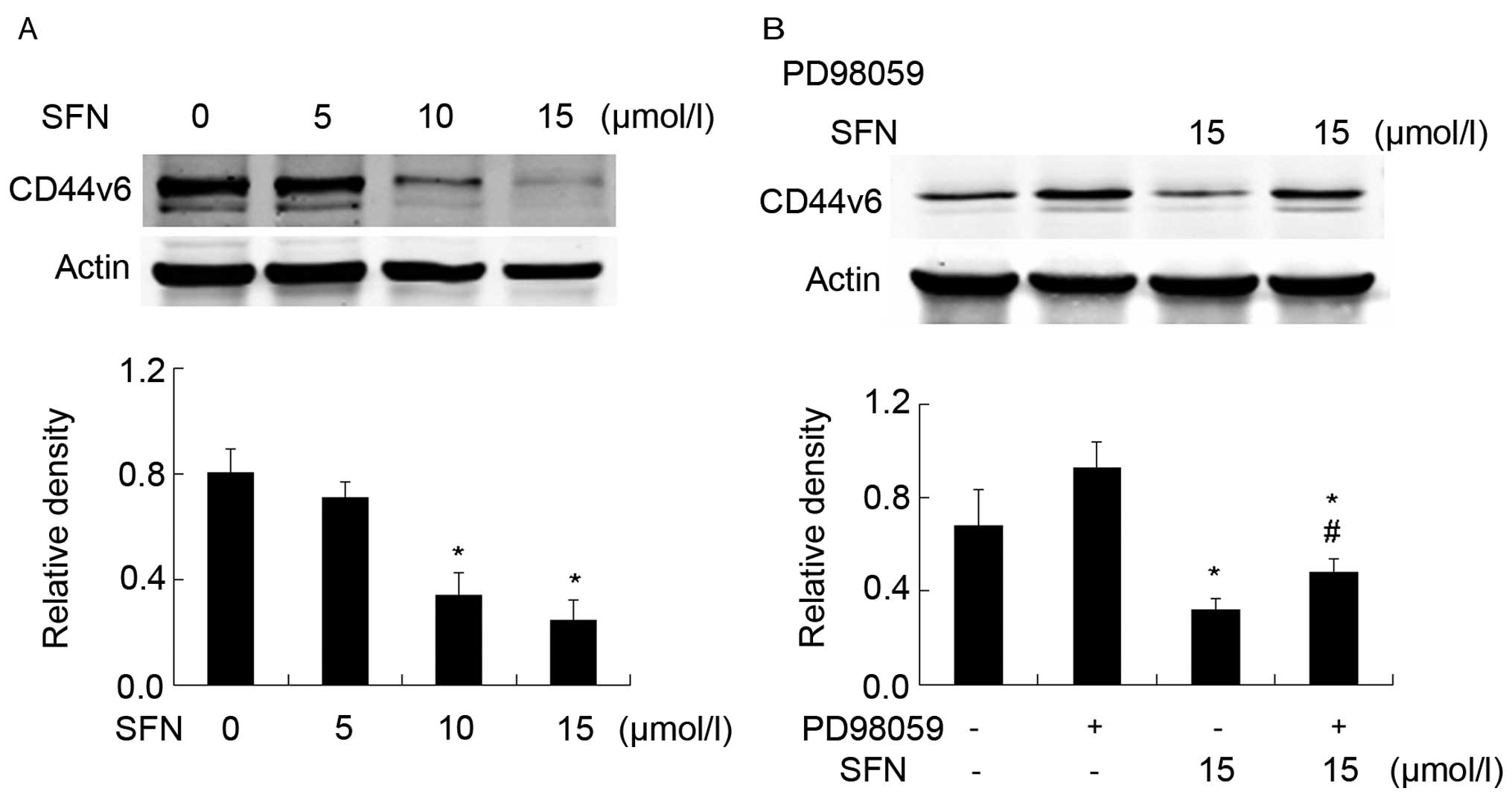
SFN downregulates CD44v6 in a sustained
manner
Western blot analysis showed that SFN downregulated
CD44v6 significantly in a dose-dependent manner (Fig. 6A). The cells were treated with
PD98059 to verify whether SFN regulated CD44v6 via ERK1/2
phosphorylation. However, western blot analysis showed that PD98059
was able to reverse the reduction of CD44v6 triggered by SFN
(Fig. 6B). This result indicated
that SFN markedly downregulated CD44v6 by phosphorylating
ERK1/2.
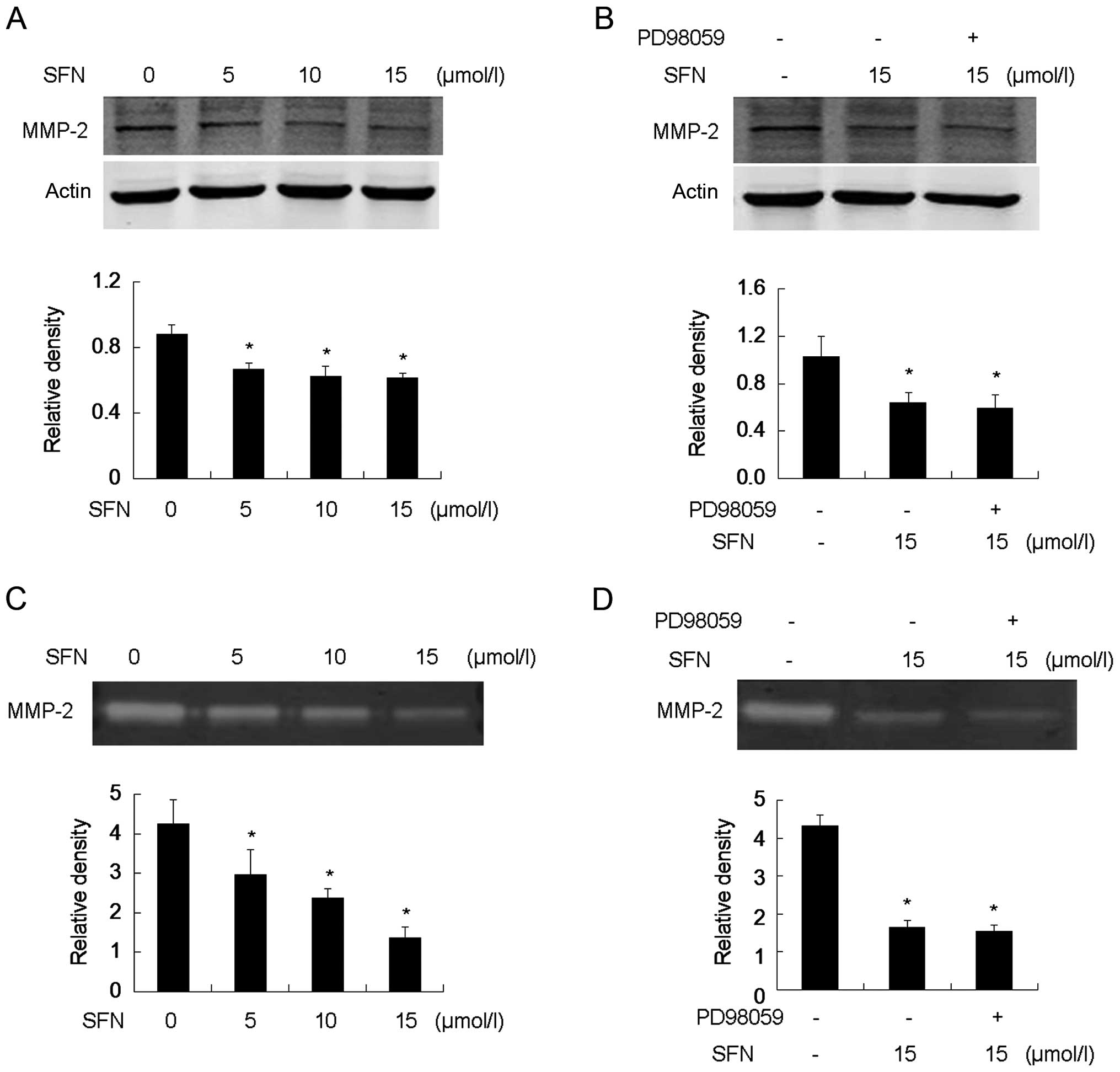
SFN downregulates pro-MMP-2 (72 kDa)
expression and decreases active MMP-2 (64 kDa) activity
Western blot analysis showed that SFN downregulated
pro-MMP-2 expression (Fig. 7A).
Gelatin zymography revealed that SFN reduced active MMP-2 in a
dose-dependent manner (Fig. 7C).
Furthermore, the cells were pretreated with PD98059 to verify
whether this regulation of MMP-2 resulted from the phosphorylation
of ERK1/2. No obvious variance between SFN-only treatment and
PD98059 plus SFN treatment was identified, suggesting that blocking
the phosphorylation of ERK1/2 did not eradicate inhibition of MMP-2
expression and activation induced by SFN. Thus, the ERK1/2
signaling pathway was dispensable in the SFN-mediated reduction of
MMP-2 expression and activation thereof. (Fig. 7B and D).
Discussion
SFN has been identified as a promising agent in
preclinical evaluation and is able to block carcinogenesis
effectively. Previous studies have reported that normal human
bronchial epithelial and normal prostate epithelial cells are more
resistant to SFN-induced apoptosis compared with human lung and
prostate cancer cells (9). Although
SFN potentially resists metastasis by suppressing cell migration
and invasion in some types of cancer, the involved mechanisms
remain to be determined (16,43–45).
In the present study, we show that SFN potently
inhibited proliferation and invasion in vitro. The results
of the MTS assay showed that SFN suppressed proliferation
significantly at a concentration of >15 µM. The
morphological observation showed that cell pseudopodia were
markedly different. These results provide an initial reference to
determine the optimal working concentration in invasion study.
The ERK1/2 signaling pathway contributes to tumor
occurrence, development and metastasis (17,21,22).
Our results indicated that SFN induced ERK1/2 phosphorylation in a
dose- and time-dependent manner. We also found that SFN suppressed
invasion through the activation of ERK1/2. These data are
consistent with those obtained in a previous study (16). One possibility for this activation
of ERK1/2 may result from the production of reactive oxygen species
(ROS) by participation of SFN (46). SFN administration to prostate cancer
cells led to ROS generation, which induced apoptosis and metastatic
inhibition (14). In addition, we
demonstrated that SFN upregulated E-cadherin and downregulated
CD44v6 via the activation of ERK1/2. These two molecules are major
players in mediating EMT. EMT is a reversible process, whereby the
epithelial cells lose their junctions and polarity, reorganize
their cytoskeleton, gain ability of motility, transit into
mesenchymal cells and develop an invasive phenotype (30). The functional loss of E-cadherin is
the typical characteristic of EMT and this contributes to the
destabilization of adheren junctions, as well as tumor invasion and
metastasis (20,30,47,48).
Therefore, SFN is capable of suppressing the process of EMT by
inducing E-cadherin. EMT is manipulated by a group of transcription
factors, including SNaIl, zinc-finger E-box-binding (ZEB) and
TWIST. TWIST can be phosphorylated by ERK1/2, which promotes its
nuclear import and functions, such as controlling the expression of
E-cadherin (30). Notably, it has
been suggested that MMPs regulate SNaIl and ZEB transcription, and
modulate the E-cadherin level (30,44).
As a feedback loop, the loss of E-cadherin induced ZEB-dependent
MMP-2 level in non-small cell lung cancer (32). Those findings are consistent with
our results whereby SFN suppressed cell invasion by inducing
E-cadherin and reducing MMP-2 in a dose-dependent manner. We also
found that SFN significantly reduced the expression of CD44v6 and
PD98059 attenuated the SFN-mediated reduction of CD44v6, suggesting
that the downregulation of CD44v6 induced was through ERK1/2
activation. These results were coincident with those in the other
studies. Overexpression of CD44v6 was found in primary prostate
cancer tissues and metastases, but not in normal prostate tissues
(3). It was also shown in the same
study that knockdown of CD44v6 by siRNA decreased prostate cancer
cell invasive and adhesive ability to hyaluronic acid (HA),
suggesting that CD44v6 is an important participant in mediating
tumor cell adhesion during metastasis (3). Another study showed that CD44 was a
downstream effector of ERK1/2 inducing cell migration and invasion
in vitro and tumor growth and metastasis in vivo in
human oral cancer (19). It was
reported that CD44v6 induced EMT in prostate cancer and the
induction of EMT was accompanied by a shift in CD44 isoforms in
breast cancer cells (49).
Therefore, SFN was suggested to suppress EMT as well as invasion
via the downregulation of CD44v6 and upregulation of
E-cadherin.
A recent study described that the CD44v6 was found
to be positively correlated with MMP-2 in papillary thyroid cancer
(50). Furthermore, CD44 promoted
invasion by anchoring MMPs on the tumor cell surface and it was
cleaved by MMPs in the extracellular of several tumors (51). Emerging evidence indicates that MMPs
can induce EMT during tumor development (39). MMP-2 is one of the most distinctive
members of this family and facilitates prostate cancer cell
invasion. Previous studies have demonstrated that MMP-2 expression
is mediated by ERK1/2 (26,27,47,52).
In the present study, we found that SFN significantly decreased
MMP-2 expression and activity. However, the SFN-triggered
inhibition of MMP-2 was not reversed by PD98059. Thus, the
downregulation of MMP-2 expression and activity modulated by SFN
was not via the ERK1/2 signaling pathway. Following the above
observations, the question concerning how SFN regulated MMP-2
expression and activity is raised. One possible mechanism leading
to decreased MMP-2 may be that SFN activates or inactivates some
transcription factors, such as nuclear factor-κB (NF-κB), SNaIl and
ZEB (30,53). In addition, MMPs can also be
regulated by their endogenous tissue inhibitors TIMPs, and the
proteolytic activity of tumor cells depends on the balance between
MMPs and TIMPs (47). Induction of
TIMP-1/-2 by SFN has been proven in human bladder cancer cells
(44). In this regard, we
speculated that the upregulation of TIMP-1/-2 following the
treatment of SFN may contribute to the inhibition of MMP-2
activity. More studies focusing on SFN-induced TIMP upregulation
should therefore be conducted.
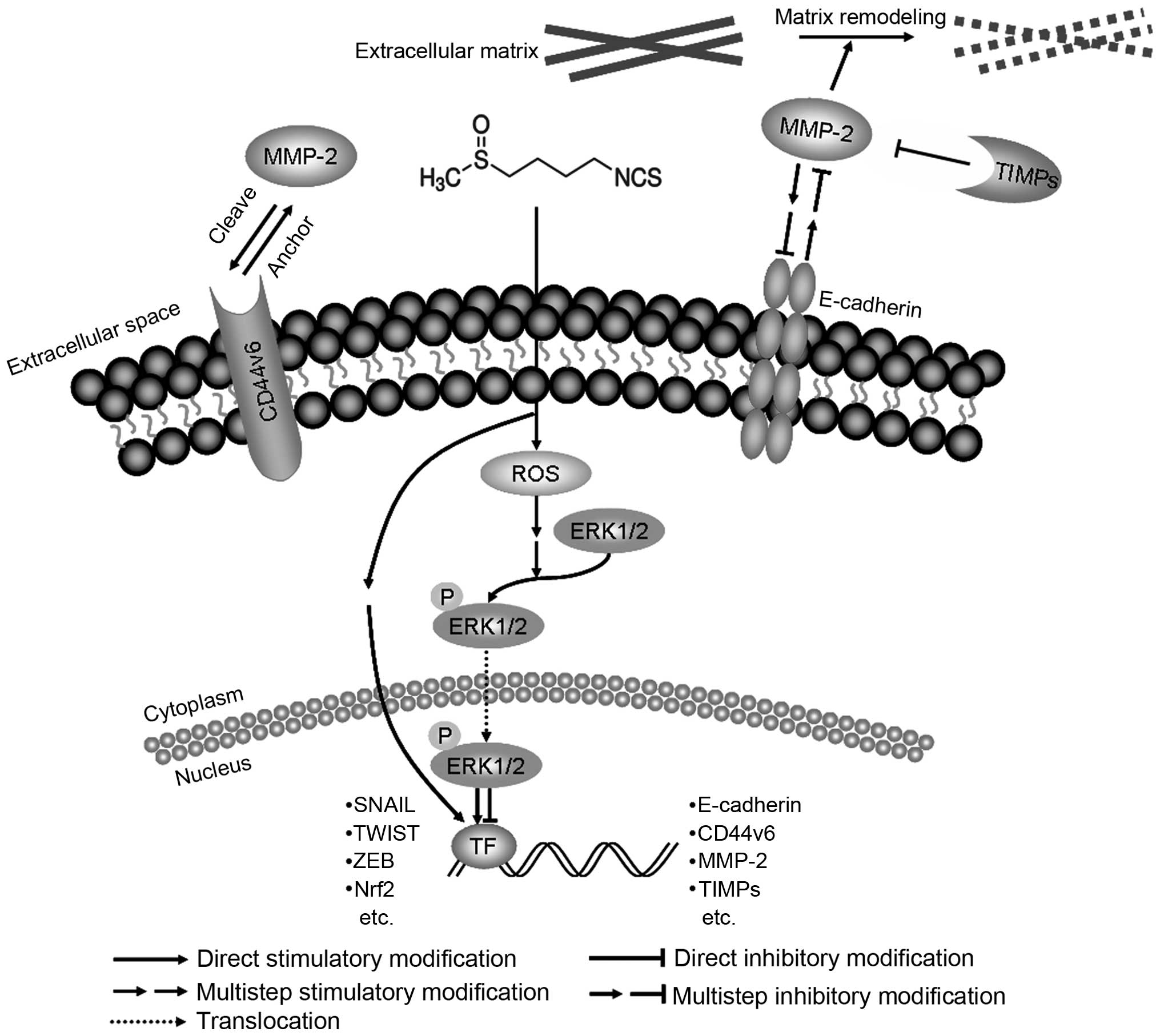
In conclusion, we have demonstrated that SFN
suppressed invasion in human DU145 prostate cancer cells by
modulating E-cadherin, CD44v6 and MMP-2, which linked to EMT
(Fig. 8). Suppression of EMT was
demonstrated to be an effective strategy against cancer metastasis
(30,47). The results suggested that SFN is a
prospective drug to prevent invasion of prostate cancer cells.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81272843) and the
Beijing Natural Science Foundation (grant no. 7132019). The funders
had no role in the study design, data collection and analysis,
decision to publish or preparation of the manuscript.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chaturvedi S and Garcia JA: Novel agents
in the management of castration resistant prostate cancer. J
Carcinog. 13:52014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ni J, Cozzi PJ, Hao JL, Beretov J, Chang
L, Duan W, Shigdar S, Delprado WJ, Graham PH, Bucci J, et al: CD44
variant 6 is associated with prostate cancer metastasis and
chemo-/radioresistance. Prostate. 74:602–617. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakano K, Komatsu K, Kubo T, Natsui S,
Nukui A, Kurokawa S, Kobayashi M and Morita T: External validation
of risk classification in patients with docetaxel-treated
castration-resistant prostate cancer. BMC Urol. 14:312014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cohen JH, Kristal AR and Stanford JL:
Fruit and vegetable intakes and prostate cancer risk. J Natl Cancer
Inst. 92:61–68. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang SM, Hunter DJ, Rosner BA,
Giovannucci EL, Colditz GA, Speizer FE and Willett WC: Intakes of
fruits, vegetables, and related nutrients and the risk of
non-Hodgkin's lymphoma among women. Cancer Epidemiol Biomarkers
Prev. 9:477–485. 2000.PubMed/NCBI
|
|
7
|
Ambrosone CB, McCann SE, Freudenheim JL,
Marshall JR, Zhang Y and Shields PG: Breast cancer risk in
premenopausal women is inversely associated with consumption of
broccoli, a source of isothiocyanates, but is not modified by GST
genotype. J Nutr. 134:1134–1138. 2004.PubMed/NCBI
|
|
8
|
Dinkova-Kostova AT and Kostov RV:
glucosinolates and isothiocyanates in health and disease. Trends
Mol Med. 18:337–347. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Choi S and Singh SV: Bax and Bak are
required for apoptosis induction by sulforaphane, a cruciferous
vegetable-derived cancer chemopreventive agent. Cancer Res.
65:2035–2043. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Talalay P and Fahey JW: Phytochemicals
from cruciferous plants protect against cancer by modulating
carcinogen metabolism. J Nutr. 131(Suppl): 3027S–3033S.
2001.PubMed/NCBI
|
|
11
|
Conaway CC, Yang YM and Chung FL:
Isothiocyanates as cancer chemopreventive agents: Their biological
activities and metabolism in rodents and humans. Curr Drug Metab.
3:233–255. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lenzi M, Fimognari C and Hrelia P:
Sulforaphane as a promising molecule for fighting cancer. Cancer
Treat Res. 159:207–223. 2014. View Article : Google Scholar
|
|
13
|
Cheung KL and Kong AN: Molecular targets
of dietary phenethyl isothiocyanate and sulforaphane for cancer
chemoprevention. AAPS J. 12:87–97. 2010. View Article : Google Scholar :
|
|
14
|
Clarke JD, Dashwood RH and Ho E:
Multi-targeted prevention of cancer by sulforaphane. Cancer Lett.
269:291–304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hecht SS: Inhibition of carcinogenesis by
isothiocyanates. Drug Metab Rev. 32:395–411. 2000. View Article : Google Scholar
|
|
16
|
Li C, Zhou Y, Peng X, Du L, Tian H, Yang
G, Niu J and Wu W: Sulforaphane inhibits invasion via activating
ERK1/2 signaling in human glioblastoma U87MG and U373MG cells. PLoS
One. 9:e905202014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deschênes-Simard X, Kottakis F, Meloche S
and Ferbeyre G: ERKs in cancer: Friends or foes? Cancer Res.
74:412–419. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou Y, Hu HY, Meng W, Jiang L, Zhang X,
Sha JJ, Lu Z and Yao Y: MEK inhibitor effective against
proliferation in breast cancer cell. Tumour Biol. 35:9269–9279.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Judd NP, Winkler AE, Murillo-Sauca O,
Brotman JJ, Law JH, Lewis JS Jr, Dunn GP, Bui JD, Sunwoo JB and
Uppaluri R: ERK1/2 regulation of CD44 modulates oral cancer
aggressiveness. Cancer Res. 72:365–374. 2012. View Article : Google Scholar :
|
|
20
|
Lau MT, So WK and Leung PC: Fibroblast
growth factor 2 induces E-cadherin down-regulation via
PI3K/akt/mToR and MAPK/ERK signaling in ovarian cancer cells. PLoS
One. 8:e590832013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Burotto M, Chiou VL, Lee JM and Kohn EC:
The MaPK pathway across different malignancies: A new perspective.
Cancer. 120:3446–3456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Marshall CJ: Specificity of receptor
tyrosine kinase signaling: Transient versus sustained extracellular
signal-regulated kinase activation. Cell. 80:179–185. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu W, Ginsburg E, Vonderhaar BK and Walker
AM: S179D prolactin increases vitamin D receptor and p21 through
up-regulation of short 1b prolactin receptor in human prostate
cancer cells. Cancer Res. 65:7509–7515. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Z, Yu X and Shaikh ZA: Rapid
activation of ERK1/2 and AKT in human breast cancer cells by
cadmium. Toxicol Appl Pharmacol. 228:286–294. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang TY, Chang GC, Chen KC, Hung HW, Hsu
KH, Sheu GT and Hsu SL: Sustained activation of ERK and
Cdk2/cyclin-A signaling pathway by pemetrexed leading to S-phase
arrest and apoptosis in human non-small cell lung cancer A549
cells. Eur J Pharmacol. 663:17–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Z, Du L, Li C and Wu W: Human chorionic
gonadotropin β induces cell motility via ERK1/2 and MMP-2
activation in human glioblastoma U87MG cells. J Neurooncol.
111:237–244. 2013. View Article : Google Scholar
|
|
27
|
Li Z, Li C, Du L, Zhou Y and Wu W: Human
chorionic gonadotropin β induces migration and invasion via
activating ERK1/2 and MMP-2 in human prostate cancer DU145 cells.
PLoS One. 8:e545922013. View Article : Google Scholar
|
|
28
|
Wheelock MJ and Johnson KR: Cadherins as
modulators of cellular phenotype. Annu Rev Cell Dev Biol.
19:207–235. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cavallaro U and Christofori G: Cell
adhesion and signalling by cadherins and Ig-CaMs in cancer. Nat Rev
Cancer. 4:118–132. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li Q and Mattingly RR: Restoration of
E-cadherin cell-cell junctions requires both expression of
E-cadherin and suppression of ERK MAP kinase activation in
Ras-transformed breast epithelial cells. Neoplasia. 10:1444–1458.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bae GY, Choi SJ, Lee JS, Jo J, Lee J, Kim
J and Cha HJ: Loss of E-cadherin activates EGFR-MEK/ERK signaling,
which promotes invasion via the ZEB1/MMP2 axis in non-small cell
lung cancer. Oncotarget. 4:2512–2522. 2013.PubMed/NCBI
|
|
33
|
Nagano O, Okazaki S and Saya H: Redox
regulation in stem-like cancer cells by CD44 variant isoforms.
Oncogene. 32:5191–5198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Khan SA, Cook AC, Kappil M, Günthert U,
Chambers AF, Tuck AB and Denhardt DT: Enhanced cell surface CD44
variant (v6, v9) expression by osteopontin in breast cancer
epithelial cells facilitates tumor cell migration: Novel
post-transcriptional, post-translational regulation. Clin Exp
Metastasis. 22:663–673. 2005. View Article : Google Scholar
|
|
35
|
Akisik E, Bavbek S and Dalay N: CD44
variant exons in leukemia and lymphoma. Pathol Oncol Res. 8:36–40.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee JL, Wang MJ, Sudhir PR, Chen GD, Chi
CW and Chen JY: Osteopontin promotes integrin activation through
outside-in and inside-out mechanisms: OPN-CD44v interaction
enhances survival in gastrointestinal cancer cells. Cancer Res.
67:2089–2097. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mulder JW, Wielenga VJ, Polak MM, van den
Berg FM, Adolf GR, Herrlich P, Pals ST and Offerhaus GJ: Expression
of mutant p53 protein and CD44 variant proteins in colorectal
tumorigenesis. Gut. 36:76–80. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jones JL and Walker RA: Control of matrix
metalloproteinase activity in cancer. J Pathol. 183:377–379. 1997.
View Article : Google Scholar
|
|
39
|
Orlichenko LS and Radisky DC: Matrix
metalloproteinases stimulate epithelial-mesenchymal transition
during tumor development. Clin Exp Metastasis. 25:593–600. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bérubé M, Deschambeault A, Boucher M,
Germain L, Petitclerc E and Guérin SL: MMP-2 expression in uveal
melanoma: Differential activation status dictated by the cellular
environment. Mol vis. 11:1101–1111. 2005.PubMed/NCBI
|
|
41
|
Sato T, Sakai T, Noguchi Y, Takita M,
Hirakawa S and Ito A: Tumor-stromal cell contact promotes invasion
of human uterine cervical carcinoma cells by augmenting the
expression and activation of stromal matrix metalloproteinases.
Gynecol Oncol. 92:47–56. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Di Nezza LA, Misajon A, Zhang J, Jobling
T, Quinn MA, Ostör AG, Nie G, Lopata A and Salamonsen LA: Presence
of active gelatinases in endometrial carcinoma and correlation of
matrix metalloproteinase expression with increasing tumor grade and
invasion. Cancer. 94:1466–1475. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gupta P, Kim B, Kim SH and Srivastava SK:
Molecular targets of isothiocyanates in cancer: Recent advances.
Mol Nutr Food Res. 58:1685–1707. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shan Y, Zhang L, Bao Y, Li B, He C, Gao M,
Feng X, Xu W, Zhang X and Wang S: Epithelial-mesenchymal
transition, a novel target of sulforaphane via COX-2/MMP2, 9/Snail,
ZEB1 and miR-200c/ZEB1 pathways in human bladder cancer cells. J
Nutr Biochem. 24:1062–1069. 2013. View Article : Google Scholar
|
|
45
|
Hahm ER, Chandra-Kuntal K, Desai D, Amin S
and Singh SV: Notch activation is dispensable for D,
L-sulforaphane-mediated inhibition of human prostate cancer cell
migration. PLoS One. 7:e449572012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jo C, Kim S, Cho SJ, Choi KJ, Yun SM, Koh
YH, Johnson GV and Park SI: Sulforaphane induces autophagy through
ERK activation in neuronal cells. FEBS Lett. 588:3081–3088. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shen KH, Liao AC, Hung JH, Lee WJ, Hu KC,
Lin PT, Liao RF and Chen PS: α-Solanine inhibits invasion of human
prostate cancer cell by suppressing epithelial-mesenchymal
transition and MMPs expression. Molecules. 19:11896–11914. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wu W and Walker AM: Human chorionic
gonadotropin beta (HCGbeta) down-regulates E-cadherin and promotes
human prostate carcinoma cell migration and invasion. Cancer.
106:68–78. 2006. View Article : Google Scholar
|
|
49
|
Brown RL, Reinke LM, Damerow MS, Perez D,
Chodosh LA, Yang J and Cheng C: CD44 splice isoform switching in
human and mouse epithelium is essential for epithelial-mesenchymal
transition and breast cancer progression. J Clin Invest.
121:1064–1074. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wu G, Zhou Y, Li T, Guo J and Zhou Z:
Immunohistochemical levels of matrix metalloproteinase-2 and CD44
variant 6 protein in the diagnosis and lateral cervical lymph node
metastasis of papillary thyroid carcinoma. J Int Med Res.
41:816–824. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yu Q and Stamenkovic I: localization of
matrix metal-loproteinase 9 to the cell surface provides a
mechanism for CD44-mediated tumor invasion. Genes Dev. 13:35–48.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chen Y, Zheng L, Liu J, Zhou Z, Cao X, Lv
X and Chen F: Shikonin inhibits prostate cancer cells metastasis by
reducing matrix metalloproteinase-2/-9 expression via AKT/mTOR and
RoS/ERK1/2 pathways. Int Immunopharmacol. 21:447–455. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang Y, Li M, Xu Y, He N, Leng L and Li Z:
Tumor necrosis factor-α regulates matrix metalloproteinase-2
expression and cell migration via ERK pathway in rat glomerular
mesangial cells. Cell Biol Int. 38:1060–1068. 2014.PubMed/NCBI
|