Introduction
Lung cancer remains the leading cause of
cancer-related mortality worldwide and non-small cell lung cancer
(NSCLC) accounts for approximately 80–85% of all cases of lung
cancer (1). The overall prognosis
and survival rate of patients with advanced NSCLC remain
unsatisfactory, with a median survival time of 8–11 months and a
one-year survival rate of 30% (2,3).
Chemotherapy plays a significant role in the management of human
lung cancer (4,5). Cisplatin (DDP) is widely used to treat
many types of solid tumors and cisplatin-based adjuvant
chemotherapy has significantly improved the progression-free
survival of cancer patients, which has translated to a 5–10%
improvement in the cure rate (6,7).
However, few patients experience complete responses to
chemotherapy, mainly because of the resistance of tumor cells
and/or tolerance of the surrounding normal tissues (8). Thus, novel approaches to enhance the
cisplatin efficacy and reverse chemoresistance are required, as
well as chemosensitizers to reduce the dose of drugs administered
and the length of time resulting in reduced side effects.
Cisplatin induces DNA crosslinks and can induce cell
apoptosis, as is the case with many other chemotherapeutic drugs
(9). Cancer cells elicits DNA
repair mechanisms including nucleotide excision repair (NER), base
excision repair (BER), double-strand break repair (DSBR) and
mismatch repair (MMR) (10–12). In addition, eukaryotic cells have a
DNA post-replication repair system that is composed of translesion
DNA synthesis (TLS) and homologous DNA recombination (HR) pathways.
The ubiquitous TLS consists of a series of specialized polymerases,
including polymerase (Pol) κ, ζ, η and ι (13). The catalytic REV3L subunit interacts
with structural REV7L to form polymerase Pol ζ. Pol ζ cannot add
nucleotides across DNA lesions, but it can extend from primers with
terminal mismatches, which makes Pol ζ crucial in translesion DNA
synthesis (TLS). Pol ζ is able to mediate DNA replication bypassing
DNA damage, which may prevent chromosome instability in cells and
be considered as a suppressor of spontaneous tumorigenesis.
However, Pol ζ lacks 3′ to 5′ exonuclease activity and can insert a
nucleotide into the lesion to complete the bypass of the lesion,
which is important for cell survival when confronted with DNA
damage (14–16). Loss of REV3L (also known as REV3 in
vetebrates) has shown increased sensitivity to exogenous insults.
For example, REV3-null mouse-derived embryonic fibroblasts
are more sensitive to UV- and γ-irradiation with increased
chromosomal abnormalities (17).
The above mentioned results indicated that REV3L is involved
in cell tolerance to various types of DNA damage.
The REV3(L) protein was partially conserved from
yeast to vertebrates. It has been previously reported that human
Pol ζ is expressed in human cancers including lung, stomach and
colorectal cancers (18,19). Moreover, REV3L suppressed the focus
formation suggesting a tumor-suppressor role. Due to its critical
role in translesion DNA synthesis, whether REV3L is involved in
chemoresistance remains unknown. In this study, we found that
cisplatin induced the expression of REV3L by recruiting Sp1 to its
promoter. Knockdown of REV3L sensitized cisplatin efficacy in H1299
cells.
Materials and methods
Reagents and plasmids
Benzo[a]pyrene (B[a]P), nicotine and
4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) were purchased
from Sigma-Aldrich (St. Louis, MO, USA). Cisplatin was purchased
from Qilu Pharmaceutical Co., Ltd. (Shandong, China). Geneticin
(G418) was obtained from Life Technologies (Gaithersburg, MD,
USA).
REV3L overexpression and R NAi
vectors
The pcDNA3.1/neo-REV3L plasmid was kindly provided
by Dr Yoshiki Murakumo, Nagoya University Graduate School of
Medicine, Nagoya, Japan. Four shRNAs targeting REV3L were designed
and constructed by GenePharma (Shanghai, China). The information of
the four shRNAs is provided in Table
I.
 | Table ITargeting sequences of shRNAs. |
Table I
Targeting sequences of shRNAs.
| Name | Targeting
sequence | Location
(REV3L) |
|---|
| shRNA-NC |
5′-GTCAATGGTCGTGTCGTGC-3′ | |
| shRNA-1 |
5′-CGAAGATTGTGACCTGAATTA-3′ | 3611–3632 |
| shRNA-2 |
5′-CTTCTGGTATGTCCTCAAAGA-3′ | 4447–4468 |
| shRNA-3 |
5′-AGGAAAGCCAAATGCCTAATA-3′ | 4672–4693 |
| shRNA-4 |
5′-CTCTAGTGATATCTCCAATTA-3′ | 6859–6880 |
Cell culture and transfection
The human H1299 lung cancer cell line was maintained
in DMEM supplemented with 10% FBS and antibiotics (Gibco, Grand
Island, NY, USA). The cells were grown in a 37°C incubator with 5%
CO2. The cells were transfected by Lipofectamine 2000
(Invitrogen Life Technologies, Carlsbad, CA, USA) with
plasmids.
To generate stable REV3L-overexpressing or
-silencing clones, the cells were grown in a 24-well culture plate
to 70–80% confluence and then transfected with 1 µg of
pcDNA3.1, pcDNA-REV3L, shRNA-NC or shRNA targeting REV3L
vector using Lipofectamine 2000 (Invitrogen Life Technologies)
according to the manufacturer's instructions. The medium was
replaced with DMEM containing 600 µg/ml G418 48 h
post-transfection. After 3–4 weeks, G418-resistant colonies were
selected and screened for REV3L expression by reverse
transcriptase-PCR (RT-PCR). The REV3L-overexpressing and -silencing
clones were cultured in DMEM supplemented with 10% fetal bovine
serum in the presence of 300 µg/ml G418 at 37°C in
humidified air with 5% CO2.
Reverse transcriptase-PCR analysis
Total RNA from lung tissues was extracted with
TRIzol (Invitrogen Life Technologies) and reverse transcribed to
cDNA using an oligo(dT)12 primer and Superscript II
(Invitrogen Life Technologies). The mRNA levels of target genes and
the internal standard glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) were measured by RT-PCR or quantitative PCR (qPCR) in
triplicate on a Prism 7500 real-time PCR machine (Applied
Biosystems, Foster City, CA, USA). The specific primers for the
genes are listed in Table II.
| Table IIPrimer sequences for RT-PCR and ChIP
analysis. |
Table II
Primer sequences for RT-PCR and ChIP
analysis.
| Gene | RT-PCR
|
|---|
| Forward primer | Reverse primer |
|---|
| GAPDH |
5′-GAAGGTGAAGGTCGGAGTC-3′ |
5′-GAAGATGGTGATGGGATTTC-3′ |
| REV3L |
5′-CGCGTCAGTTGGGACTTAAG-3′ |
5′-ACTATCGCCAACCTCAATGC-3′ |
|
| ChIP
|
| Gene | Forward primer | Reverse primer |
|
| Region 1 |
5′-GAAGGTGAAGGTCGGAGTC-3′ |
5′-GAAGATGGTGATGGGATTTC-3′ |
| Region 2 |
5′-CGCGTCAGTTGGGACTTAAG-3′ |
5′-ACTATCGCCAACCTCAATGC-3′ |
Immunostaining
H1299 cells were fixed with 4% paraformaldehyde,
washed with PBS, and permeabilized with 1% Triton X-100 in PBS. The
cells were blocked with blocking buffer (PBS, 1% Triton X-100, and
5% BSA) and incubated at 4°C with the REV3L antibody (1:1000;
Abnova, Taiwan) overnight. FITC-conjugated goat anti-mouse (1:100)
was incubated for 30 min at room temperature. Nuclear
counter-staining was performed using 4,6-diamidino-2-phenylindole
(DAPI). Cells not treated with primary antibody served as the
negative control.
Cell viability assay
Cell viability was evaluated using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT) assay. Cells were plated in 96-well plates. The following
day, the cells were transfected with plasmids or shRNAs according
to the experimental design. The cells were then incubated with 20
µl MTT (5 mg/ml) for 4 h. After the medium was removed, 100
µl DMSO was added and the optical density (OD) at 490 nm was
measured using a Microplate Reader (Bio-Rad, Hercules, CA, USA).
The viability index was calculated as experimental OD value/control
OD value. Three independent experiments were performed in
quadruplicate.
Western blot analysis
The cells were lysed in lysis buffer (Promega,
Madison, WI, USA) and centrifuged at 4°C for 10 min. The
supernatant was collected and subjected to western blotting.
Protein (50 µg) from each lysate was fractionated by 10%
SDS-PAGE and transferred to polyvinylidene difluoride membranes
(Millipore, Bedford, MA, USA). After blocking with 5% non-fat milk
in PBS Tween-20 for 1 h at room temperature, the membranes were
blotted with the appropriate Bax, Bcl2 or GAPDH primary antibody
(All from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) at a
1:1000 dilution. The membranes were then incubated with the
appropriate horseradish peroxidase-conjugated secondary antibody at
a 1:2000 dilution for 1 h at room temperature. After TBST washes,
the blot was incubated in the ECL detection kit (Amersham
Bioscience, Freiburg, Germany).
Plasmid reactivation assay
A plasmid reactivation assay was performed as
previously reported (20). The
pGL3-promoter mammalian expression vector (Promega) containing the
Firefly luciferase gene expressed high-level Firefly
luciferase in mammalian cells. The plasmid DNA was dissolved in
buffer containing 10 mM Tris and 1 mM EDTA (pH 7.4) and incubated
with different concentrations of cisplatin for 1 h. The platinated
DNA was then purified by ethanol precipitation and unbound free
cisplatin was removed. This procedure resulted in plasmid DNA that
was >90% supercoiled as verified by gel electrophoresis. Similar
levels of platination have previously been shown not to affect the
efficiency of transfection (21).
For the luciferase assay, the pGL3-Promoter reporter vector plus
pRL-TK (Promega) was co-transfected with pcDNA3.1-REV3L or
shRNA-targeting REV3L. Luciferase activity was measured with the
Dual-Luciferase Reporter Assay System (Promega). Promoter
activities were expressed as the ratio of Firefly luciferase
to Renilla luciferase activities.
Quantitative chromatin
immunoprecipitation (ChIP)
H1299 cells were used for ChIP assays. The EZ ChIP
kit (Upstate Biotechnology, Inc, Lake Placid, NY, USA) was used
according to the manufacturer's instructions as previously reported
(22). Briefly, 5×106
cells were cross-linked with 1% formaldehyde and sonicated to ~500
bp fragments. ChIP was conducted with antibodies against PAX2 and
IgG. Input control DNA or immunoprecipitated DNA was amplified in a
20-µl reaction volume consisting of 4 µl eluted DNA
template and primers specific for REV3L promoter (Table II). The immunoprecipitated
fragments and the inputs were amplified by RT-PCR and qPCR. Results
for the immunoprecipitated fragments were calculated compared to
the Ct values obtained for the input samples in each case and were
expressed as a percentage of the input.
Measurement of apoptosis
Cells were transfected with shRNA-NC or shRNA-REV3L
for 24 h prior to treatment with cisplatin. Apoptosis was measured
using propidium iodide (PI)/Annexin V double staining as per the
manufacturer's instructions (Nanjing KeyGen Biotech. Co., Ltd.,
Nanjing, China). The cells were harvested 24 h after treatment with
cisplatin and apoptotic fractions were measured using flow
cytometry (Beckman Coulter, Miami, FL, USA). The Annexin
V+/PI− cells indicated early apoptosis, while
the Annexin V+/PI+ cells indicated late
apoptosis. The percentage of the two types of cells was
calculated.
Statistical analysis
Data were presented as the mean ± standard error of
the mean (SEM) of at least three independent experiments. Standard
error bars were included for all the data points. The data were
then analyzed using the Student's t-test when only two groups were
present or assessed by one-way analysis of variance (ANOVA) when
more than two groups were compared. Statistical analysis was
performed using SPSS software (Release 17.0, SPSS Inc.). Data were
considered significant when P<0.05.
Results
Cisplatin induces the expression of
REV3L
It has been suggested that cisplatin can induce an
increase in REV3L mRNA levels in both normal and cancer cells.
Thus, we investigated whether cisplatin also increases REV3L mRNA
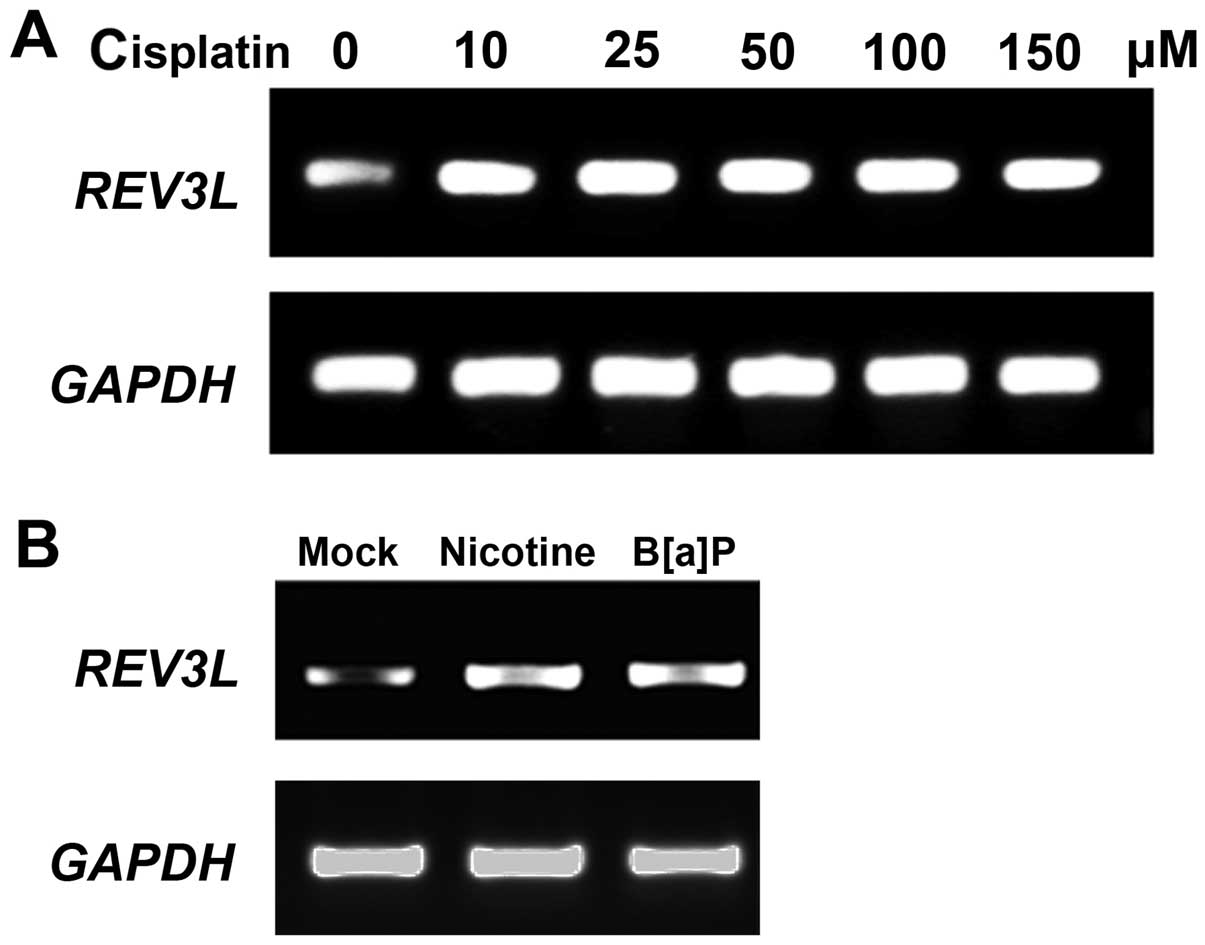
levels in human H1299 NSLSC cells. As shown in Fig. 1A, 10 µM cisplatin increased
the REV3L mRNA level while higher concentrations of
cisplatin did not further increase REV3L expression. Moreover, DNA
damage reagents B[a]P and nicotine upregulated REV3L expression in
H1299 cells, indicating that REV3L was increased in response to DNA
damage.
SP1 is recruited in the REV3L promoter
with cisplatin
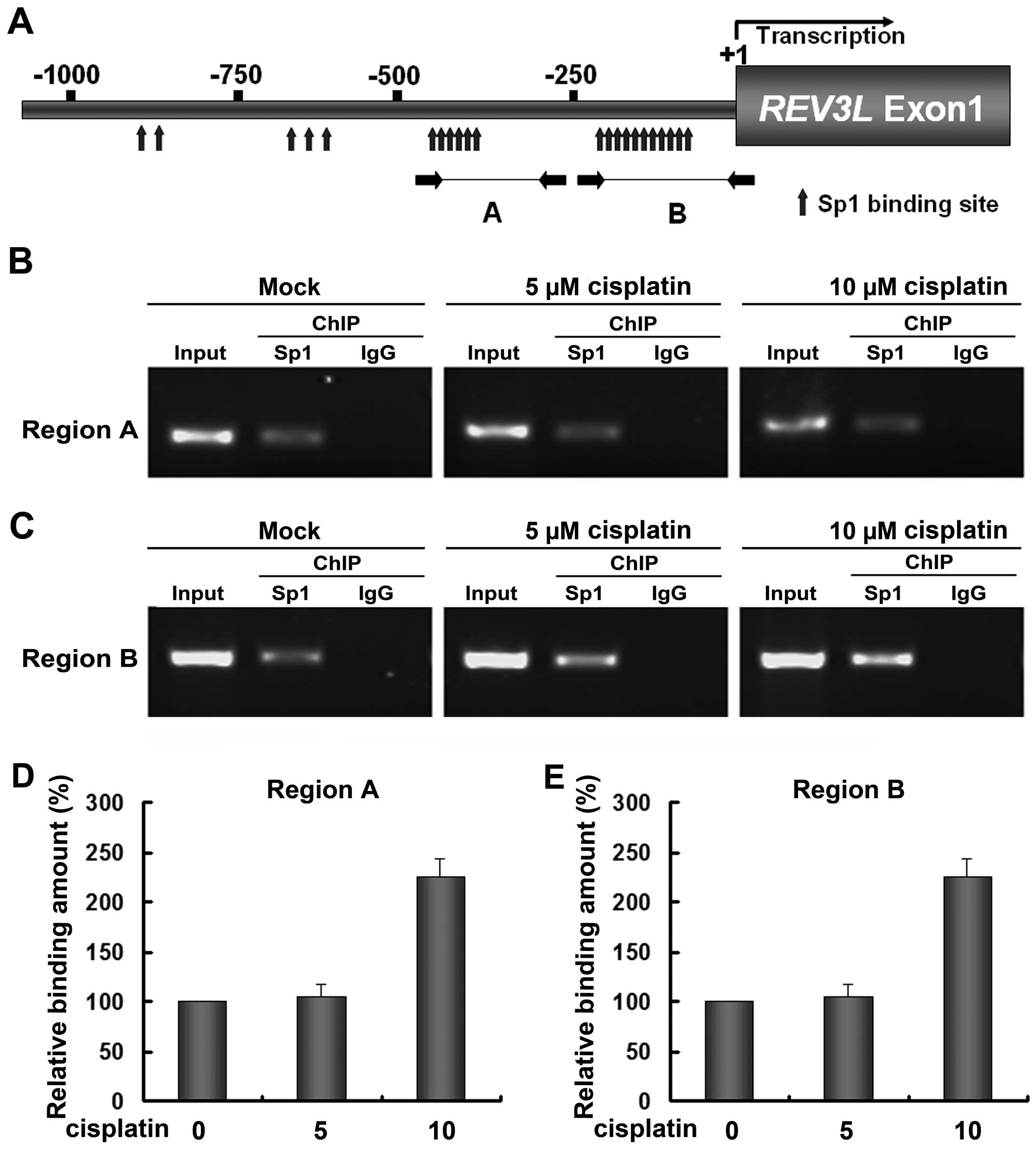
Bioinformatic tools predicted that the promoter of
REV3L harbors multiple binding sites of transcription factor Sp1
(Fig. 2A). Two dense Sp1 binding
regions were observed, the upstream one located between −450 and
−360 bp (Region A) and the downstream one located between −200 and
−75 bp (Region B, relative to the transcription start site). To
investigate whether the interaction between Sp1 and the
REV3L promoter conferred the cisplatin-induced increase of
REV3L, quantitative ChIP was performed. In H1299 cells, the
REV3L promoter was specifically precipitated with a Sp1
antibody but not with the control IgG in the two Sp1 dense regions
(Fig. 2B and C), indicating the
presence of Sp1 bound to the REV3L promoter in vivo. Results
also showed that 5 µM cisplatin did not change the relative
binding amount of Sp1 to the REV3L promoter, whereas 10
µM cisplatin significantly increased the amount of Sp1 bound
to the REV3L promoter in the two regions (Fig. 3B–E). These results also indicated
that changes in REV3L expression result in a corresponding
change in Sp1 binding to the REV3L promoter.
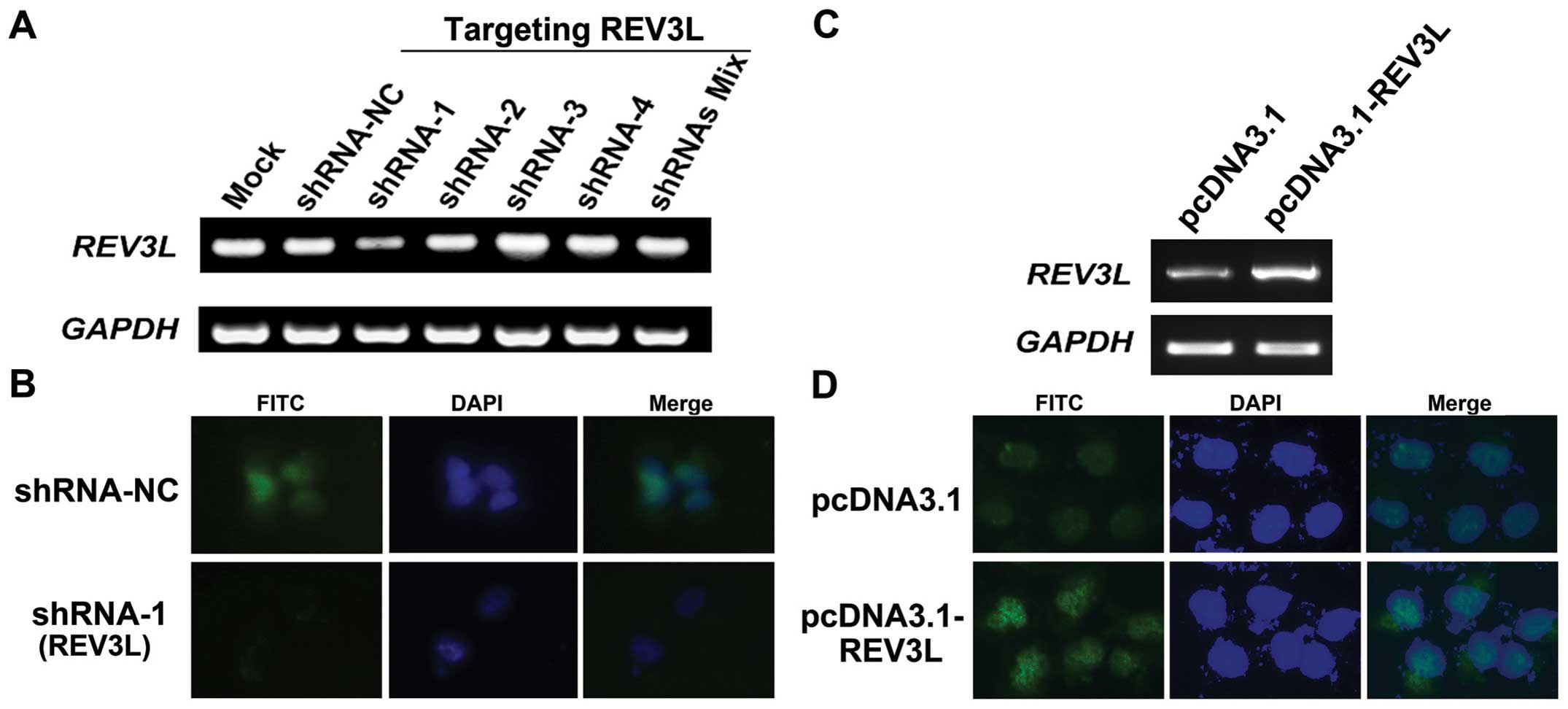
Establishment of H1299 cell lines with
overexpression and knockdown of REV3L
The function of REV3L in human lung cancer
H1299 cells. We genetically manipulated REV3L expression in
a cell line derived from H1299 was investigated. pcDNA3.1-REV3L was
stably transfected into H1299 cells to generate
REV3L-overexpressing cells. We also designed and constructed four
shRNA vectors targeting different locations of the REV3L
mRNA. REV3L cell lines stably transfected with the control,
overexpression or shRNA vectors were screened by G418. As shown in
Fig. 3A, compared with mock- or
shRNA-NC-transfected H1299 cells, transfection of REV3L targeting
shRNA-1 showed reduced REV3L transcripts (inhibition rate
71.4%, relative to shRNA-NC-transfected cells). Other shRNAs did
not show obvious silencing ability of REV3L. To confirm this
result, immunostaining was performed and the results demonstrated
that in shRNA-1-transfected cells, the nuclear staining of REV3L
was obviously reduced (Fig. 3B),
validating the RT-PCR results. In addition, REV3L expression was
increased in REV3L-overexpressing stable cells, compared to
the pcDNA3.1-transfected cells (Fig. 3C
and D).
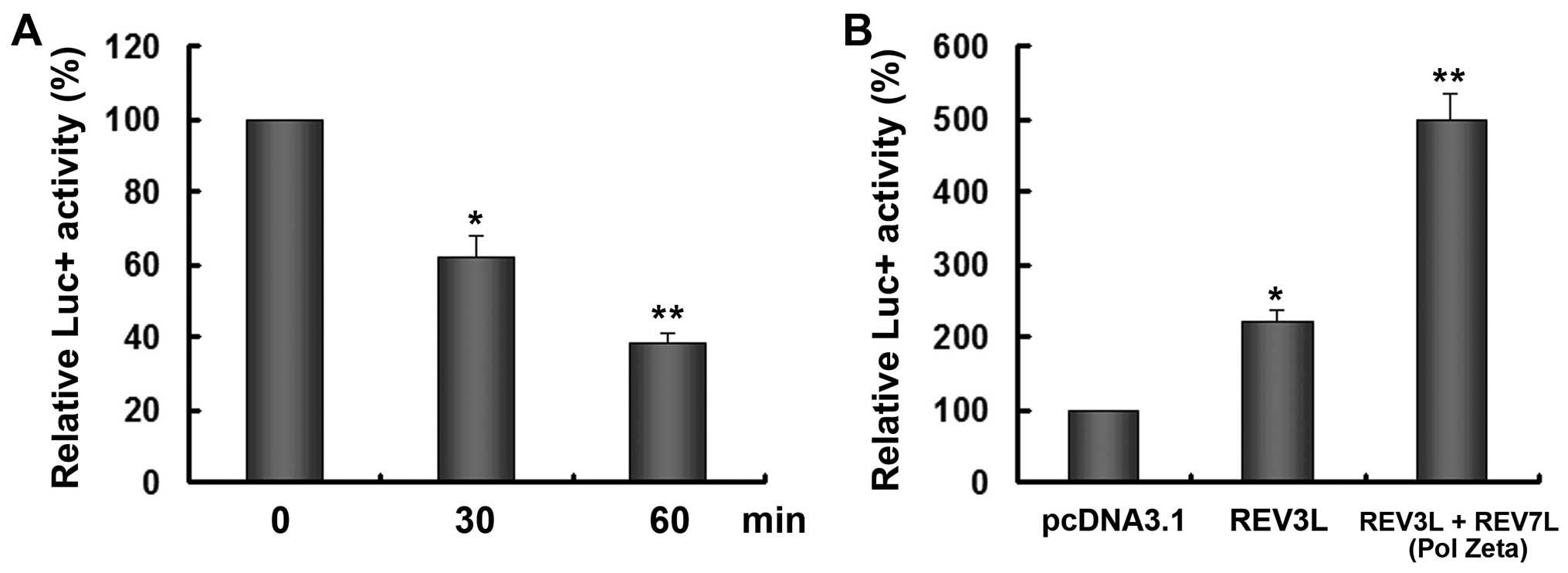
REV3L overexpression abolishes
cisplatin-induced DNA damage
DNA is thought to be the primary biological target
of cisplatin. The platinum atom of cisplatin forms covalent bonds
with the N7 position of purine bases to form 1,2- or
1,3-intrastrand crosslinks and a lower percentage of interstrand
crosslinks, both of which interfere with DNA transcription
(23). To investigate whether REV3L
affects cisplatin-treated DNA damage, the pGL3-promoter reporter
was treated with cisplatin and then transfected into H1299 cells.
The ability of cells in sweeping away cisplatin-induced adducts was
assessed by determining the ability of the cell to successfully
express the firefly luciferase from the extensively platinated
reporter by treatment with cisplatin before transfection. As shown
in Fig. 4A, the treatment of
cisplatin decreased the reporter activity in a time-dependent
manner possibly due to the crosslink of the plasmid DNA. After 30
min treatment of cisplatin, the reporter was used for subsequent
experiments. Co-transfection of the reporter with REV3L
overexpression vector significantly enhanced the reporter activity
by 2.23-fold. Moreover, the addition of REV3L and REV7L, which was
able to form the complete Pol ζ, significantly increased the
reporter expression by >5-fold.
REV3L knockdown sensitizes H1299 cells to
cisplatin
We investigated the effect of REV3L expression on
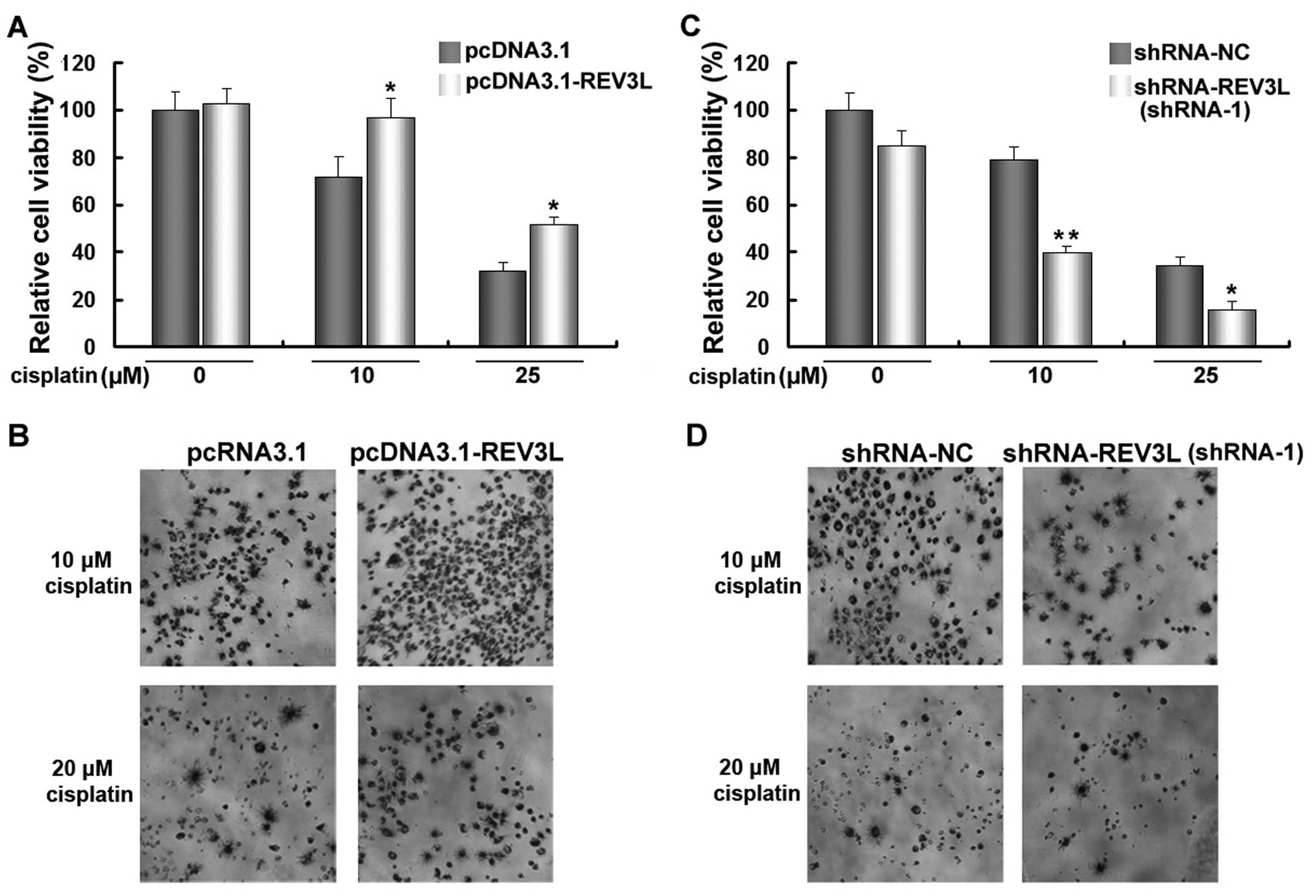
cell sensitivity to cisplatin treatment. As shown in Fig. 5A and C, transfection of
pcDNA3.1-REV3L or shRNA-REV3L did not cause an obvious change of
cell viability in H1299 cells. The cells were transfected with
pcDNA3.1-REV3L, shRNA-REV3L or control vectors for 24 h, and then
treated with 10 of 25 µM cisplatin for another 48 h. The
results revealed that a forced expression of REV3L significantly
increased cell viability (Fig. 5A and
B), while the knockdown of REV3L significantly decreased the
viability of H1299 cells treated with cisplatin (Fig. 5C and D), compared with cisplatin
treatment alone. These results demonstrated that silencing of REV3L
effectively enhanced the anticancer efficacy of cisplatin.
REV3L silencing promotes
cisplatin-induced apoptosis
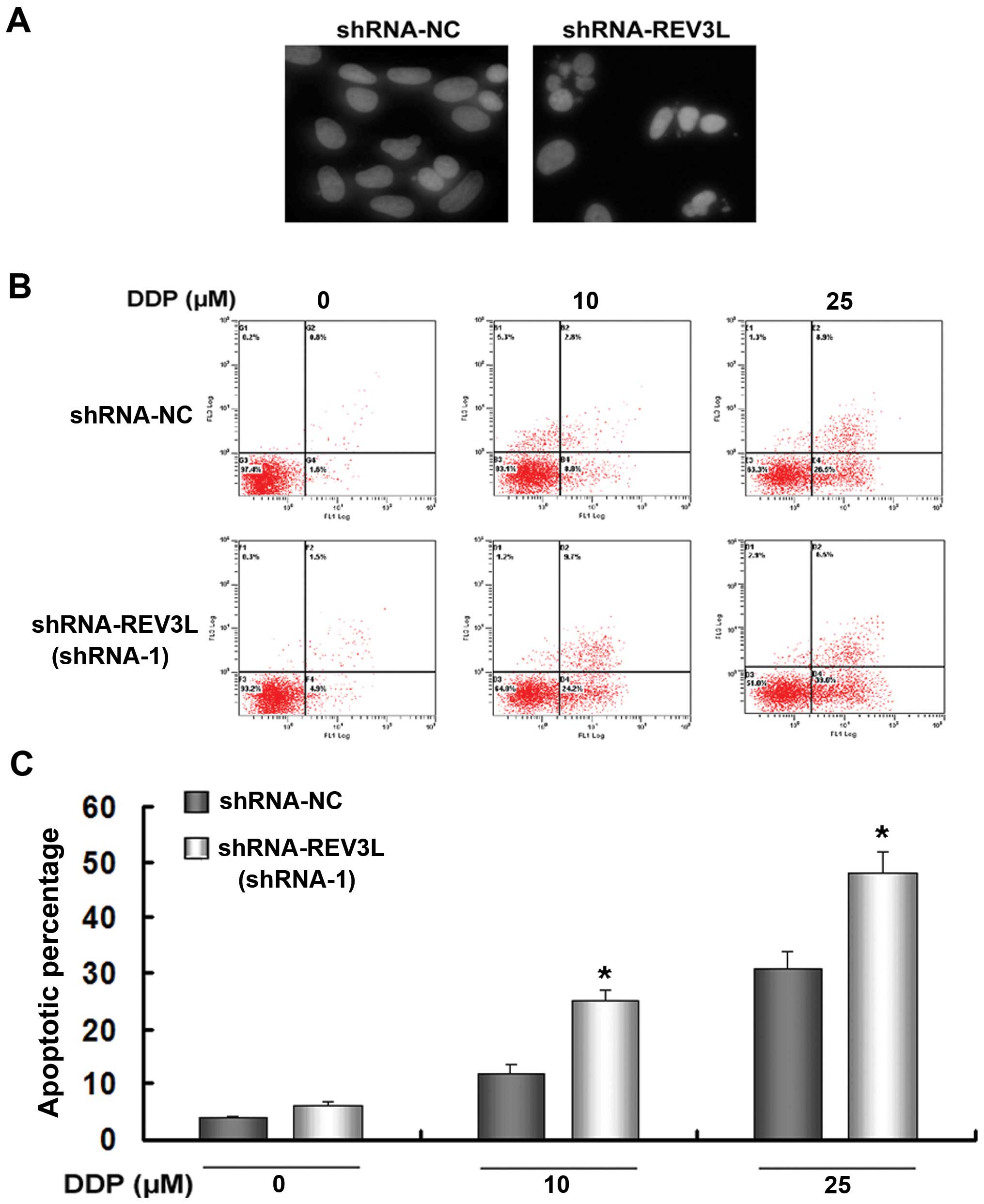
DAPI staining of nucleus was used to observe
morphological changes such as nuclear condensation and
fragmentation. The results showed that the nuclear condensation and
fragmentation of shRNA-REV3L cells were more pronounced than
shRNA-NC H1299 cells after 10 µM cisplatin exposure for 48 h
(Fig. 6A).
We investigated whether silencing of REV3L affected
cell apoptosis following treatment with cisplatin. As shown in
Fig. 6B and C, knockdown of REV3L
did not induce apoptosis (Annexin V+/PI− plus
Annexin V+/PI+ cells). However, silencing of
REV3L enhanced the response of H1299 cells to 10 or 25 µM
cisplatin (shRNA-NC + 10 µM cisplatin 12.41% vs. shRNA-REV3L
+ 10 µM cisplatin 25.37%, P<0.05; (shRNA-NC + 25
µM cisplatin 31.84% vs. shRNA-REV3L + 25 µM cisplatin
46.68%, P<0.05). Taken together, these results demonstrated that
REV3L inhibition enhances apoptotic cell death of H1299 cells in
response to cisplatin.
Discussion
Repair of cisplatin-induced DNA damage has focused
on the NER MMR and HR pathways (11). In a study conducted to explore the
contribution of DNA damage response pathways in tolerance to
cross-linking agents in vertebrates, a panel of gene-disrupted
clones from chicken DT40 cells was used to measure the
sensitivities of chicken DT40 cells to cross-linking agents,
including cisplatin, mitomycin C, and melphalan. It was found that
cells harboring defects in the TLS pathway, Fanconi anemia
complementation groups (FANC) or homologous recombination pathway
exhibited marked hypersensitivity to all the cross-linking agents,
whereas NER played only a minor role (24). In particular, cells deficient in
REV3 showed the highest sensitivity and markedly increased
chrosomal abberrations to cisplatin (24).
The role of low-fidelity TLS polymerases in
tumorigenesis remains controversial, because their existence is
beneficial for the survival of human cells, while it accumulates
mutations during DNA replication (25). Given the importance of TLS
participants, studies have attempted to increase the efficacy of
chemotherapy by inhibiting this pathway. Albertella et al
found that Pol η is migrated to nucleus in response to cisplatin
and its deficiency significantly decreased cell focus formation
after cisplatin treatment (26).
DNA polymerase η could replicate across intrastrand cross-link
between cisplatin and two adjacent G residues (27). Wu et al found that cisplatin
induced a concentration- and time-dependent increase in hREV3 mRNA
and suppression of REV3L by transfection of a vector-expressing
hREV3 antisense mRNA increased cisplatin sensitivity in human
fibroblast (28).
hREV1, another important member in translesional
replication and a Pol ζ interaction molecule, was also upregulated
in response to cisplatin in human ovarian carcinoma 2008 cells
(29). Inactivation of REV1 causes
it to become hypersensitive to a wide variety of DNA-damaging
agents including cisplatin in human ovarian cancer cells (29,30).
Similar results were obtained in colon cancer HCT-116 cells where
the introduction of a shRNA against REV3L reduced the mutagenic
bypass of cisplatin and cisplatin resistance. Comparatively,
inhibition of the DNA MMP pathway did not have a significant effect
on reporter expression, which emphasizes the importance of REV3L
(17). Suppression of the structual
unit of Pol ζ-REV7L (MAD2B) conferred hypersensitivity to a range
of DNA-damaging agents, especially DNA cross-linkers such as
cisplatin and γ-irradiation (31),
suggesting that each of the TLS enzymes may have specificity to the
chemotherapeutic drugs. We have previously reported that the
downregulation of REV3L sensitized glioma cells to cisplatin via
mitochondria-mediated apoptosis (32). Taken together, those results
indicate that TLS enzymes are activated in response to chemotherapy
as endogenous adaptive mechanisms and inhibition of these
polymerases contributes to the efficacy of cisplatin.
In this study, we also found that transcription
factor Sp1 is involved in cisplatin-induced REV3L overexpression.
In addition, increased Sp1 binding was found in the proximal region
of the REV3L promoter. To the best of our knowledge, this is
the first study to show the transcriptional control of REV3L
following cisplatin exposure. Sp1 is a ubiquitously expressed zinc
finger-containing DNA-binding protein that can activate or repress
gene transcription in response to various physiologic and
pathological stimuli (33). Sp1
binds to GC-rich recognition elements (GC-boxes) through its
C-terminal zinc finger motifs (34,35).
Sp1 has been reported to be activated in response to oxidative
stress and regulate neuronal survival in cortical neurons (36). DNA damage induces transient
elevation in its DNA binding activity and phosphorylation of Ser-56
and Ser-101 residues on Sp1 in an ATM-dependent manner (35,37).
In cancer cells, Sp1 is involved in the transcriptional control of
pro-apoptotic NOXA following cisplatin exposure (38). Our results identified a new
downstream target of Sp1 in response to cisplatin.
Taken together, we have demonstrated that cisplatin
induced the expression of REV3L by recruiting Sp1 to its promoter.
The knockdown of REV3L sensitized cisplatin efficacy in human lung
cancer H1299 cells. Similar results were obtained when the ability
of the cells to express luciferase from a platinated plasmid was
measured.
Acknowledgments
This study is supported by the National Natural
Science Foundation of China (81472920, 81472917, 31400720 and
81372433), Suzhou Administration of Science and Technology
(SYS201416) and the Priority Academic Program Development of
Jiangsu Higher Education Institutions (PAPD).
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Langer CJ, Mok T and Postmus PE: Targeted
agents in the third-/fourth-line treatment of patients with
advanced (stage III/IV) non-small cell lung cancer (NSCLC). Cancer
Treat Rev. 39:252–260. 2013. View Article : Google Scholar
|
|
3
|
de Boer RH, Arrieta Ó, Yang CH, Gottfried
M, Chan V, Raats J, de Marinis F, Abratt RP, Wolf J, Blackhall FH,
et al: Vandetanib plus pemetrexed for the second-line treatment of
advanced non-small-cell lung cancer: A randomized, double-blind
phase III trial. J Clin Oncol. 29:1067–1074. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Rourke N, Roqué I, Figuls M, Farré
Bernadó N and Macbeth F: Concurrent chemoradiotherapy in non-small
cell lung cancer. Cochrane Database Syst Rev.
6:CD0021402010.PubMed/NCBI
|
|
5
|
Barr MP, Gray SG, Hoffmann AC, Hilger RA,
Thomale J, O'Flaherty JD, Fennell DA, Richard D, O'Leary JJ and
O'Byrne KJ: Generation and characterisation of cisplatin-resistant
non-small cell lung cancer cell lines displaying a stem-like
signature. PLoS One. 8:e541932013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Einhorn LH: First-line chemotherapy for
non-small-cell lung cancer: Is there a superior regimen based on
histology? J Clin Oncol. 26:3485–3486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rossi A, Di Maio M, Chiodini P, Rudd RM,
Okamoto H, Skarlos DV, Früh M, Qian W, Tamura T, Samantas E, et al:
Carboplatin- or cisplatin-based chemotherapy in first-line
treatment of small-cell lung cancer: The COCIS meta-analysis of
individual patient data. J Clin Oncol. 30:1692–1698. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar
|
|
9
|
Wang D and Lippard SJ: Cellular processing
of platinum anticancer drugs. Nat Rev Drug Discov. 4:307–320. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Martin LP, Hamilton TC and Schilder RJ:
Platinum resistance: The role of DNA repair pathways. Clin Cancer
Res. 14:1291–1295. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Basu A and Krishnamurthy S: Cellular
responses to Cisplatin-induced DNA damage. J Nucleic Acids.
2010:2013672010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Friedberg EC, Lehmann AR and Fuchs RP:
Trading places: How do DNA polymerases switch during translesion
DNA synthesis? Mol Cell. 18:499–505. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gan GN, Wittschieben JP, Wittschieben BØ
and Wood RD: DNA polymerase zeta (pol zeta) in higher eukaryotes.
Cell Res. 18:174–183. 2008. View Article : Google Scholar
|
|
15
|
Okada T, Sonoda E, Yoshimura M, Kawano Y,
Saya H, Kohzaki M and Takeda S: Multiple roles of vertebrate REV
genes in DNA repair and recombination. Mol Cell Biol. 25:6103–6111.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shen X, Jun S, O'Neal LE, Sonoda E, Bemark
M, Sale JE and Li L: REV3 and REV1 play major roles in
recombination-independent repair of DNA interstrand cross-links
mediated by monoubiquitinated proliferating cell nuclear antigen
(PCNA). J Biol Chem. 281:13869–13872. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wittschieben JP, Reshmi SC, Gollin SM and
Wood RD: Loss of DNA polymerase zeta causes chromosomal instability
in mammalian cells. Cancer Res. 66:134–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan Q, Fang Y, Xu Y, Zhang K and Hu X:
Down-regulation of DNA polymerases kappa, eta, iota, and zeta in
human lung, stomach, and colorectal cancers. Cancer Lett.
217:139–147. 2005. View Article : Google Scholar
|
|
19
|
Zhang S, Chen H, Zhao X, Cao J, Tong J, Lu
J, Wu W, Shen H, Wei Q and Lu D: REV3L 3′UTR 460 T>C
polymorphism in microRNA target sites contributes to lung cancer
susceptibility. Oncogene. 32:242–250. 2013. View Article : Google Scholar
|
|
20
|
Lin X, Trang J, Okuda T and Howell SB: DNA
polymerase zeta accounts for the reduced cytotoxicity and enhanced
mutagenicity of cisplatin in human colon carcinoma cells that have
lost DNA mismatch repair. Clin Cancer Res. 12:563–568. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eastman A, Jennerwein MM and Nagel DL:
Characterization of bifunctional adducts produced in DNA by
transdiamminedichloroplatinum(II). Chem Biol Interact. 67:71–80.
1988. View Article : Google Scholar
|
|
22
|
Zhang S, Lu J, Zhao X, Wu W, Wang H, Lu J,
Wu Q, Chen X, Fan W, Chen H, et al: A variant in the CHEK2 promoter
at a methylation site relieves transcriptional repression and
confers reduced risk of lung cancer. Carcinogenesis. 31:1251–1258.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jamieson ER and Lippard SJ: Structure,
recognition, and processing of cisplatin-DNA adducts. Chem Rev.
99:2467–2498. 1999. View Article : Google Scholar
|
|
24
|
Nojima K, Hochegger H, Saberi A, Fukushima
T, Kikuchi K, Yoshimura M, Orelli BJ, Bishop DK, Hirano S, Ohzeki
M, et al: Multiple repair pathways mediate tolerance to
chemotherapeutic cross-linking agents in vertebrate cells. Cancer
Res. 65:11704–11711. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lange SS, Takata K and Wood RD: DNA
polymerases and cancer. Nat Rev Cancer. 11:96–110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Albertella MR, Green CM, Lehmann AR and
O'Connor MJ: A role for polymerase eta in the cellular tolerance to
cisplatin-induced damage. Cancer Res. 65:9799–9806. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alt A, Lammens K, Chiocchini C, Lammens A,
Pieck JC, Kuch D, Hopfner KP and Carell T: Bypass of DNA lesions
generated during anticancer treatment with cisplatin by DNA
polymerase eta. Science. 318:967–970. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu F, Lin X, Okuda T and Howell SB: DNA
polymerase zeta regulates cisplatin cytotoxicity, mutagenicity, and
the rate of development of cisplatin resistance. Cancer Res.
64:8029–8035. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Okuda T, Lin X, Trang J and Howell SB:
Suppression of hREV1 expression reduces the rate at which human
ovarian carcinoma cells acquire resistance to cisplatin. Mol
Pharmacol. 67:1852–1860. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin X, Okuda T, Trang J and Howell SB:
Human REV1 modulates the cytotoxicity and mutagenicity of cisplatin
in human ovarian carcinoma cells. Mol Pharmacol. 69:1748–1754.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cheung HW, Chun AC, Wang Q, Deng W, Hu L,
Guan XY, Nicholls JM, Ling MT, Chuan Wong Y, Tsao SW, et al:
Inactivation of human MAD2B in nasopharyngeal carcinoma cells leads
to chemosensitization to DNA-damaging agents. Cancer Res.
66:4357–4367. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang H, Zhang SY, Wang S, Lu J, Wu W, Weng
L, Chen D, Zhang Y, Lu Z, Yang J, et al: REV3L confers
chemoresistance to cisplatin in human gliomas: The potential of its
RNAi for synergistic therapy. Neurooncol. 11:790–802. 2009.
|
|
33
|
Tan NY and Khachigian LM: Sp1
phosphorylation and its regulation of gene transcription. Mol Cell
Biol. 29:2483–2488. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fojas de Borja P, Collins NK, Du P,
Azizkhan-Clifford J and Mudryj M: Cyclin A-CDK phosphorylates Sp1
and enhances Sp1-mediated transcription. EMBO J. 20:5737–5747.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Iwahori S, Yasui Y, Kudoh A, Sato Y,
Nakayama S, Murata T, Isomura H and Tsurumi T: Identification of
phosphorylation sites on transcription factor Sp1 in response to
DNA damage and its accumulation at damaged sites. Cell Signal.
20:1795–1803. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ryu H, Lee J, Zaman K, Kubilis J, Ferrante
RJ, Ross BD, Neve R and Ratan RR: Sp1 and Sp3 are oxidative
stress-inducible, antideath transcription factors in cortical
neurons. J Neurosci. 23:3597–3606. 2003.PubMed/NCBI
|
|
37
|
Meighan-Mantha RL, Riegel AT, Suy S,
Harris V, Wang FH, Lozano C, Whiteside TL and Kasid U: Ionizing
radiation stimulates octamer factor DNA binding activity in human
carcinoma cells. Mol Cell Biochem. 199:209–215. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Grande L, Bretones G, Rosa-Garrido M,
Garrido-Martin EM, Hernandez T, Fraile S, Botella L, de Alava E,
Vidal A, Garcia del Muro X, et al: Transcription factors Sp1 and
p73 control the expression of the proapoptotic protein NOXA in the
response of testicular embryonal carcinoma cells to cisplatin. J
Biol Chem. 287:26495–26505. 2012. View Article : Google Scholar : PubMed/NCBI
|