Introduction
Bladder cancer is the fifth most common cancer in
Western countries, responsible for more than 130,000 deaths
worldwide annually, and is the first leading cause of death among
urinary malignancies in China (1,2).
Cigarette smoking is estimated to cause ~50% of the bladder cancer
cases in males and 25% of the bladder cancers in females in the US.
At any point in time, 2.7 million people have a history of bladder
cancer (3).
Smoking, specific industrial chemicals, dietary
nitrates and arsenic represent the most important exogenous risk
factors for bladder cancer (4).
Cigarette smoke (CS) is the main risk factor for bladder cancer
(4,5). In a meta-analysis of 43 published
case-control and cohort studies, Zeegers et al concluded
that current cigarette smokers have an ~3-fold higher risk of
developing bladder cancer than non-smokers (6). There are more than 60 established
carcinogens, which occur in mainstream smoke, side-stream smoke,
and the particulate phase of cigarette smoking extract (CSE), such
as polycyclic aromatic hydrocarbons (PAHs), benzo[a]pyrene (BaP),
nitrosamines and aromatic amines (7). Although enormous progress has been
made in exploring how CS leads to bladder cancer development, the
molecular pathogenesis remains largely unknown.
Epithelial-mesenchymal transition (EMT) is a
multistep process in which epithelial cells lose their epithelial
characteristics and gain mesenchymal characteristics, such as
motility and invasive properties (8). Numerous in vitro and in
vivo studies suggest that EMT is associated with cancer cell
invasion and metastasis in various malignancies, including bladder
cancer. In addition to facilitating tumor invasion and metastasis,
EMT is also critically involved in the initiation of tumorigenesis
by promoting cell malignant transformation (9–12). It
has been reported that changes related to EMT are observed in the
very earliest stages of oral malignancy and become more severe as
lesions progress (13).
Nonetheless, the underlying mechanisms by which CS induces EMT are
poorly understood.
The mitogen-activated protein kinases (MAPKs)
include four major subfamilies: extracellular regulated protein
kinases 1 and 2 (ERK1/2), the Jun N-terminal kinases (JNKs), p38
and ERK5 (14,15). ERK5, also termed big MAPK1 (BMK1),
is the least studied member of the MAPK family. MAPKs are
responsible for the phosphorylation and activation of jun and fos
proteins. The MEK5/ERK5 pathway is implicated in important cellular
processes, including gene expression, proliferation, apoptosis,
angiogenesis, cell motility and differentiation (16–19).
While some studies have suggested the functions of ERK5 in cancer
oncogenesis, its role in EMT regulation has not been well
explored.
To date, no studies have been carried out to examine
the action of ERK5 in CS-induced urocystic EMT. The purpose of the
present study was to investigate CS-induced EMT in normal human
urothelial cells. Furthermore, we discussed the potential role of
the ERK5 pathway in CS-induced EMT, which may contribute to a
better understanding of carcinogenesis related to CS, including
bladder cancer.
Materials and methods
Materials
An SV-40 immortalized human urothelial cell line
(SV-HUC-1) was purchased from the Chinese Academy of Typical
Culture Collection Cell Bank. F12K medium was purchased from Gibco
(New York, NY, USA). Fetal bovine serum (FBS) was obtained from PAA
Laboratories (Pasching, Austria).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
was purchased from Sigma-Aldrich. Phosphorylated ERK5,
phosphorylated c-jun (p-c-jun), phosphorylated c-fos (p-c-fos),
snail, slug, E-cadherin, N-cadherin and vimentin were obtained from
Cell Signaling Technology (Beverly, MA, USA). The antibody for ZO-1
was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
The GAPDH antibody was obtained from Biogot Technology (Nanjing,
China). Primers for E-cadherin, ZO-1, N-cadherin, vimentin and
GAPDH were synthesized by Invitrogen (Carlsbad, CA, USA). XMD8–92
was purchased from Beyotime (Shanghai, China). Sources of other
materials are noted accordingly in the study.
Cell culture and treatment
SV-HUC-1 cells were cultured under an atmosphere of
5% Co2 at 37°C in F12K medium containing antibiotics
(100 U/ml penicillin and 100 μg/ml streptomycin). The cells
were seeded in 10 cm2 flasks. The medium was changed
every other day until cells reached 80–90% confluence, and then
treated with various concentrations of CS extract or with XMD8–92
(5 μM).
Preparation of CSE
CSE was freshly prepared for each experiment by
combusting one commercial cigarette according to the reported
method (20,21). Commercial cigarettes (Hongtashan
filter-tipped cigarettes made in Yunnan, China; each contain 12 mg
tar and 1.1 mg nicotine) were smoked. By application of a vacuum,
mainstream smoke was drawn through 10 ml of pre-warmed (37°C)
FBS-free F12K medium supplemented with penicillin and streptomycin
at a rate of 5 min/cigarette. The obtained solution was referred to
as having 100% strength. Then the CSE stock solution was filtered
through a 0.22-μm pore size filter and diluted to the
desired concentration with treatment medium. The resulting CSE was
applied to epithelial cell cultures within 30 min of preparation.
The control solution was prepared using the same protocol, except
that the cigarettes were unlit.
Cell toxicity assay
SV-HUC-1 cells were seeded in 96-well plates at a
plating density of 2×103 cells/well in 200 μl of
medium. Then the cells were exposed to various concentrations of
CSE prepared as previously outlined for 5 days, and the cell
viability was determined by MTT assay. For the present study, media
containing various concentrations of CSE were changed every day.
Five days later, MTT stock solution was added to each well to
solubilize the formazan crystals, and plates were incubated for an
additional 4 h at 37°C. Afterwards, MTT solution in the medium was
removed and the crystals were solubilized in dimethylsulfoxide
(DMSO). Absorbance was measured at 490 nm using a microplate
reader. All measurements were performed in triplicate.
Transwell assay
The invasion assays were performed in a 24-well
Boyden chamber with an 8-μm pore size polycarbonate membrane
(Millipore, Billerica, MA, USA) coated with Matrigel to form a
matrix barrier. A total of 100 μl of serum-free medium
(containing 1×104 cells) was added to the upper
compartment of the chamber, whereas the lower compartment was
filled with 800 μl of F12K supplemented with 10% FBS. After
incubation at 37°C for 48 h, the SV-HUC-1 cells remaining inside
the upper chamber were removed with cotton swabs. The cells on the
lower surface of the membrane were stained with 0.1% crystal violet
after fixation with methanol and then imaged under a light
microscope. Subsequently, the cells were bleached with glacial
acetic acid and the absorbance of the eluant was measured at 570
nm.
Western blot analysis
SV-HUC-1 cells were harvested, washed with ice-cold
phosphate-buffered saline (PBS) and lysed in RIPA buffer (Thermo
Scientific, USA). Concentrations of the precipitated proteins in
the cell lysates were measured with BCA protein assay (Pierce,
Rockford, IL, USA). Afterwards, the proteins were diluted to equal
concentrations, boiled for 5 min and separated by 7.5–10% SDS-PAGE,
transferred onto polyvinylidene difluoride membranes (Millipore).
After blocking with 5% milk, the membranes were blocked and
incubated with the indicated primary antibodies and secondary
antibodies. The blots were subsequently developed using an enhanced
chemiluminescence detection kit (Amersham Biosciences, USA) and
exposed to film. GAPDH served as the loading control.
Quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
Total RNA was isolated by RNAiso Plus according to
the manufacturer's instructions. (Takara, Japan). Then total RNA
was transcribed into cDNA using AMV reverse transcriptase (Takara)
following the manufacturer's protocol. qRT-PCR was performed using
the Power SYBR-Green Master Mix (Takara) and an ABI 7300 Real-Time
PCR Detection System (Applied Biosystems). All of the primers were
synthesized by Invitrogen. Respectively, normalization was achieved
by dividing the expression level of mRNA by its respective GAPDH
expression level. Fold-change in the expression of each gene was
calculated by a comparative threshold cycle (Ct) method using the
formula 2−ΔΔCt. The primers used were: E-cadherin
forward, 5′-TCGACACCCGA TTCAAAGTGG-3′ and reverse,
5′-TTCCAGAAACGGAGGC CTGAT-3′; vimentin forward, 5′-CCTTGACATTGAGATT
GCCA-3′ and reverse, 5′-GTATCAACCAGAGGGAGTGA-3′; ZO-1 forward,
5′-GCAGCCACAACCAATTCATAG-3′ and reverse,
5′-GCAGACGATGTTCATAGTTTC-3′; N-cadherin forward,
5′-ATCAAGTGCCATTAGCCAAG-3′ and reverse, 5′-CTGAGCAGTGAATGTTGTCA-3′;
GAPDH forward, 5′-GCTGCCCAACGCACCGAATA-3′ and reverse, 5′-GAGT
CAACGGATTTGGTCGT-3′.
Statistical analysis
Statistical analyses were performed with SPSS 16.0.
All data are expressed as mean ± standard deviation. One-way ANOVA
was used for comparison of statistical differences among multiple
groups, followed by the LSD significant difference test. In case of
comparison between two groups, an unpaired Student's t-test was
used. A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
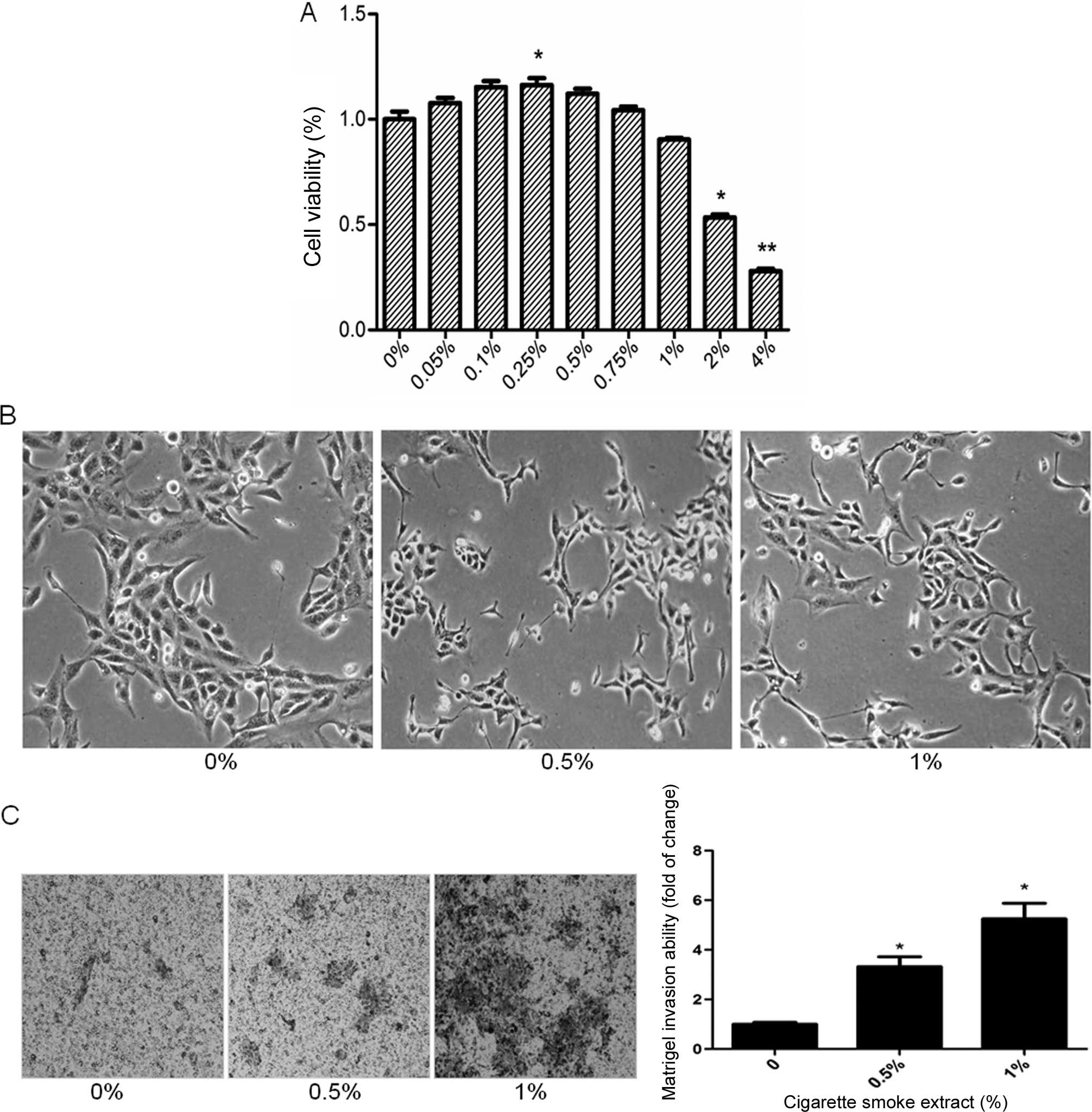
CS induces EMT in SV-HUC-1 cells
CS is the most important risk factor for bladder
cancer, and CS-induced EMT is critically involved in CS-associated
malignant transformation. To study the cell viability of the
SV-HUC-1 cells after treatment with CSE, the cells were incubated
with CSE (0, 0.05, 0.1, 0.25, 0.5, 0.75, 1, 2 and 4%) for 5 days
and examined by MTT assay. The results showed that the cell
viability decreased below 80% when the cells were exposed to 2% or
higher CSE concentrations, which proved to be toxic to the SV-HUC-1
cells (Fig. 1A). Therefore, 1% CSE
was selected as the maximum concentration for the following
experiments.
The process of EMT is manifested by alterations in
cell morphology and invasive capacity, as well as expression of
epithelial and mesenchymal markers. Treatment of the SV-HUC-1 cells
with CSE for 5 days resulted in significant morphological change
from a urothelial oblate-shape to a spindle-like mesenchymal form
(Fig. 1B). Additionally, the cells
became dispersive and some SV-HUC-1 cells even generated a
tail-like change after treatment with CSE. To further examine the
effect of CSE on EMT, Transwell assays were carried out to analyze
SV-HUC-1 cell invasive capacities in response to CSE. Invasion of
the cells through reconstituted Matrigel matrices was enhanced by
CSE (Fig. 1C). To determine whether
molecular alterations of EMT occurred in the CSE-treated cells, the
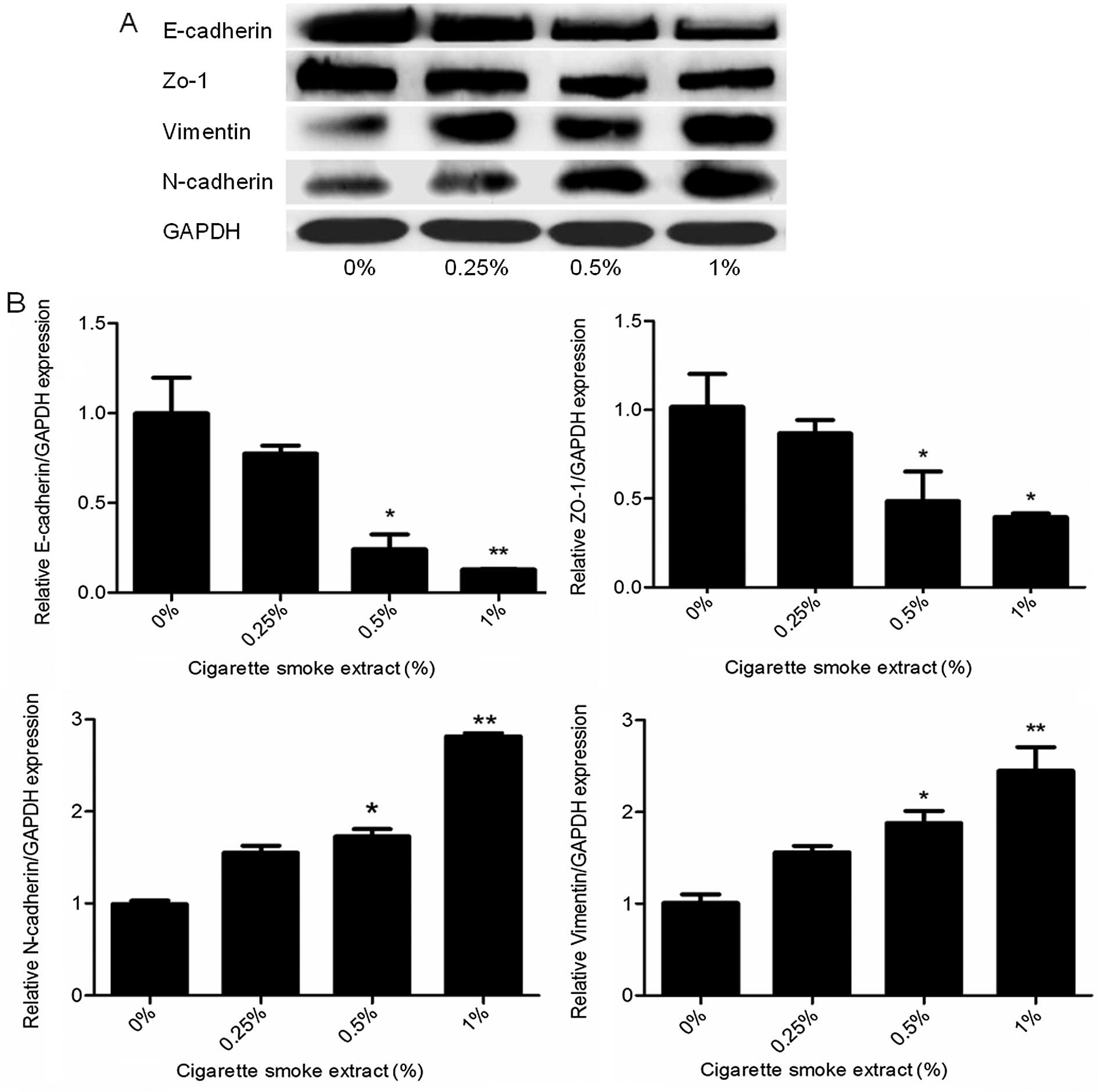
expression levels of EMT markers were determined. Exposure of the
SV-HUC-1 cells to CSE resulted in decreased protein expression of
the epithelial markers E-cadherin and ZO-1. In contrast, the
protein levels of mesenchymal markers vimentin and N-cadherin were
increased, as shown by western blot analyses (Fig. 2A). Moreover, qRT-PCR analyses
revealed similar changes in the mRNA levels of epithelial and
mesenchymal markers in the SV-HUC-1 cells exposed to CSE (Fig. 2B). Moreover, the higher the
concentration of CSE, the more obvious was the observed change.
Collectively, data from the morphological, invasive and molecular
changes demonstrated that CS exposure induced EMT in human
urothelial cells in a dose-dependent manner.
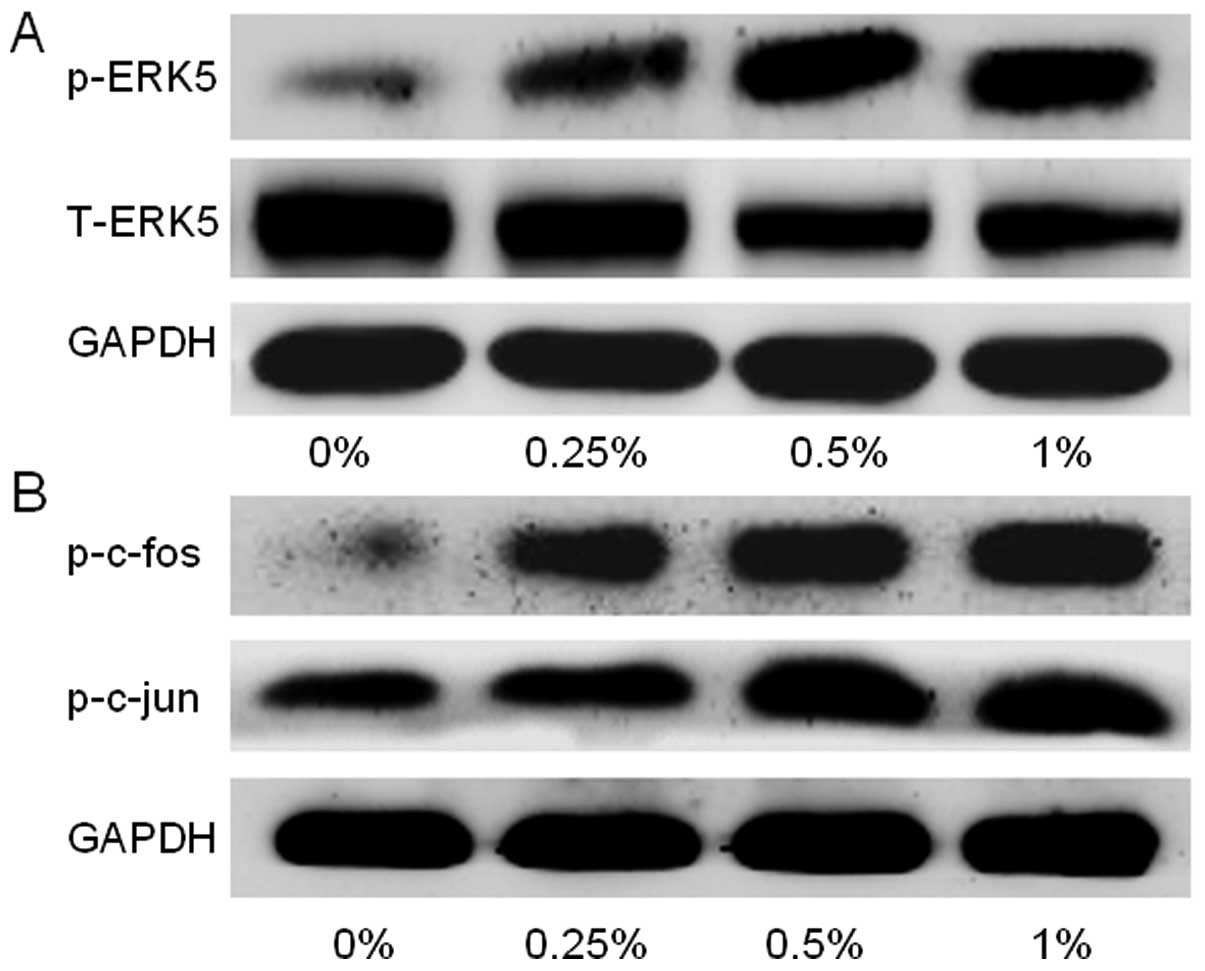
CS-induced urocystic EMT is associated
with ERK5 activation
As the lesser studied member of the MAPK family,
ERK5 is implicated in cancer oncogenesis. The function of ERK5 in
EMT regulation has not been well explored, and several lines of
evidence suggest that a differential regulatory role of ERK5 in EMT
exists. To determine whether CS-elicited urothelial EMT is
associated with a change in ERK5 activation, the level of
phosphorylated ERK5, an indicator of ERK5 activation was measured.
Exposure of the SV-HUC-1 cells to CSE for 5 days dose-dependently
activated the level of phosphorylated ERK5 (p-ERK5) and
simultaneously restrained the level of total ERK5, suggesting a
stimulative effect of CS on ERK5 activity (Fig. 3A). Meanwhile, CS increased the
activation of AP-1 protein in the SV-HUC-1 cells, as indicated by
elevated levels of p-c-jun and p-c-fos (Fig. 3B).
Inhibition of ERK5 attenuates CS-induced
EMT in SV-HUC-1 cells
Since the above results revealed that CS-induced EMT
was associated with ERK5 activation in SV-HUC-1 cells, we next
explored the role of ERK5 in urocystic EMT regulation. SV-HUC-1
cells were treated for 5 days with 5 μM XMD8–92, a highly
specific ERK5 inhibitor which suppresses ERK5 autophosphorylation
and does not inhibit ERK1/2 activity (22,23).
As expected, XMD8–92 downregulated p-ERK5 and levels of p-c-jun and
p-c-fos in the SV-HUC-1 cells (Fig.
4A). XMD8–92 treatment reversed the mensenchymal-like
morphological change in the cells (Fig.
4B). Moreover, XMD8–92 also restrained the invasive capacity of
the SV-HUC-1 cells, as determined by Transwell assays (Fig. 4C). Furthermore, inhibition of ERK5
by XMD8–92 resulted in upregulation of the protein and mRNA levels
of the epithelial markers E-cadherin and ZO-1, as well as
downregulation of the mesenchymal markers vimentin and N-cadherin
(Fig. 4D and E). Therefore, these
results suggest that ERK5 activity plays an important role in
CSE-induced EMT in SV-HUC-1 cells.
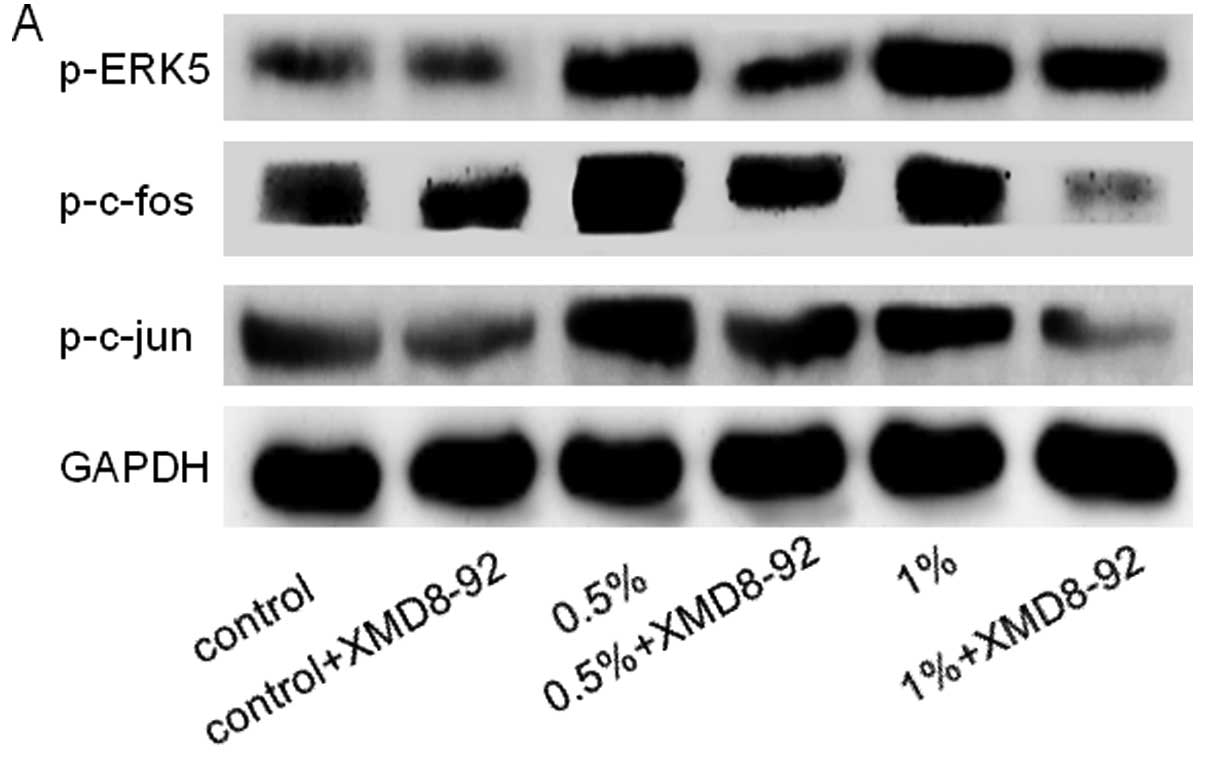 | Figure 4Inhibition of ERK5 attenuates
CS-induced EMT in SV-HUC-1 cells. (A) ERK5 inhibitor XMD8–92
suppressed ERK5 activation in the SV-HUC-1 cells. SV-HUC-1 cells
were treated with various concentrations of CSE together with a
highly specific ERK5 inhibitor (XMD8–92) for 5 days, and western
blot analyses were performed for the measurements of p-ERK5,
p-c-fos and p-c-Jun. The control group represented 0% CSE + DMSO.
XMD8–92 suppressed the activation of p-ERK5, p-c-fos and p-c-Jun
induced by CS exposure. Moreover, XMD8–92 had little effect on the
control group. (B) XMD8–92 attenuated the morphological change of
SV-HUC-1 cells. Compared with the CSE group, the SV-HUC-1 cells
became collective and full after treatement with XMD8–92. (C)
XMD8–92 decreased the invasive capacity of the SV-HUC-1 cells, as
determined by the invasion assay. Results showed that the enhanced
invasion capacity of the SV-HUC-1 cells triggered by 1% CSE was
reversed by XMD8–92. (D) Treatment of ERK5 inhibitor XMD8–92 along
with 0.5% CSE or 1% CSE resulted in enhanced protein levels of
E-cadherin and ZO-1, and decreased protein levels of vimentin and
N-cadherin. (E) Treatment with the ERK5 inhibitor XMD8–92 along
with 0.5% CSE or 1% CSE increased the expression of E-cadherin and
ZO-1 mRNAs, and decreased vimentin and N-cadherin mRNAs. All the
data are expressed as mean ± SD. *P<0.05 and
**P<0.01, CSE group compared with the control group.
#p<0.05, XMD8–92 group compared with the srespective
CSE group. CS, cigarette smoke; EMT, epithelial-mesenchymal
transition; DMSO, dimethylsulfoxide. |
Discussion
Bladder cancer is one of the leading causes of
cancer-related death worldwide. The relationship between the
occurrence of urocystic cancer and cigarette smoke (CS) has been
established (24,25). CS is the primary cause of bladder
cancer, which promotes the initiation and progression of bladder
tumorigenesis. The underlying molecular mechanisms by which CS
causes bladder cancer development remain to be established. In the
present study, we revealed that CS induced EMT in SV-HUC-1 cells.
Most importantly, we demonstrated for the first time that ERK5
positively regulated CS-mediated EMT, as evidenced by the findings
that CS promoted ERK5 activation and that CS-triggered EMT
phenotypic alteration was reversed by ERK5 inhibition. These
findings suggest the important role of ERK5 activity in
CS-associated EMT and provide critical information concerning the
molecular mechanisms of CS-related bladder tumorigenesis as well as
the search for potential targets of bladder cancer
intervention.
Characterized by changes in cell morphology,
migration and invasion capacity, as well as the expression profile
of epithelial and mesenchymal markers, EMT is a crucial process in
cancer development. Evidence has revealed that exposure of cells to
carcinogens induces EMT during transformation and tumor formation
(10,11,26),
suggesting the important role of EMT in the initiation of
tumorigenesis by promoting cell malignant transformation. Likewise,
Cavallaro et al put forward the cadherin switch theory. That
is, epithelial cells undergo mesenchymal cell phenotype conversion,
accompanied by loss of E-cadherin and gain of N-cadherin, which
strongly supports the view that malignant tumors develop from
benign tumors (27). E-cadherin
plays an essential role in epithelial cell-to-cell interactions
since it mediates the connections between adjacent epithelial cells
and maintains the phenotype and apical-base polarity of epithelial
cells (28). It has been documented
that CS promotes EMT, resulting in loss of cellular polarity,
down-regulation of epithelial cadherin, loss of cell-cell adhesion
and increased mobility of epithelial cells (29). In agreement with previous studies,
we showed in the present study that exposure to CS induced EMT in
SV-HUC-1 cells, as manifested by morphological change from an
epithelial to a mesenchymal form, increased invasive capacity, as
well as alterations in the expression of EMT markers, including
decreased E-cadherin and increased N-cadherin, which powerfully
verify the cadherin switch theory. Taken together, our data proved
that CS triggers EMT in vitro settings.
To date, no studies have been carried out to
investigate the function of ERK5 in CS-induced EMT or in bladder
cancer cell invasion and metastasis. In the present study, we
showed that CS-induced EMT was associated with activation of ERK5
in vitro. To determine the role of ERK5 in urocystic EMT
regulation, the inhibitory effect of ERK5 on CS was mimicked with a
highly specific ERK5 inhibitor. Inhibition of ERK5 reversed the
mesenchymal-like morphological changes triggered by CS. Moreover,
the enhanced invasive capacities as well as alterations in EMT
marker expression were attenuated by inhibition of ERK5.
Collectively, these data clearly indicated that ERK5 positively
regulated CS-induced urocystic EMT.
In regards to proto-oncogenic signaling, the MAPK
pathways are generally thought to promote cancer development; while
differential biological functions exist for individual MAPK
pathways. Among the MAPK family, ERK5 is twice the size of the
other members. The N-terminal half of ERK5 contains the kinase
domain which is similar to that of ERK1/2 and has the TEY
activation motif. Unlike other MAPKs, the C-terminal region of ERK5
contains a long non-catalytic domain which has a unique function.
Upon activation, ERK5 phosphorylates and activates downstream
target molecules, including transcription factors such as members
of the AP-1 proteins (30).
Activation of ERK5 also results in autophos-phorylation of the
C-terminus, which alone has the ability to increase transcriptional
activity (31,32). Therefore, ERK5 differs from other
MAPKs in possessing transcriptional activation activity. It is able
to regulate the transcription of downstream molecules in two ways,
i.e., through either the phosphorylation or the enhancement of the
transcription activity of target molecules (16–19).
Nevertheless, only a small number of molecules have been identified
as target genes of ERK5. Notably, it has been found that ERK5
transcriptionally regulates gene expression in a tissue-specific
manner (33). In skeletal muscle
cells, activation of ERK5 significantly inhibits TNF-α-mediated
NF-κB activity (34). In lung
microvascular endothelial cells, activation of ERK5 suppresses
HIF1α expression and inhibits angiogenesis (35). In the present study, we showed that
CS-mediated ERK5 activation induced the activation of AP-1 proteins
c-fos and c-Jun. Correspondingly, inhibition of ERK5 decreased
c-fos and c-Jun activation, suggesting the positive modulation of
ERK5 on the AP-1 pathway. It is noteworthy that the precise
mechanism by which ERK5 regulates AP-1 activation is unknown at the
present time, and future studies are warranted to define the
underlying mechanism of urocystic ERK5 regulation on AP-1
protein.
As previously mentioned, XMD8–92 is a highly
specific inhibitor of ERK5. XMD8–92 can be competitively combined
with ATP which effectively inhibits ERK5 autophosphorylation and
does not affect the activity of ERK1/2. In animal experiments,
treating lung xenograft tumors of mouse with XMD8–92, effectively
inhibits tumor growth. This inhibition is accomplished through
inhibition of ERK5 phosphorylation and inhibition of the PML gene
(35). Notably, research has also
shown that XMD8–92 significantly inhibits the activity of CDKI,
thus as to promote the proliferation of human breast cancer cells
(36). At present, there is no
study on the application of XMD8–92 in bladder urothelial cells.
Thus, the present study is the first to confirm that ERK5
positively regulates EMT in urothelial cells triggered by CS. Based
on the important role of the ERK5 signaling pathway in the
development of cancer, a new choice has been provided for tumor
treatment. Nevertheless, how to block the ERK5 signaling pathway in
a reasonable manner requires further exploration.
In summary, exposure to CS induced EMT in normal
urothelial cells and activated the ERK5 pathway and the
transcription factor AP-1, while the ERK5 inhibitor inhibited AP-1
activity and afterwards restrained the EMT triggered by CS. The
present study effectively elucidated the underlying molecular
mechanisms involved during the occurrence of bladder tumors related
to CS. Furthermore, inhibition of the EKR5 pathway may be a
therapeutic target for the treatment of early stage bladder
cancer.
Acknowledgments
The present study was supported by grants from the
National Natural Science foundation of China (nos. 81373005,
81072330 and 81202194), and by the Priority Academic Program
Development of Jiangsu Higher Education Institutions (PAPD).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ploeg M, Aben KK and Kiemeney LA: The
present and future burden of urinary bladder cancer in the world.
World J Urol. 27:289–293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Volanis D, Kadiyska T, Galanis A, Delakas
D, Logotheti S and Zoumpourlis V: Environmental factors and genetic
susceptibility promote urinary bladder cancer. Toxicol Lett.
193:131–137. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Boffetta P: Tobacco smoking and risk of
bladder cancer. Scand J Urol Nephrol Suppl. 42(s218): S45–S54.
2008. View Article : Google Scholar
|
|
6
|
Zeegers MP, Tan FE, Dorant E and van Den
Brandt PA: The impact of characteristics of cigarette smoking on
urinary tract cancer risk: A meta-analysis of epidemiologic
studies. Cancer. 89:630–639. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hecht SS: Tobacco carcinogens, their
biomarkers and tobacco-induced cancer. Nat Rev Cancer. 3:733–744.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun JL, Chen DL, Hu ZQ, Xu YZ, Fang HS,
Wang XY, Kan L and Wang SY: Arsenite promotes intestinal tumor cell
proliferation and invasion by stimulating epithelial-to-mesenchymal
transition. Cancer Biol Ther. 15:1312–1319. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu W, Ji J, Xu Y, Liu Y, Shi L, Liu Y, Lu
X, Zhao Y, Luo F, Wang B, et al: MicroRNA-191, by promoting the EMT
and increasing CSC-like properties, is involved in neoplastic and
metastatic properties of transformed human bronchial epithelial
cells. Mol Carcinog. 54(Suppl 1): E148–E161. 2015. View Article : Google Scholar
|
|
11
|
Tellez CS, Juri DE, Do K, Bernauer AM,
Thomas CL, Damiani LA, Tessema M, Leng S and Belinsky SA: EMT and
stem cell-like properties associated with miR-205 and miR-200
epigenetic silencing are early manifestations during
carcinogen-induced transformation of human lung epithelial cells.
Cancer Res. 71:3087–3097. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu Y, Luo F, Xu Y, Wang B, Zhao Y, Xu W,
Shi L, Lu X and Liu Q: Epithelial-mesenchymal transition and cancer
stem cells, mediated by a long non-coding RNA, HoTAIR, are involved
in cell malignant transformation induced by cigarette smoke
extract. Toxicol Appl Pharmacol. 282:9–19. 2015. View Article : Google Scholar
|
|
13
|
Danielsson K, Wahlin YB, Coates PJ and
Nylander K: Increased expression of Smad proteins, and in
particular Smad3, in oral lichen planus compared to normal oral
mucosa. J oral Pathol Med. 39:639–644. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sangrar W, Shi C, Mullins G, LeBrun D,
Ingalls B and Greer PA: Amplified Ras-MAPK signal states correlate
with accelerated EGFR internalization, cytostasis and delayed HER2
tumor onset in Fer-deficient model systems. Oncogene.
27:3402014.
|
|
16
|
Drew BA, Burow ME and Beckman BS:
MEK5/ERK5 pathway: The first fifteen years. Biochim Biophys Acta.
1825:37–48. 2012.
|
|
17
|
Nishimoto S and Nishida E: MAPK
signalling: ERK5 versus ERK1/2. EMBO Rep. 7:782–786. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hayashi M, Fearns C, Eliceiri B, Yang Y
and Lee JD: Big mitogen-activated protein kinase 1/extracellular
signal-regulated kinase 5 signaling pathway is essential for
tumor-associated angiogenesis. Cancer Res. 65:7699–7706.
2005.PubMed/NCBI
|
|
19
|
Wang X and Tournier C: Regulation of
cellular functions by the ERK5 signalling pathway. Cell Signal.
18:753–760. 2006. View Article : Google Scholar
|
|
20
|
Gál K, Cseh A, Szalay B, Rusai K, Vannay
A, Lukácsovits J, Heemann U, Szabó AJ, Losonczy G, Tamási L, et al:
Effect of cigarette smoke and dexamethasone on Hsp72 system of
alveolar epithelial cells. Cell Stress Chaperones. 16:369–378.
2011. View Article : Google Scholar :
|
|
21
|
Tian D, Zhu M, Chen WS, Li JS, Wu RL and
Wang X: Role of glycogen synthase kinase 3 in squamous
differentiation induced by cigarette smoke in porcine
tracheobronchial epithelial cells. Food Chem Toxicol. 44:1590–1596.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang Q, Deng X, Lu B, Cameron M, Fearns C,
Patricelli MP, Yates JR III, Gray NS and Lee JD: Pharmacological
inhibition of BMK1 suppresses tumor growth through promyelocytic
leukemia protein. Cancer Cell. 14:258–267. 2010. View Article : Google Scholar
|
|
23
|
Wang X, Pesakhov S, Weng A, Kafka M, Gocek
E, Nguyen M, Harrison JS, Danilenko M and Studzinski GP: ERK 5/MAPK
pathway has a major role in 1α,25-(OH)2 vitamin
D3-induced terminal differentiation of myeloid leukemia cells. J
Steroid Biochem Mol Biol. 144(Pt A): 223–227. 2014. View Article : Google Scholar
|
|
24
|
Freedman ND, Silverman DT, Hollenbeck AR,
Schatzkin A and Abnet CC: Association between smoking and risk of
bladder cancer among men and women. JAMA. 306:737–745. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alguacil J, Kogevinas M, Silverman DT,
Malats N, Real FX, García-Closas M, Tardón A, Rivas M, Torà M,
García-Closas R, et al: Urinary pH, cigarette smoking and bladder
cancer risk. Carcinogenesis. 32:843–847. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao Y, Xu Y, Li Y, Xu W, Luo F, Wang B,
Pang Y, Xiang Q, Zhou J, Wang X, et al: NF-κB-mediated inflammation
leading to EMT via miR-200c is involved in cell transformation
induced by cigarette smoke extract. Toxicol Sci. 135:265–276. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cavallaro U, Schaffhauser B and
Christofori G: Cadherins and the tumour progression: Is it all in a
switch? Cancer Lett. 176:123–128. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rangel MC, Karasawa H, Castro NP, Nagaoka
T, Salomon DS and Bianco C: Role of Cripto-1 during
epithelial-to-mesen-chymal transition in development and cancer. Am
J Pathol. 180:2188–2200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Veljkovic E, Jiricny J, Menigatti M,
Rehrauer H and Han W: Chronic exposure to cigarette smoke
condensate in vitro induces epithelial to mesenchymal
transition-like changes in human bronchial epithelial cells,
BEAS-2B. Toxicol In Vitro. 25:446–453. 2011. View Article : Google Scholar
|
|
30
|
Kasler HG, Victoria J, Duramad O and
Winoto A: ERK5 is a novel type of mitogen-activated protein kinase
containing a transcriptional activation domain. Mol Cell Biol.
20:8382–8389. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nagathihalli NS, Massion PP, Gonzalez AL,
Lu P and Datta PK: Smoking induces epithelial-to-mesenchymal
transition in non-small cell lung cancer through HDAC-mediated
downregulation of E-cadherin. Mol Cancer Ther. 11:2362–2372. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nithianandarajah-Jones GN, Wilm B,
Goldring CE, Müller J and Cross MJ: ERK5: Structure, regulation and
function. Cell Signal. 24:2187–2196. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sohn SJ, Li D, Lee LK and Winoto A:
Transcriptional regulation of tissue-specific genes by the ERK5
mitogen-activated protein kinase. Mol Cell Biol. 25:8553–8566.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Woo CH, Massett MP, Shishido T, Itoh S,
Ding B, McClain C, Che W, Vulapalli SR, Yan C and Abe J: ERK5
activation inhibits inflammatory responses via peroxisome
proliferator-activated receptor delta (PPARdelta) stimulation. J
Biol Chem. 281:32164–32174. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pi X, Garin G, Xie L, Zheng Q, Wei H, Abe
J, Yan C and Berk BC: BMK1/ERK5 is a novel regulator of
angiogenesis by destabilizing hypoxia inducible factor 1alpha. Circ
Res. 96:1145–1151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Perez-Madrigal D, Finegan KG, Paramo B and
Tournier C: The extracellular-regulated protein kinase 5 (ERK5)
promotes cell proliferation through the down-regulation of
inhibitors of cyclin dependent protein kinases (CDKs). Cell Signal.
24:2360–2368. 2012. View Article : Google Scholar : PubMed/NCBI
|