Introduction
The treatment options for breast cancer are based in
part upon the classification of the tumor molecular subtype. Of the
three molecular subtypes of invasive breast cancer, triple-negative
(TN) is the most aggressive and is associated with the worst
prognosis for patients (1–3). Moreover, patients with this form of
breast cancer are typically not candidates for hormonal therapy or
human epidermal growth factor 2 (HER2)-targeted therapy (4). Another molecular classification is
estrogen receptor-positive (ER+) breast cancer. This
molecular subtype of breast cancer is associated with greater
treatment options than TN, such as treatment with the
anti-estrogen, tamoxifen (5,6).
Furthermore, patients diagnosed with ER+ breast cancer
have an improved prognosis as compared to patients with TN breast
cancer (7). This includes greater
response rates to chemotherapeutic treatments, higher median
survival rates, and increased lifespan (8,9).
However, a complicating factor in the treatment of patients with
ER+ breast cancer, as well as those with TN breast
cancer, is the phenomenon that certain TN and ER+ breast
tumors are resistant to chemotherapeutic treatments. While there
are several mechanisms in which such breast tumors fail to respond
to chemotherapeutic treatments, one in particular is the ability to
evade the apoptotic cell death induced by chemotherapeutic agents
via inhibition of pro-apoptotic mediators or activation of
anti-apoptotic proteins (10–12).
Therefore, improved therapies for TN and ER+ breast
cancers, and breast tumors resistant to current therapies, are
paramount in order to successfully treat these devastating
conditions and improve the prognosis for breast cancer patients in
the future.
An emerging target for the improved treatment of
breast cancer is the transient receptor potential, melastatin-2
(TRPM2) channel. TRPM2 is a non-specific cation channel activated
by oxidative stress (13). Once
activated, it gates sodium, potassium and calcium ions into the
cell (14). Among these cations,
calcium has been studied the most extensively in regards to cell
death. Calcium influx due to TRPM2 activation has been shown to
facilitate apoptosis and caspase-independent cell death pathways
(15–17). Due to this positive regulation of
cell death pathways by TRPM2 in noncancerous cells, TRPM2 is thus
targeted for pharmacologic inhibition for the protective effects
such agents provide in these cells (16,18,19).
However, in cancer cells, TRPM2 appears to have a unique role. Two
TRPM2 mRNA transcripts, one antisense transcript and one truncated
TRPM2 transcript were shown to be increased in metastatic melanoma
cell lines (20). TRPM2 isoforms
were shown to be overexpressed in several cancers, including
melanoma, breast, and lung cancer (20,21).
In prostate and breast cancer cells, TRPM2 appears to play a
protective role. Inhibition or RNAi silencing of TRPM2 in prostate
cancer cells led to decreased proliferation, while equivalent
treatments failed to decrease proliferation in noncancerous
prostate cells (22). Our previous
study demonstrated that TRPM2 inhibition or RNAi silencing caused
decreased proliferation and increased levels of DNA damage in
breast adenocarcinoma cells, yet no significant effects were noted
in non-cancerous mammary epithelial cells (23). Furthermore, a recent study
demonstrated that RNAi knockdown of TRPM2 led to decreased growth
of human xenograft neuroblastoma tumors in athymic nude mice, which
thus demonstrated that TRPM2 modulates tumor growth in
neuroblastoma (21). Thus, there is
increasing evidence which demonstrates that inhibition of TRPM2
provides chemotherapeutic effects in cancer cells, with little or
no harmful effects in non-cancerous cells. TRPM2 therefore appears
to be a unique target in multiple cancers, where its pharmacologic
inhibition can potentially provide an innovative strategy to
selectively eradicate the tumors associated with those types of
cancers.
However, in breast cancer cells, a comprehensive
understanding of the chemotherapeutic effects elicited by TRPM2
inhibition is not completely known. Furthermore, since TRPM2
exacerbates cell death in normal cells (24) but has a novel protective role in
breast cancer cells (23), a
complete understanding of the cell death mechanisms initiated after
TRPM2 inhibition in breast cancer cells is not known. In the
present study, we investigated the chemotherapeutic effects
produced in breast adenocarcinoma cells via TRPM2 inhibition or
RNAi silencing, with a particular focus on cell death pathways, DNA
damage levels, and the ability of TRPM2 inhibition to enhance the
cytotoxicity of currently used chemotherapeutic agents-of-choice in
different molecular subtypes of breast cancer. The present study
found that TRPM2 inhibition enhanced the cytotoxicity of
chemotherapeutic agents currently utilized to treat TN and
ER+ breast cancer. Also, TRPM2 inhibition induced
alternative pathways of cell death in these breast cancer cells.
Since these results have not been previously reported in breast
cancer cells, we thus report important findings which further
identify the targeting of TRPM2 as a potential strategy to improve
the chemotherapeutic treatment of breast cancer patients.
Materials and methods
Chemicals
N-(p-amylcinnamoyl)anthranilic acid
(ACA), maintained as a 50 mM stock solution in dimethylsulfoxide
(DMSO), 2-aminoethoxydiphenyl borate (2-APB; 75 mM stock solution
in DMSO), aristolochic acid (75 mM stock solution in DMSO) and
3-methyladenine (3-MA; 50 mM stock solution in DMSO) were purchased
from Sigma (St. Louis, MO, USA).
N-Methyl-N'-nitro-N-nitrosoguanidine (MNNG; 0.5 M stock solution in
DMSO) was purchased from AccuStandard (New Haven, CT, USA).
Doxorubicin hydrochloride and tamoxifen citrate were purchased from
Thermo Scientific (Waltham, MA, USA). Q-VD-OPh was purchased from
R&D Systems (Minneapolis, MN, USA) and maintained as a 50 mM
stock solution in DMSO. Propidium iodide (PI) (10 mg/ml solution)
was purchased from Thermo Scientific. ApoScreen Annexin
V-fluorescein isothiocyanate (FITC) was purchased from Southern
Biotech (Birmingham, AL, USA).
Cell lines and cell culture reagents
MCF-10A (human mammary epithelial), MCF-7 (human
breast adenocarci-noma), and MDA-MB-231 (human breast
adenocarcinoma) cell lines were purchased from the American Type
Culture Collection (ATCC; Manassas, VA, USA). The human mammary
epithelial cell (HMEC) line was purchased from Lonza (Walkersville,
MD, USA). Dulbecco's modified Eagle's medium (DMEM) and fetal
bovine serum (FBS; US origin, certified) were purchased from
HyClone (Logan, UT, USA). Mammary epithelial growth medium (MEGM),
which is specialized growth medium for HMEC and MCF-10A cells, was
purchased from Lonza. Trypsin-EDTA (0.05%/0.53 mM),
penicillin-streptomycin solution (10,000 IU/ml penicillin, 10,000
μg/ml streptomycin), and 200 mM L-glutamine solution were
purchased from Corning (Manassas, VA, USA).
Other reagents
Opti-MEM reduced serum medium and Lipofectamine 2000
reagent were purchased from Invitrogen (Carlsbad, CA, USA).
Protease inhibitor cocktail tablets (Complete Mini EDTA-free) were
purchased from Roche (Mannheim, Germany). Primary antibodies
utilized were poly-clonal rabbit anti-human TRPM2 antibody (cat.
#A300-414A; Bethyl Laboratories, Montgomery, TX, USA), polyclonal
rabbit anti-human β-actin (cat. #600-401-886), polyclonal rabbit
anti-human apoptosis-inducing factor (AIF) (cat. #200-401-985)
(both from Rockland Immunochemicals, Limerick, PA, USA) and
monoclonal mouse anti-human poly(ADP-ribose) glycohydrolase (PARG)
clone D8B10 (cat. #MABS61; Millipore). Two secondary antibodies,
horseradish peroxidase (HRP)-conjugated goat anti-rabbit and
HRP-conjugated rabbit anti-mouse were purchased from Jackson
ImmunoResearch (West Grove, PA, USA). The CometAssay kit, which
includes alkaline lysis solution, LMAgarose, 2-well CometSlides and
SYBR-Green was purchased from Trevigen (Gaithersburg, MD, USA).
Cell culture
MDA-MB-231 and MCF-7 cells were grown and maintained
in DMEM supplemented with 10% FBS and 2 mM L-glutamine.
Non-cancerous human mammary cells (MCF-10A and HMEC) were cultured
in MEGM specialty medium which contains mammary epithelial cell
basal medium (MEBM), 5 μg/ml bovine pituitary extract, 0.01
μg/ml human epidermal growth factor, 0.5 μg/ml
hydrocortisone, GA-1000 (60 μg/ml gentamicin and 0.03
μg/ml amphotericin B) and 5 μg/ml insulin. All
cultures were incubated at 37°C in 5% CO2 until
treatments and analyses. Every two days in culture, cells were
washed once with phosphate-buffered saline pH 7.2 (PBS) and
cultured in fresh growth medium.
RNA interference
The silencing of TRPM2 by RNAi was performed as
previously reported using small-interfering RNA (siRNA) specific to
nucleotides 4574–4594 in human T R PM2
(5′-AUAGAUCAGGAACUCCGUCUC-3′) (22). This RNA oligo was purchased as
duplexed RNA (Integrated DNA Technologies, Coralville, IA, USA),
resuspended in RNase-free water at a final concentration of 40
μM and stored at -20°C. The silencing of AIF by RNAi was
performed as previously described using sense
(5′-CUUGUUCCAGCGAUGGCAUtt-3′) and antisense
(5′-AUGCCAUCGCUGGAACAAGtt-3′) RNA oligos that target nucleotides
151–171 in human AIF (25). The
silencing of PARG by RNAi was performed as previously described
using RNA oligos (5′-AAATGGGACTTTACAGCTTTG-3′) that target
nucleotides 1960-1980 in human PARG (26). Oligos 5′-AGACAGAAGACAGAUAGGCtt-3′
(sense) and 5′-GCCUAUCUGUCUUCUGUCUtt-3′ (antisense) were used for
all negative controls (25).
Anti-AIF and anti-PARG RNA oligos were purchased as duplexed RNA
from Sigma-Aldrich, resuspended in RNase-free H2O at a
final concentration of 20 μM and stored at -20°C.
For RNAi transfections, cells were plated in 24-well
plates containing 0.5 ml medium/well without antibiotics. On the
day of transfection, cells were ~50–70% confluent. For RNAi
transfection, duplex siRNA was added to 50 μl Opti-MEM
medium and 1 μl Lipofectamine 2000 was added to 50 μl
Opti-MEM. The two solutions were gently mixed and incubated at room
temperature for 20 min. Final concentrations of siRNA were 100 nM
for TRPM2, 20 nM for AIF and 40 nM for PARG. The mixtures were
added dropwise to each well and cells were cultured for an
additional 48 h.
Whole cell lysate extraction
Transfected cells were harvested by trypsinization,
centrifuged at 112 × g for 5 min at 4°C, and washed once with 0.2
ml ice-cold PBS. The pellet was resuspended in 0.2 ml lysis buffer
containing 25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM
EGTA, 1% NP-40 and protease inhibitors (Complete Mini EDTA-free
tablets). Suspensions were incubated for 30 min on ice and vortexed
every 10 min. The cell lysates were cleared by centrifugation at
16,000 × g for 10 min. The protein concentration for all sample
lysates was then obtained using the Pierce BCA protein assay kit
(Thermo Scientific). After BCA quantification, sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer
(50 mM Tris-HCl pH 6.8, 1% SDS, 2.5% glycerol, 0.005% bromophenol
blue and 100 mM dithiothreitol) were added to the supernatants.
Samples were then heated for 2 min at 95°C in a digital dry bath
incubator (Labnet International, Edison, NJ, USA).
Cell death assay
Thirty minutes after the cells were pretreated with
TRPM2 inhibitors or cell death inhibitors, or 48 h after cells were
transfected with siRNA, the cells were treated with doxorubicin,
MNNG or tamoxifen at the indicated doses and exposures.
Approximately 16–20 h after treatment, the cells were harvested,
washed with PBS and resuspended in Annexin V binding buffer (10 mM
HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) to
~1×106 cells/ml. For each 100 μl of cell
suspension, 4 μl of ApoScreen Annexin V-FITC-conjugated
solution (Southern Biotech) and 1 μl of 100 μg/ml PI
solution were added. The cells were then incubated at room
temperature for 15 min and 400 μl Annexin V binding buffer
was added. Cell death was then measured by quantifying the
percentage of cells that exhibited Annexin V-FITC and/or PI
fluorescence using a flow cytometer (BD Accuri, Ann Arbor, MI,
USA). Total cell death was quantified by adding the percentage of
cells detected in the upper left (PI), upper right (PI + Annexin
V-FITC), and lower right (Annexin V-FITC) quadrants in the FACS dot
plots. For pretreatment with cell death inhibitors, the cells were
treated with 5 mM 3-MA or 40 μM Q-VD-OPh 30 min prior to
chemotherapeutic treatments. Treatment with 3-MA or Q-VD-OPh
continued during all treatments and until FACS analyses were
performed.
Immunoblotting
Approximately 40 μg of total protein from
each lysate was separated on a 7.5% SDS-PAGE gel. The proteins were
transferred to 0.45 μm nitrocellulose (Protran BA85 85; GE
Healthcare, Pittsburgh, PA, USA) by semi-dry transfer at 25 V for 1
h using a Trans-blot SD apparatus (Bio-Rad, Hercules, CA, USA).
Membranes were blocked with PBS containing 0.05% Tween-20 (PBST)
and 5% milk at room temperature for 1 h, and then incubated with
primary antibodies (1:1,000 anti-TRPM2, 1:2,000 β-actin, 1:2,000
anti-AIF, or 1:2,000 anti-PARG) in PBST + 5% milk overnight at 4°C
with shaking. Membranes were then washed with PBST three times and
incubated with HRP-conjugated goat anti-rabbit (1:5,000) or
HRP-conjugated rabbit anti-mouse antibody (1:5,000) for 1 h. The
membranes were washed as before and chemiluminescence was initiated
using the SuperSignal West Pico Chemiluminescent Substrate or
SuperSignal West Femto Chemiluminescent Substrate (Thermo Fisher
Pierce). Chemiluminescent detection was then accomplished on a
ChemiDoc XRS gel imaging system (Bio-Rad).
Comet assays
Comet assays were performed as previously described
using the CometAssay ES system and the manufacturer's protocol for
alkaline electrophoresis (27).
Briefly, MCF-7 cells in 24-well tissue culture plates were
incubated overnight in 0.5 ml growth medium, and then treated the
next day with 2 μM doxorubicin with or without a 30-min
pretreatment with 100 μM 2-APB. After collection by
trypsinization 24 h later, a cell concentration of 1×105
cells/ml was combined with low melting point agarose at a ratio of
1:10 (v/v). Approximately 50 μl of the agarose/cell mix was
transferred onto a CometSlide and placed in the dark at 4°C for 30
min. The slide containing the solidified gel was then placed in
lysis solution at 4°C overnight. The next day, the slide was
removed and then placed in alkaline unwinding solution for 20 min
at room temperature. Pre-chilled alkaline electrophoresis solution
was added to the CometAssay ES electrophoresis unit and the slide
was placed into the chamber, where electrophoresis was performed at
18 V for 30 min. When complete, the slide was washed twice in water
and once in 70% ethanol for 5 min each. The slide was dried at 37°C
for 15 min and then stored at room temperature with a desiccant
until ready for staining. The CometSlide was stained for 30 min
using 100 μl of SYBR-Green solution at room temperature.
Afterwards, the CometSlide was dried and then imaged using a Zeiss
Axio Observer Z1 inverted fluorescence microscope (Thornwood, NY,
USA) with Hamamatsu ORCA-ER digital camera and AxioVision software.
The values for 'Tail moment', a standard comet assay value that
represents DNA damage and calculated as the product of the tail
length and the fraction of total DNA in the tail (28,29),
were then calculated from the images using CometScore software
(Tritek Corporation, Sumerduck, VA, USA). A minimum of 200 cells
for each treatment group were scored for quantification of tail
moment.
Statistical analyses
All error bars for calculated cell death and comet
assay quantifications represent the standard error of the mean
(SEM). Statistical analyses were accomplished by one-way analysis
of variance (ANOVA) followed by the Tukey's and unpaired Student's
t-tests. Statistical significance was defined as p<0.05.
Results
TRPM2 inhibition enhances cytotoxicity in
MDA-MB-231 breast adenocarcinoma cells treated with
chemotherapeutic agents
As we previously demonstrated that inhibition of
TRPM2 led to decreased proliferation in breast adenocarcinoma cells
(23), we next investigated the
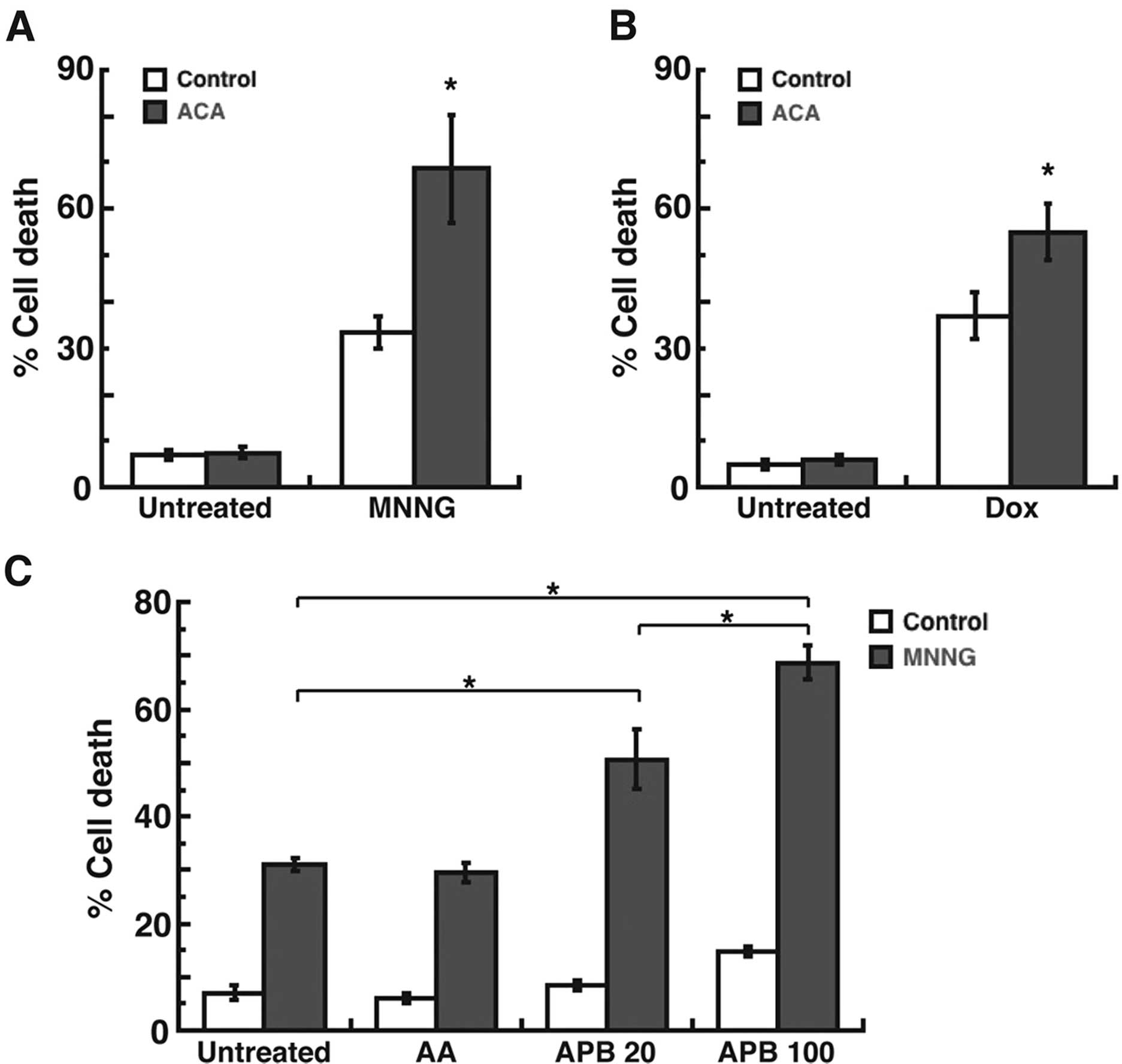
ability of TRPM2 inhibition to induce cell death. Treatment with
the DNA methylating agent, N-methyl-N′-nitro-N-nitrosoguanidine
(MNNG) (30), led to a significant
level of cell death (35%) in the MDA-MB-231 breast adenocarcinoma
cells (Fig. 1A). However,
pretreatment with the TRPM2 inhibitor,
N-(p-amylcinnamoyl) anthranilic acid (ACA) (31), enhanced the level of cell death to
68% in response to MNNG. Similar results were observed after
treatment with doxorubicin, where the level of cell death caused by
doxorubicin (38%) was significantly increased due to pretreatment
with ACA (56%) (Fig. 1B). Since ACA
was also reported to inhibit phospholipase A2 (32), we next utilized the phospholipase
A2 inhibitor, aristolochic acid (33) in our studies. The results
demonstrated that pretreatment of MDA-MB-231 breast adenocarcinoma
cells with aristolochic acid, followed by treatment with MNNG, led
to a cell death level of 29% (Fig.
1C). This level of cell death was not calculated to be
significantly different than the level of cell death induced by
MNNG alone (32%) (Fig. 1C). We also
used an additional TRPM2 inhibitor, 2-aminoethoxydiphenyl borate
(2-APB) (34), to further
demonstrate the effects of TRPM2 inhibition on cell death in the
MDA-MB-231 breast adenocarcinoma cells. The results demonstrated
that pretreatment of these cells with 2-APB led to a dose-dependent
increase in cell death after MNNG treatment, where 20 μM
2-APB caused 48% cell death and 100 μM caused 68% cell death
in the MDA-MB-231 cells (Fig. 1C).
Thus, the results of these studies indicated that inhibition of
TRPM2 caused increased levels of cell death in the MDA-MB-231
breast adenocarcinoma cells after chemotherapeutic treatments.
Moreover, this effect was most likely not mediated through
phospholipase A2 inhibition. These results therefore
demonstrated improved efficacy in the induction of cytotoxicity in
breast cancer cells due to TRPM2 inhibition.
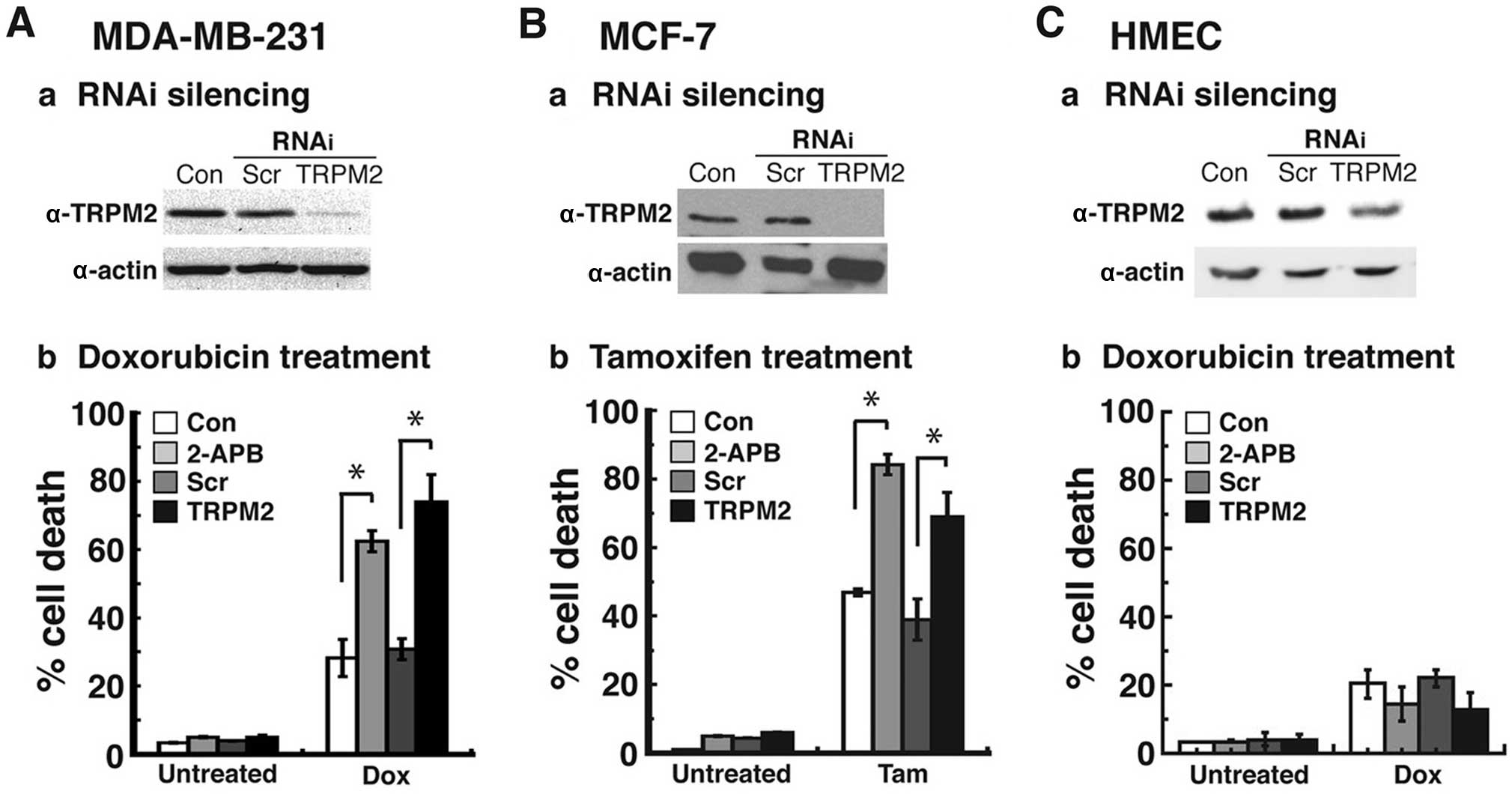
TRPM2 RNAi silencing enhances
cytotoxicity in triple-negative and estrogen receptor-positive
breast adenocarcinoma cells treated with chemotherapeutic
agents
To demonstrate the specificity of chemotherapeutic
effects due to decreased TRPM2 function, we utilized RNA
interference to knock down the expression levels of TRPM2. We
successfully decreased TRPM2 protein levels in the MDA-MB-231
breast adenocarcinoma cells by RNAi silencing (Fig. 2A–a). Treatment of this
triple-negative (TN) breast cancer cell line with doxorubicin led
to a 71% level of cell death (Fig.
2A–b). This level of cell death was similar to the level
observed after 2-APB treatment in these cells (62%) (Fig. 2A–b). Furthermore, it was a
significant increase from the level of cell death that resulted
from doxorubicin treatment in the cells transfected with negative
control scrambled (non-silencing) siRNA (30%).
We next investigated the effects of RNAi silencing
of TRPM2 in the MCF-7 breast adenocarcinoma cell line, which is an
estrogen receptor-positive breast cancer cell line (Fig. 2B). After successful RNAi silencing
of TRPM2 in these cells (Fig.
2B–a), treatment with tamoxifen led to a 66% level of cell
death, which was lower than the amount of cell death observed after
2-APB treatment (83%), yet significantly higher than the cell death
exhibited by cells transfected with scrambled siRNA and treated
with tamoxifen (36%) (Fig. 2B–b).
Finally, in order to determine the effects of RNAi silencing in
normal, non-cancerous breast cells, we successfully decreased TRPM2
protein levels in human mammary epithelial cells (HMECs) (Fig. 2C–a). Treatment of TRPM2-silenced
HMECs with doxorubicin led to a 16% level of cell death, which was
comparable to the 18% cell death observed in these cells after
2-APB treatment (Fig. 2C–b). These
cell death levels were not significantly higher than the levels of
cell death observed in the negative control groups (no pretreatment
with a TRPM2 inhibitor or transfection with scrambled siRNA
oligos). Moreover, the cell death levels in the TRPM2-silenced
HMECs were significantly lower than the cell death levels in the
TRPM2-silenced MDA-MB-231 and MCF-7 cells. Taken together, the
results demonstrated that RNAi knockdown of TRPM2 levels caused
increased levels of cell death in the MDA-MB-231 and MCF-7 cells
after chemotherapeutic treatments. Furthermore, TRPM2 inhibition or
RNAi silencing did not cause increased cell death in the
non-cancerous HMECs. These studies therefore showed that decreased
TRPM2 function led to enhanced cytotoxicity after chemotherapy in a
TN breast cancer cell line and an ER+ breast cancer cell line, but
not in a non-cancerous breast cell line.
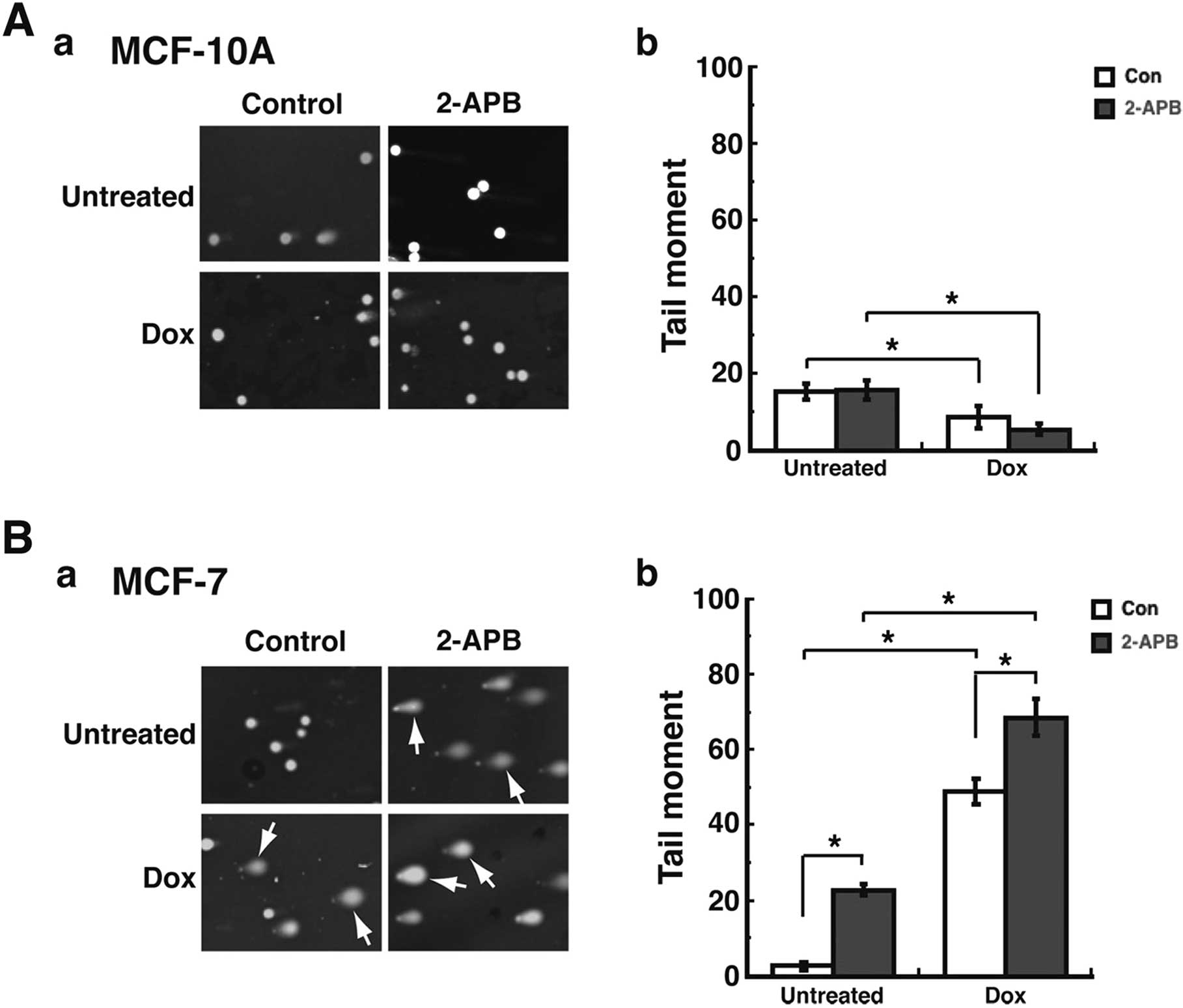
TRPM2 inhibition enhances DNA damage
levels in MCF-7 breast adenocarcinoma cells treated with
doxorubicin
We previously demonstrated that TRPM2 inhibition or
RNAi silencing caused increased levels of DNA damage in breast
adenocarcinoma cells (23). We next
investigated the potential ability of TRPM2 inhibition to increase
the DNA damage levels induced by chemotherapy. To accomplish this,
we utilized the single-cell gel electrophoresis (comet) assay. In
the comet assay, cells are embedded in agarose on a microscope
slide and then lysed to form nucleoids containing supercoiled DNA.
Electrophoresis in alkaline conditions results in structures
resembling comets, as observed by fluorescence microscopy. The
intensity of the comet tail relative to the head corresponds to the
amount of DNA strand breaks (35).
Treatment of the non-cancerous MCF-10A cells with 2-APB with or
without doxorubicin resulted in minimal levels of comets (Fig. 3A–a), which indicates minimal levels
of DNA damage. This was quantified by calculating the 'Tail moment'
values for each experimental group. Tail moment, a common
quantification of comet assay results, is calculated as the product
of the tail length and the fraction of total DNA in the tail
(28,29). Thus, using CometScore software and
analyzing a minimum of 200 cells/treatment group, the Tail moment
values for the untreated MCF-10A cells and those pretreated with
2-APB were ~18 for each cohort (Fig.
3A–b). Treatment of these cells with doxorubicin led to
decreased Tail moment values, which indicates decreased levels of
DNA damage in MCF-10A cells. However, in the MCF-7 breast
adenocarcinoma cells, increased levels of DNA damage were observed
(Fig. 3B–b) after 2-APB
pretreatment. This increased level of DNA damage was confirmed
after quantification of Tail moment values, where the Tail moment
value for untreated cells was 3, while the value for 2-APB
pretreatment was 22 (Fig. 3B–b).
Furthermore, while treatment with doxorubicin caused increased
levels of DNA damage in these cells, pretreatment with 2-APB
enhanced the level of DNA damage observed after doxorubicin
treatment (Fig. 3B–a). This was
also quantified, as the Tail moment value for doxorubicin treatment
was 44, while the value for doxorubicin + 2-APB was 65 (Fig. 3B–b). Moreover, these enhanced levels
of DNA damage due to 2-APB and doxorubicin treatment were
statistically significant when compared to the DNA damage levels
due to doxorubicin alone or 2-APB alone (Fig. 3B–b). These results thus indicated
that the inhibition of TRPM2 caused increased levels of DNA damage
in the MCF-7 breast adenocarcinoma cells after chemotherapeutic
treatment. Furthermore, the results also demonstrated that TRPM2
inhibition did not lead to increased DNA damage levels in the
non-cancerous breast cells.
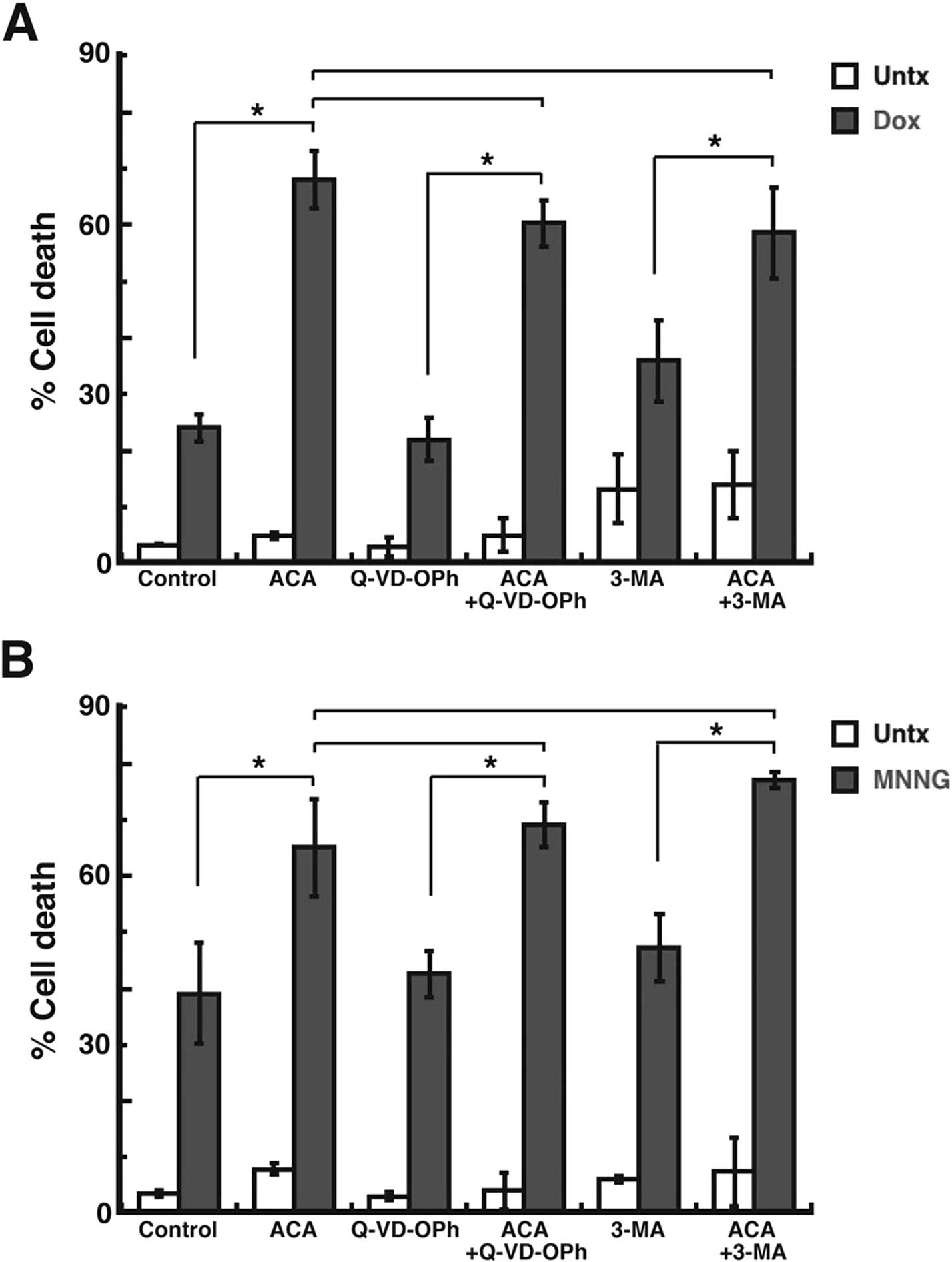
Increased cell death in MDA-MB-231 breast
adenocarcinoma cells due to TRPM2 inhibition is not primarily
mediated via apoptosis or autophagy
Since the aforementioned studies demonstrated
increased cell death in breast adenocarcinoma cells after TRPM2
inhibition, we next investigated the potential cell death pathways
induced after TRPM2 inhibition. To investigate apoptosis, we
utilized Q-VD-OPh, an inhibitor of apoptosis due to its ability to
function as a pan-caspase inhibitor (36). Pretreatment of the MDA-MB-231 breast
adenocarcinoma cells with Q-VD-OPh led to a cell death level of 59%
after ACA and doxorubicin treatment (Fig. 4A). However, this level of cell death
was not significantly decreased when compared to the cell death
induced by ACA and doxorubicin without Q-VD-OPh pretreatment (63%)
(Fig. 4A). To investigate
autophagy, we utilized the autophagy inhibitor, 3-methyladenine
(3-MA) (37). Pretreatment with
3-MA led to 57% cell death in the MDA-MB-231 breast adenocarcinoma
cells after ACA and doxorubicin treatment (Fig. 4A). This level of cell death was also
not significantly different from the amount of cell death induced
by ACA and doxorubicin without 3-MA pretreatment (Fig. 4A). Similar results were observed
when the chemotherapeutic agent utilized was MNNG. Q-VD-OPh
pretreatment led to 65% cell death after ACA and MNNG treatment,
which was not significantly different than the amount of cell death
caused by ACA + MNNG alone (63%) (Fig.
4B). Pretreatment with 3-MA led to 77% cell death in the
MDA-MB-231 cells after ACA + MNNG. However, when compared to the
cell death induced by ACA + MNNG (63%), this difference in cell
death was not found to be statistically significant. The results
thus demonstrated that – and 3-MA both failed to significantly
decrease the amount of cell death observed after TRPM2 inhibition
and chemotherapeutic treatments. Therefore, the studies suggest
that apoptosis and autophagy are not the primary pathways of cell
death induced by TRPM2 inhibition in breast adenocarcinoma cells
after chemotherapy.
Increased cell death in MDA-MB-231 breast
adenocarcinoma cells due to TRPM2 inhibition is not primarily due
to caspase-independent cell death mediated by poly(ADP-ribose) and
apoptosis-inducing factor
We continued our cell death analyses by
investigating caspase-independent cell death. One
caspase-independent pathway is cell death associated with
poly(ADP-ribose) (PAR) and mediated by apoptosis-inducing factor
(AIF) (38). PAR is an essential
biopolymer to the cell that is synthesized by the PAR polymerases
(PARPs) in response to DNA damage (39,40).
High levels of PAR, due to high levels of DNA damage or the absence
of PAR glycohydrolase (PARG) - the enzyme required to catabolize
PAR - leads to caspase-independent cell death mediated by the
pro-cell death protein, AIF (41,42).
In addition, the activation and function of TRPM2 channels are
known to be mediated by the metabolism of PA R (43). We thus utilized RNAi to knock down
the expression of AIF and PARG in order to determine whether this
pathway of caspase-independent cell death was a primary contributor
to the increased cell death caused by TRPM2 inhibition. We
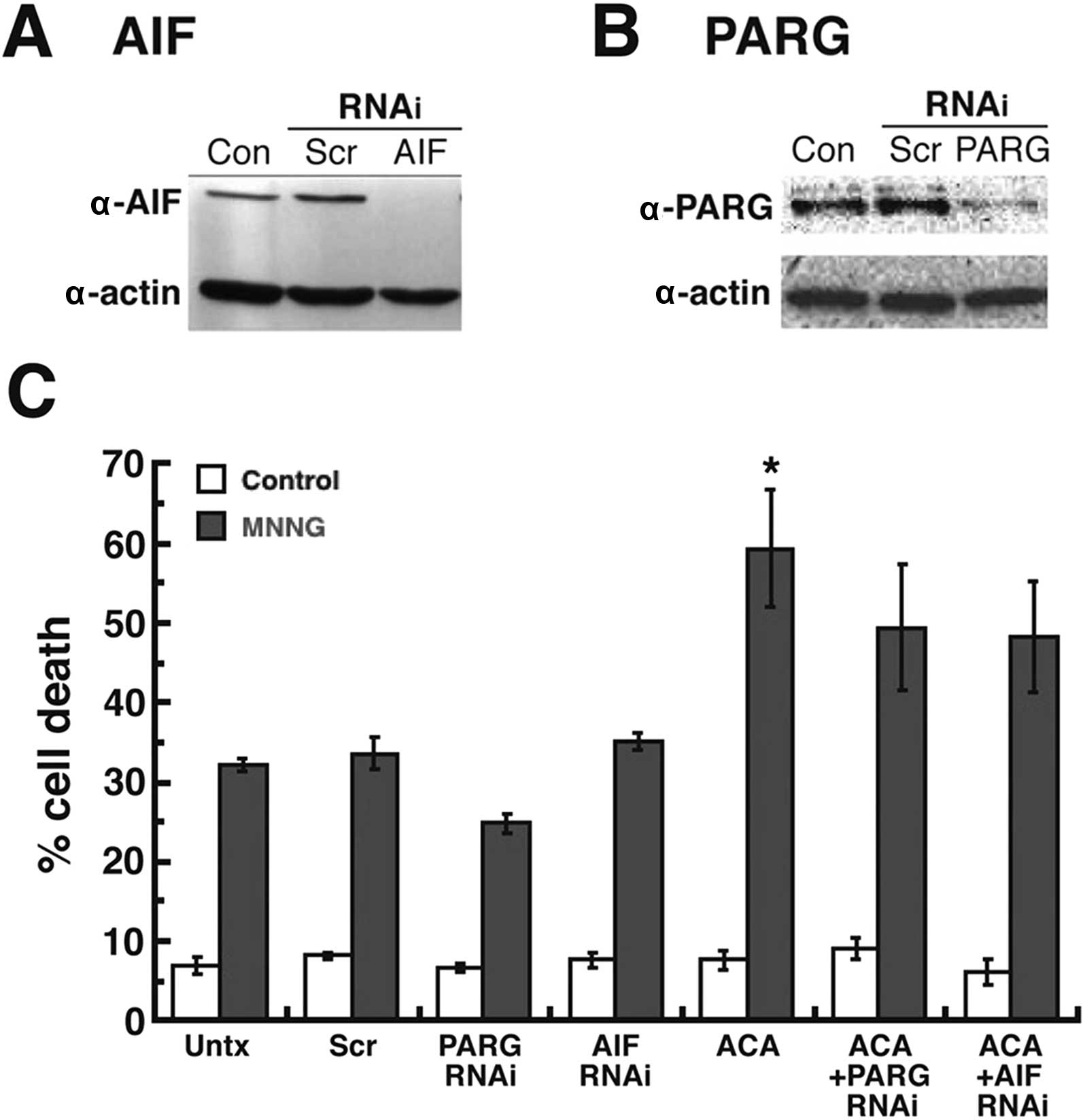
successfully decreased AIF (Fig.
5A) and PARG (Fig. 5B) protein
levels in the MDA-MB-231 breast adenocarcinoma cells by RNAi.
Treatment of these cells with ACA and MNNG produced 50% cell death
in the AIF-silenced cells and 49% cell death in the PARG-silenced
cells (Fig. 5C). Both of these cell
death values were lower than the cell death observed in the
untransfected MDA-MB-231 cells treated with ACA + MNNG (59%), yet
the difference was not found to be statistically significant. Thus,
the results demonstrated that the RNAi knockdown of PARG and AIF
expression did not significantly decrease the amount of cell death
observed after TRPM2 inhibition and chemotherapeutic treatments.
Therefore, the studies indicated that caspase-independent cell
death mediated by PAR and AIF was not a primary pathway of cell
death induced by TRPM2 inhibition in the breast adeno-carcinoma
cells after chemotherapy.
Discussion
We presented new data that further demonstrates the
therapeutic potential of inhibiting TRPM2 function in the treatment
of breast cancer. While we previously demonstrated a novel role for
TRPM2 in breast adenocarcinoma cells, where it was shown to mediate
DNA damage levels and cell proliferation, we expand upon these
findings by reporting increased cell death due to inhibition of
TRPM2. Furthermore, this was demonstrated in TN and ER+
breast cancer cell lines. Thus, the present study presents the
possibility that targeting TRPM2 is expected to provide an
additional strategy to successfully treat these molecular subtypes
of breast cancer tumors. This is particularly important for TN
breast cancer patients, as treatment options are limited, and the
efficacy and prognosis of currently available treatments are not as
favorable as those for patients with other molecular breast cancer
subtypes. Therefore, the ability of TRPM2 inhibition to enhance
cytotoxicity in TN and ER+ breast adenocarcinoma cells
suggests that targeting TRPM2 may provide improved treatment
options for TN and ER+ breast cancer patients in the
future.
Our studies also provided important mechanistic and
therapeutic insight into the cell death pathways induced by TRPM2
inhibition after chemotherapeutic treatments in breast
adenocarcinoma cells. Increased cell death was observed in TN and
ER+ breast adenocarcinoma cells after TRPM2 inhibition.
This was corroborated with increased DNA damage levels in breast
adenocarcinoma cells after chemotherapeutic treatments.
Furthermore, the increased cell death observed was not found to be
primarily mediated by apoptotic, autophagic or caspase-independent
cell death pathways. It was particularly unexpected that
caspase-independent cell death induced by PAR and mediated by AIF
was not a primary pathway that was triggered by TRPM2 inhibition.
This is because TRPM2 function is regulated by PAR metabolism,
where ADP-ribose moieties generated due to the catabolism of PAR
polymers by PARG have been shown to activate the gating of cations
by TRPM2 channels. Due to this regulation of TRPM2 channels by PAR,
along with the ability of activated TRPM2 channels to facilitate
cell death in non-cancerous cells, the lack of caspase-independent
cell death after TRPM2 inhibition in breast adenocarcinoma cells
was unexpected. However, our studies demonstrated that TRPM2
appears to have a novel role in breast cancer cells, while Zeng
et al previously demonstrated a potentially novel role for
TRPM2 in prostate cancer cells (22). Furthermore, our observation of the
lack of PAR-mediated cell death in breast cancer cells after TRPM2
inhibition, along with the observation by Zeng et al of the
failure of PAR to mediate TRPM2 function in prostate cancer cells,
appears to corroborate this novel role in both breast and prostate
cancer cells. Thus, it is conceivable that the novel role for TRPM2
in cancer cells is the basis for the observation that inhibition of
TRPM2 produces novel chemotherapeutic effects in cancer cells, with
minimal deleterious effects in non-cancerous cells.
Additional therapeutic insight gained from these
results is that TRPM2 inhibition has the potential to eradicate
breast cancer cells that are resistant to chemotherapy due to their
evasion of apoptosis. Our preliminary findings indicate that TRPM2
inhibition is expected to induce alternative cell death pathways in
breast adenocarcinoma cells. It is therefore possible that TRPM2
inhibition could provide the same effects in breast cancer cells
that are refractive to chemotherapy, particularly those that evade
apoptotic cell death, and thus survive after chemotherapy. This is
a significant finding, since breast tumors that are not responsive
to chemotherapy are a cause for significant morbidity and mortality
in breast cancer patients. The ability to overcome this resistance
to chemotherapy would clearly lead to improvements in breast cancer
chemotherapeutic treatments, and the overall survival and prognosis
of breast cancer patients in the future. Thus, our results offer
the possibility that targeting TRPM2 in breast tumors refractive to
chemotherapeutic treatments may lead to the improved eradication of
such tumors.
Future studies will be required to identify the
primary cell death pathway(s) induced by TRPM2 inhibition. The lack
of a primary role for apoptosis, autophagy or PAR-mediated
caspase-independent cell death in breast adenocarcinoma cells after
TRPM2 inhibition and chemotherapeutic treatments suggests that
necrosis is the primary cell death pathway induced. This is a
viable possibility, as a previous study demonstrated the
exacerbation of necrotic cell death due to TRPM2 activation
(24). However, this study was
accomplished in non-cancerous cells. Furthermore, the clinical
significance of other potential alternative cell death pathways are
beginning to emerge. For example, TRPM2 inhibition in cardiac and
neuroblastoma cells resulted in the upregulation of mitophagy
(21,44). Thus, more studies are required in
order to determine the primary cell death pathway(s) involved in
breast adenocarcinoma cells after TRPM2 inhibition.
Future studies will also be required to characterize
and identify the cellular effects of TRPM2 in breast cancer cells.
These mechanistic studies will be particularly important in order
to determine whether TRPM2 has different roles, not only in
cancerous vs. non-cancerous cells, but also among different types
of cancers. Current data are suggestive, yet not conclusive, that
TRPM2 may indeed have different roles in various types of cancers.
Our previous study in breast cancer cells, along with the study by
Zeng et al that investigated TRPM2 in prostate cancer cells,
determined that TRPM2 has a nuclear localization in breast and
prostate cancer cells. This localization was in contrast to the
currently known localization of TRPM2, where it functions as an ion
channel in the plasma membrane and lysosomal membrane. However, in
a well-designed recent report, the differential role of TRPM2 was
found to be dependent on the activities of TRPM2 isoforms, where a
truncated TRPM2 isoform (TRPM2-S) was found to decrease levels of
the transcription factors, HIF-1 and HIF-2 (21). This led to cytotoxic effects in
neuroblastoma cells. However, these studies demonstrated a
localization of TRPM2 in the plasma and lysosomal membranes. Based
on the studies to date in three different types of cancer, it is
possible that TRPM2 may have different localizations and different
roles in various types of cancer. Future studies will be required
to determine whether TRPM2 does indeed have novel roles in
different types of cancer.
In conclusion, we demonstrated that TRPM2 inhibition
leads to increased cell death, increased DNA damage, and the
induction of alternative pathways of cell death. This enhanced and
novel induction of alternative cell death appears to be possible in
TN and ER+ breast cancer cells. It also has the
potential to lead to the targeting and successful eradication of
chemotherapy-resistant breast cancer cells in the future. Thus, the
present study provides compelling evidence that TRPM2 is a novel
target that can lead to improved outcomes in the treatment of
breast cancer patients in the future.
Acknowledgments
The present study was supported in part by the
Bower, Bennet and Bennet Endowed Chair Research Award at Ohio
Northern University to D.W.K., the James and Diann Robbers Student
Research Fund at Washington State University (WSU) to X.F., and the
NIH/NIGMS Training Grant T32GM08336 for the NIH Protein
Biotechnology Program at WSU to M.M.H.
Abbreviations:
|
2-APB
|
2-aminoethoxydiphenyl borate
|
|
3-MA
|
3-methyladenine
|
|
AA
|
aristolochic acid
|
|
ACA
|
N-(p-amylcinnamoyl) anthranilic
acid
|
|
AIF
|
apoptosis-inducing factor
|
|
ER+
|
estrogen receptor-positive
|
|
MNNG
|
N-methyl-N'-nitro-N-nitrosoguanidine
|
|
PAR
|
polyadenosine diphosphoribose
biopolymer
|
|
PARG
|
poly-(ADP-ribose) glycohydrolase
|
|
TN
|
triple-negative
|
|
TRP
|
transient receptor potential
superfamily of cation channels
|
|
TRPM
|
transient receptor potential,
melastatin subfamily
|
References
|
1
|
Batist G, Harris L, Azarnia N, Lee LW and
Daza-Ramirez P: Improved anti-tumor response rate with decreased
cardiotoxicity of non-pegylated liposomal doxorubicin compared with
conventional doxorubicin in first-line treatment of metastatic
breast cancer in patients who had received prior adjuvant
doxorubicin: Results of a retrospective analysis. Anticancer Drugs.
17:587–595. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Harris LN, Broadwater G, Lin NU, Miron A,
Schnitt SJ, Cowan D, Lara J, Bleiweiss I, Berry D, Ellis M, et al:
Molecular subtypes of breast cancer in relation to paclitaxel
response and outcomes in women with metastatic disease: Results
from CALGB 9342. Breast Cancer Res. 8:R662006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sotiriou C, Neo SY, McShane LM, Korn EL,
Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL and Liu ET: Breast
cancer classification and prognosis based on gene expression
profiles from a population-based study. Proc Natl Acad Sci USA.
100:10393–10398. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
André F and Zielinski CC: Optimal
strategies for the treatment of metastatic triple-negative breast
cancer with currently approved agents. Ann Oncol. 23(Suppl 6):
vi46–vi51. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Early Breast Cancer Trialists'
Collaborative Group (EBCTCG): Effects of chemotherapy and hormonal
therapy for early breast cancer on recurrence and 15-year survival:
An overview of the randomised trials. Lancet. 365:1687–1717. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fisher B, Anderson S, Tan-Chiu E, Wolmark
N, Wickerham DL, Fisher ER, Dimitrov NV, Atkins JN, Abramson N,
Merajver S, et al: Tamoxifen and chemotherapy for axillary
node-negative, estrogen receptor-negative breast cancer: Findings
from National Surgical Adjuvant Breast and Bowel Project B-23. J
Clin Oncol. 19:931–942. 2001.PubMed/NCBI
|
|
7
|
Dunnwald LK, Rossing MA and Li CI: Hormone
receptor status, tumor characteristics, and prognosis: A
prospective cohort of breast cancer patients. Breast Cancer Res.
9:R62007. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lumachi F, Brunello A, Maruzzo M, Basso U
and Basso SM: Treatment of estrogen receptor-positive breast
cancer. Curr Med Chem. 20:596–604. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Parise CA and Caggiano V: Breast cancer
survival defined by the ER/PR/HER2 subtypes and a surrogate
classification according to tumor grade and immunohistochemical
biomarkers. J Cancer Epidemiol. 2014:4692512014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cory S and Adams JM: The Bcl2 family:
Regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Aird KM, Ghanayem RB, Peplinski S, Lyerly
HK and Devi GR: X-linked inhibitor of apoptosis protein inhibits
apoptosis in inflammatory breast cancer cells with acquired
resistance to an ErbB1/2 tyrosine kinase inhibitor. Mol Cancer
Ther. 9:1432–1442. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang MY, Chen PS, Prakash E, Hsu HC, Huang
HY, Lin MT, Chang KJ and Kuo ML: Connective tissue growth factor
confers drug resistance in breast cancer through concomitant
up-regulation of Bcl-xL and cIAP1. Cancer Res. 69:3482–3491. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nadler MJ, Hermosura MC, Inabe K, Perraud
AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg
AM, et al: LTRPC7 is a Mg·ATP-regulated divalent cation channel
required for cell viability. Nature. 411:590–595. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Perraud AL, Fleig A, Dunn CA, Bagley LA,
Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, et al:
ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed
by Nudix motif homology. Nature. 411:595–599. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Blenn C, Wyrsch P, Bader J, Bollhalder M
and Althaus FR: Poly(ADP-ribose)glycohydrolase is an upstream
regulator of Ca2+ fluxes in oxidative cell death. Cell
Mol Life Sci. 68:1455–1466. 2011. View Article : Google Scholar :
|
|
16
|
Sun L, Yau HY, Wong WY, Li RA, Huang Y and
Yao X: Role of TRPM2 in H2O2-induced cell
apoptosis in endothelial cells. PLoS One. 7:e431862012. View Article : Google Scholar
|
|
17
|
Zhang W, Chu X, Tong Q, Cheung JY, Conrad
K, Masker K and Miller BA: A novel TRPM2 isoform inhibits calcium
influx and susceptibility to cell death. J Biol Chem.
278:16222–16229. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lange I, Yamamoto S, Partida-Sanchez S,
Mori Y, Fleig A and Penner R: TRPM2 functions as a lysosomal
Ca2+-release channel in beta cells. Sci Signal.
2:ra232009. View Article : Google Scholar
|
|
19
|
Zhang W, Hirschler-Laszkiewicz I, Tong Q,
Conrad K, Sun SC, Penn L, Barber DL, Stahl R, Carey DJ, Cheung JY,
et al: TRPM2 is an ion channel that modulates hematopoietic cell
death through activation of caspases and PARP cleavage. Am J
Physiol Cell Physiol. 290:C1146–C1159. 2006. View Article : Google Scholar
|
|
20
|
Orfanelli U, Wenke AK, Doglioni C, Russo
V, Bosserhoff AK and Lavorgna G: Identification of novel sense and
antisense transcription at the TRPM2 locus in cancer. Cell Res.
18:1128–1140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen SJ, Hoffman NE, Shanmughapriya S, Bao
L, Keefer K, Conrad K, Merali S, Takahashi Y, Abraham T,
Hirschler-Laszkiewicz I, et al: A splice variant of the human ion
channel TRPM2 modulates neuroblastoma tumor growth through
hypoxia-inducible factor (HIF)-1/2α. J Biol Chem. 289:36284–36302.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zeng X, Sikka SC, Huang L, Sun C, Xu C,
Jia D, Abdel-Mageed AB, Pottle JE, Taylor JT and Li M: Novel role
for the transient receptor potential channel TRPM2 in prostate
cancer cell proliferation. Prostate Cancer Prostatic Dis.
13:195–201. 2010. View Article : Google Scholar :
|
|
23
|
Hopkins MM, Feng X, Liu M, Parker LP and
Koh DW: Inhibition of the transient receptor potential melastatin-2
channel causes increased DNA damage and decreased proliferation in
breast adenocarcinoma cells. Int J Oncol. 46:2267–2276.
2015.PubMed/NCBI
|
|
24
|
Hiroi T, Wajima T, Negoro T, Ishii M,
Nakano Y, Kiuchi Y, Mori Y and Shimizu S: Neutrophil TRPM2 channels
are implicated in the exacerbation of myocardial
ischaemia/reperfusion injury. Cardiovasc Res. 97:271–281. 2013.
View Article : Google Scholar
|
|
25
|
Liu T, Biddle D, Hanks AN, Brouha B, Yan
H, Lee RM, Leachman SA and Grossman D: Activation of dual apoptotic
pathways in human melanocytes and protection by survivin. J Invest
Dermatol. 126:2247–2256. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Blenn C, Althaus FR and Malanga M:
Poly(ADP-ribose) glycohydrolase silencing protects against
H2O2-induced cell death. Biochem J.
396:419–429. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou Y, Feng X and Koh DW: Enhanced DNA
accessibility and increased DNA damage induced by the absence of
poly(ADP-ribose) hydrolysis. Biochemistry. 49:7360–7366. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hartmann A, Agurell E, Beevers C,
Brendler-Schwaab S, Burlinson B, Clay P, Collins A, Smith A, Speit
G, Thybaud V, et al 4th International Comet Assay Workshop:
Recommendations for conducting the in vivo alkaline Comet assay.
4th International Comet Assay Workshop. Mutagenesis. 18:45–51.
2003. View Article : Google Scholar
|
|
29
|
Olive PL, Banáth JP, Durand RE and Banath
JP: Heterogeneity in radiation-induced DNA damage and repair in
tumor and normal cells measured using the 'comet' assay. Radiat
Res. 122:86–94. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pinsky SD, Tew KD, Smulson ME and Woolley
PV III: Modification of L1210 cell nuclear proteins by
1-methyl-1-nitro-sourea and 1-methyl-3-nitro-1-nitrosoguanidine.
Cancer Res. 39:923–928. 1979.PubMed/NCBI
|
|
31
|
Kraft R, Grimm C, Frenzel H and Harteneck
C: Inhibition of TRPM2 cation channels by
N-(p-amylcinnamoyl)anthranilic acid. Br J Pharmacol. 148:264–273.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Konrad RJ, Jolly YC, Major C and Wolf BA:
Inhibition of phospholipase A2 and insulin secretion in
pancreatic islets. Biochim Biophys Acta. 1135:215–220. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rosenthal MD, Lattanzio KS and Franson RC:
The effects of the phospholipase A2 inhibitors
aristolochic acid and PGBx on A23187-stimulated mobilization of
arachidonate in human neutrophils are overcome by diacylglycerol or
phorbol ester. Biochim Biophys Acta. 1126:319–326. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Togashi K, Inada H and Tominaga M:
Inhibition of the transient receptor potential cation channel TRPM2
by 2-aminoethoxy-diphenyl borate (2-APB). Br J Pharmacol.
153:1324–1330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Singh NP, McCoy MT, Tice RR and Schneider
EL: A simple technique for quantitation of low levels of DNA damage
in individual cells. Exp Cell Res. 175:184–191. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Caserta TM, Smith AN, Gultice AD, Reedy MA
and Brown TL: Q-VD-Oph, a broad spectrum caspase inhibitor with
potent antiapoptotic properties. Apoptosis. 8:345–352. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Seglen PO and Gordon PB: 3-Methyladenine:
Specific inhibitor of autophagic/lysosomal protein degradation in
isolated rat hepatocytes. Proc Natl Acad Sci USA. 79:1889–1892.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yu SW, Wang H, Poitras MF, Coombs C,
Bowers WJ, Federoff HJ, Poirier GG, Dawson TM and Dawson VL:
Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by
apoptosis-inducing factor. Science. 297:259–263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dantzer F, de La Rubia G, Ménissier-De
Murcia J, Hostomsky Z, de Murcia G and Schreiber V: Base excision
repair is impaired in mammalian cells lacking poly(ADP-ribose)
polymerase-1. Biochemistry. 39:7559–7569. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Trucco C, Oliver FJ, de Murcia G and
Ménissier-de Murcia J: DNA repair defect in poly(ADP-ribose)
polymerase-deficient cell lines. Nucleic Acids Res. 26:2644–2649.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou Y, Feng X and Koh DW: Activation of
cell death mediated by apoptosis-inducing factor due to the absence
of poly(ADP-ribose) glycohydrolase. Biochemistry. 50:2850–2859.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW,
Sasaki M, Klauss JA, Otsuka T, Zhang Z, Koehler RC, et al:
Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad
Sci USA. 103:18308–18313. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Buelow B, Song Y and Scharenberg AM: The
Poly(ADP-ribose) polymerase PARP-1 is required for oxidative
stress-induced TRPM2 activation in lymphocytes. J Biol Chem.
283:24571–24583. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Miller BA, Hoffman NE, Merali S, Zhang XQ,
Wang J, Rajan S, Shanmughapriya S, Gao E, Barrero CA,
Mallilankaraman K, et al: TRPM2 channels protect against cardiac
ischemia-reperfusion injury: Role of mitochondria. J Biol Chem.
289:7615–7629. 2014. View Article : Google Scholar : PubMed/NCBI
|