Introduction
Renal cell carcinoma (RCC) is one of the most common
malignant tumors of the urinary system, accounting for 80–90% of
kidney neoplasms. The American Cancer Society estimated that the
number of new cancer cases and deaths caused by cancer of the
kidney and renal pelvis were 63,920 and 13,860 in 2014 in the US
(1). Surgical resection is
considered to be the first choice for the curative treatment of
localized RCC and some locally advanced RCC. However, ~25–30%
patients initially present with metastases, and a substantial part
of patients have subclinical metastases at that time (2,3). Some
patients undergoing surgical treatment relapse to metastatic renal
cell carcinoma (mRCC). mRCC is usually resistant to radiotherapy
and chemotherapy, while immunotherapy shows limited response rates
of 15–20% (4–6). Therefore, understanding the mechanisms
involved in the progression of RCC especially mRCC and exploring
more effective targeted therapies are urgently needed.
The hedgehog (Hh) pathway was firstly emphasized in
embryonic developmental studies (7,8).
Recent research has demonstrated that the Hh pathway is
constitutively activated in cancer compared with normal tissue and
plays a crucial role in cancer progression (9–13).
Inhibition of Hh signaling was found to significantly induce
apoptotic cell death in pancreatic cancer cell lines both in
vitro and in vivo (9).
Hh signaling-mediated survival of gastric cancer cells is known to
be associated with the regulation of apoptosis inhibition protein
BCL2 (12). Selective
downregulation of GLI family zinc finger 1 (GLI1) by antisense
oligodeoxynucleotides results in decreased BCL2 expression and
cancer cell survival (13).
Clinical trials suggest benefits for patients administered specific
Hh pathway inhibitors. GDC-0449, a small-molecule inhibitor of SMO,
has shown encouraging antitumor activity in locally advanced or
metastatic solid tumors in two separated phase I trials (14,15).
Another Hh inhibitor itraconazole decreased cancer cell
proliferation by 45%, Hh pathway activity by 65%, and reduced the
tumor area by 24% in phase II trials (16). Dormoy et al also reported
that the sonic Hh signaling pathway is reactivated in RCC compared
with normal tissue, and inhibition of this pathway dramatically
decreases tumor cell proliferation and induces apoptosis in
vitro and in vivo (17).
Thus, the Hh pathway may be a promising target for RCC especially
mRCC therapy.
Silibinin is a natural flavonoid which is widely
used for the treatment of drug- or alcohol-induced liver injury and
cirrhosis. Recent studies have shown the significant anticancer
activity of silibinin. For many tumors such as prostate (18,19),
bladder (20,21), lung (22) and colon (23,24)
cancers, silibinin exhibits growth inhibitory and anti-inflammatory
effects, cell cycle arrest, apoptosis induction,
chemosensitization, inhibition of angiogenesis, reversal of
multi-drug resistance and inhibition of invasion and metastasis,
which involves regulation of receptor tyrosine kinases, the
androgen receptor, and cell cycle regulatory and apoptotic
signaling pathways (25). Cheung
et al showed that oral administration of silibinin
suppressed local and metastatic tumor growth in an orthotopic
xenograft model of RCC. This anti-neoplastic action of silibinin
might involve insulin-like growth factor-binding protein 3
(IGFBP-3) (26). Our previous
studies demonstrated the apoptosis induction and metastatic
inhibitory effects of silibinin against RCC by targeting the
epidermal growth factor receptor (eGFR) pathway (27,28).
Recently, we found that autophagy induction by silibinin positively
contributes to its anti-metastatic capacity via the adenosine
5′-monophos-phate-activated protein kinase (aMPk)/mammalian target
of rapamycin (mTOR) pathway in RCC cells (29). However, whether the Hh pathway is
regulated by silibinin followed by induction of apoptosis remains
to be explored.
In the present study, 769-P, 786-O, OS-RC-2 (cell
lines derived from primary clear cell adenocarcinoma, which have
the similar genetic and molecular features of RCC) and ACHN (a cell
line derived from the malignant pleural effusion, which has the
similar genetic and molecular feature of mRCC) cell lines were
applied as the model system. We first document that the
mTOR-GLI1-BCL2 pathway is crucial for silibinin-induced apoptosis
in RCC cells, and imply that GLI1 is a novel regulator for the
potential therapeutic application of silibinin against RCC.
Materials and methods
Cell culture and reagents
Human RCC cell lines 769-P, 786-O, ACHN and OS-RC-2
were purchased from the american Type Culture Collection (ATCC;
Manassas, VA, USA), and cultured in Dulbecco's modified eagle's
medium (DMEM)/F-12 medium supplemented with 10% fetal bovine serum
(FBS) (Gibco-BRL, New York, NY, USA) at 37°C, in humidified air
containing 5% CO2. Silibinin, cyclopamine, LY294002,
rapamycin and 3-(4,5-dimeth-ylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO,
USA). Antibodies for western blot analysis against poly(ADP-ribose)
polymerase (PARP), cleaved caspase-3, p-mTOR, mTOR, p-eRk, eRk,
p-akT (Thr308), p-akT (Ser473) and AKT were purchased from Cell
Signaling Technology, Inc. (Beverly, MA, USA). anti-GLI1 was from
Bioss, Inc. (Beijing, China). Anti-BCL2 was from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). anti-glyceraldehyde
3-phosphate dehydroge-nase (GAPDH) was purchased from KangChen
Bio-tech, Inc. (Shanghai, China). Antibodies for
immunohistochemistry against p-mTOR and BCL2 were from Abcam
(Cambridge, MA, USA), and the antibody against GLI1 was from Santa
Cruz Biotechnology, Inc.
Cell viability assay
Cell viability was measured by a tetrazolium-based
assay. Briefly, cells were seeded in a 96-well plate and treated
with doses of silibinin from 0 to 200 µM for the indicated
time. The medium was then removed and cells were incubated with 0.5
mg/ml MTT for 4 h and the formazan products were resolved with
dimethyl sulfoxide (DMSO) (150 µl/well). The OD value of
each well was measured by a microplate reader at a wavelength of
490 nm. The experiments were performed in triplicate.
Colony formation assay
Cells were seeded in a 6-well plate (1,000
cells/well) and incubated for 48–72 h, followed by treatment with
different doses of silibinin. The mediums and drugs were replaced
every 3 days. Ten days later when a single cell formed a colony
(≥50 cells), the plates were washed with phosphate-buffered saline
(PBS), fixed in 10% formalin, and then stained with 0.1% crystal
violet solution. Cell colony numbers were counted under a micro
scope. The experiments were performed in triplicate.
Quantitative detection of apoptosis
To evaluate the apoptosis induced by the reagents,
flow cytometry was performed. Cells were treated with the indicated
doses of silibinin for 48 h, and collected for Annexin V and
propidium iodide (PI) staining (Roche Diagnostics GmbH, Mannheim,
Germany) following the manufacturer's instructions, and the
apoptotic cells were detected and analyzed by flow cytometry
(FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA).
Western blot analysis
Cell lysates were prepared in lysis buffer [10 mM of
Tris-HCl (pH 7.4), 150 mM of naCl, 0.1% of SDS, 1 mM of EDTa, 1 mM
of EGTA, 0.3 mM of PMSF, 0.2 mM of sodium orthovanadate, 1% of
nP-40, 10 mg/ml of leupeptin and 10 mg/ml of aprotinin]. Proteins
were separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) on 10 or 12% Tris-glycine gels, and
transferred onto nitrocellulose membranes. The membranes were
blocked with 5% skim milk in TBS for 1 h at room temperature, and
probed the with primary antibody overnight at 4°C followed by
secondary antibody incubation for 1 h at room temperature. The
protein expression was visualized with the ECL detection system or
Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE,
USA).
Reverse transcription and real-time
PCR
Total RNA was isolated with RNAfast 200 reagents
(Fastagen Biotechnology Co., Ltd., Shanghai, China) following the
manufacturer's instructions and was quantitated by absorbance at
260 nm. The RNA (0.5 µg) sample was used for reverse
transcription with PrimeScript™ RT Master Mix according to the
manufacturer's instructions, and quantitative PCR was performed
with SYBR-Green PCR Master Mix (both from Takara Bio, Inc., Dalian,
China) and GAPDH mRNA was used as the internal control. The primer
sequences were: GLI1 forward, 5′-GATGATCCCACATCCTCAGTCC-3′ and
reverse, 5′-ACTTGCCAACCAGCATGTCC-3′; GAPDH forward,
5′-ATGGGGAAGGTGAAGGTCGG-3′ and reverse,
5′-GACGGTGCCATGGAATTTGC-3′.
Plasmid, siRNA transfection
GLI1 cDNA was cloned into the pcDNA3.1 vector. Small
interfering RNAs were designed and synthesized by Gene Pharma
(Shanghai, China). Cells achieved 70–80% confluence for plasmid
transfection or 30–50% confluence for siRNA transfection, and were
transfected with X-tremeGENE HP DNA or X-tremeGene siRNA
transfection reagents (Roche Diagnostics GmbH) for 2–3 days, and
harvested for subsequent experiments.
Xenograft model
Cells (786-O) were mixed with Matrigel (5:1, v/v)
and injected subcutaneously into the right hind flank of male
BALB/c (nu/nu) mice. Twelve days after injection, the mice were
randomly divided into two groups (5 mice/group), and were fed by
oral gavage with saline or silibinin (200 mg/kg). The volume of
tumors were measured with a vernier caliper every three days.
Thirty days after implantation, the mice were sacrificed and the
tumors were weighed with electronic scales and fixed in 10%
formalin and embedded in paraffin. Animal experiments were approved
by the Institutional animal Care and use Committee of Xi'an
Jiaotong University (permit no. SCXK2014-0155, 5 March 2014).
Immunohistochemistry
Immunohistochemical staining was performed with the
EnVision™ system (Dako, Carpinteria, Ca, USA), and the slices were
de-paraffinized, rehydrated, followed by 5 min antigen retrieval,
10 min of endogenous enzyme block and incubated with the primary
antibody overnight at 4°C. Then the slices were incubated with
EnVision-HRP secondary antibody for 1 h, and the signals were
detected by diaminobenzidine (DAB) followed by hematoxylin
counter-staining. The results were observed by a microscope.
Stained cells were quantified as the number of positive cells ×
100/total number of cells in 10 random microscopic (magnification,
×200) fields in each slice.
Statistical analysis
All the statistical analyses were performed by
GraphPad Prism version 5.0 software, and the Student's t-test was
used for two-group comparisons. P<0.05 was considered
statistically significant.
Results
Silibinin inhibits the proliferation and
colony formation of RCC cells
To evaluate the effects of silibinin on RCC cells,
four cell lines 769-P, 786-O, ACHN and OS-RC-2 were treated with
different doses of silibinin. The results showed that silibinin
significantly inhibited the proliferation of RCC cells in a dose-
and time-dependent manner as detected by MTT assay (Fig. 1a). Similarly, silibinin treatment
also decreased the colony numbers in the 769-P, 786-O and ACHN RCC
cells as determined by the colony formation assay (Fig. 1B and C).
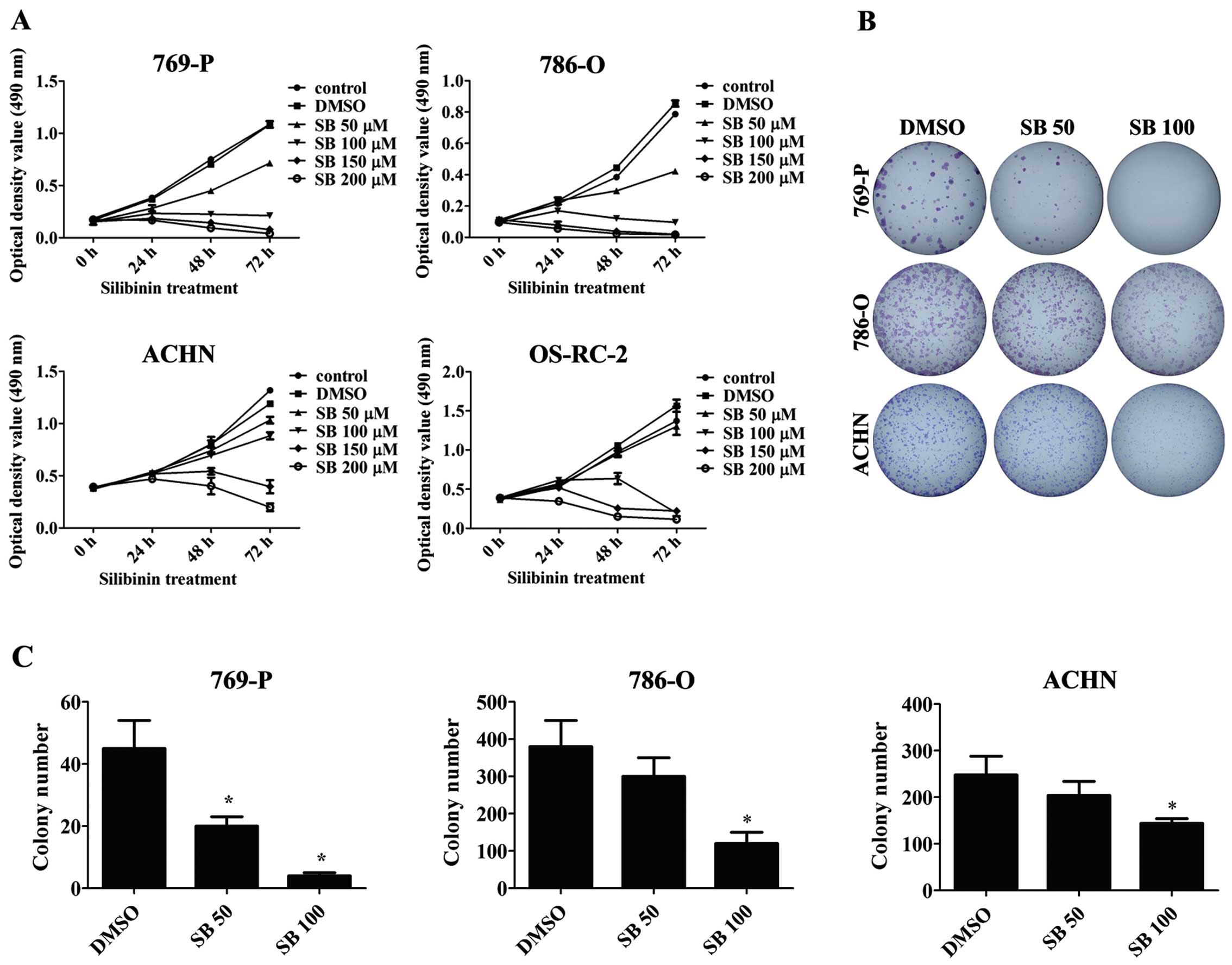 | Figure 1Silibinin inhibits the proliferation
and colony formation of RCC cells. (A) RCC 769-P, 786-O, ACHN and
OS-RC-2 cells were treated with different doses of silibinin (SB)
(50, 100, 150 and 200 µM) or DMSO (control) for 0, 24, 48
and 72 h, and MTT assay was performed to detect cell viability. (B)
The 769-P, 786-O and ACHN cells were treated with DMSO (control) or
silibinin (50 and 100 µM), and the colony formation assay
was performed after 10 days of treatment. The results were recorded
by photographing. (C) Quantitative results of the colony numbers.
The values represent the mean ± SD of three independent experiments
(*P<0.05). RCC, renal cell carcinoma. |
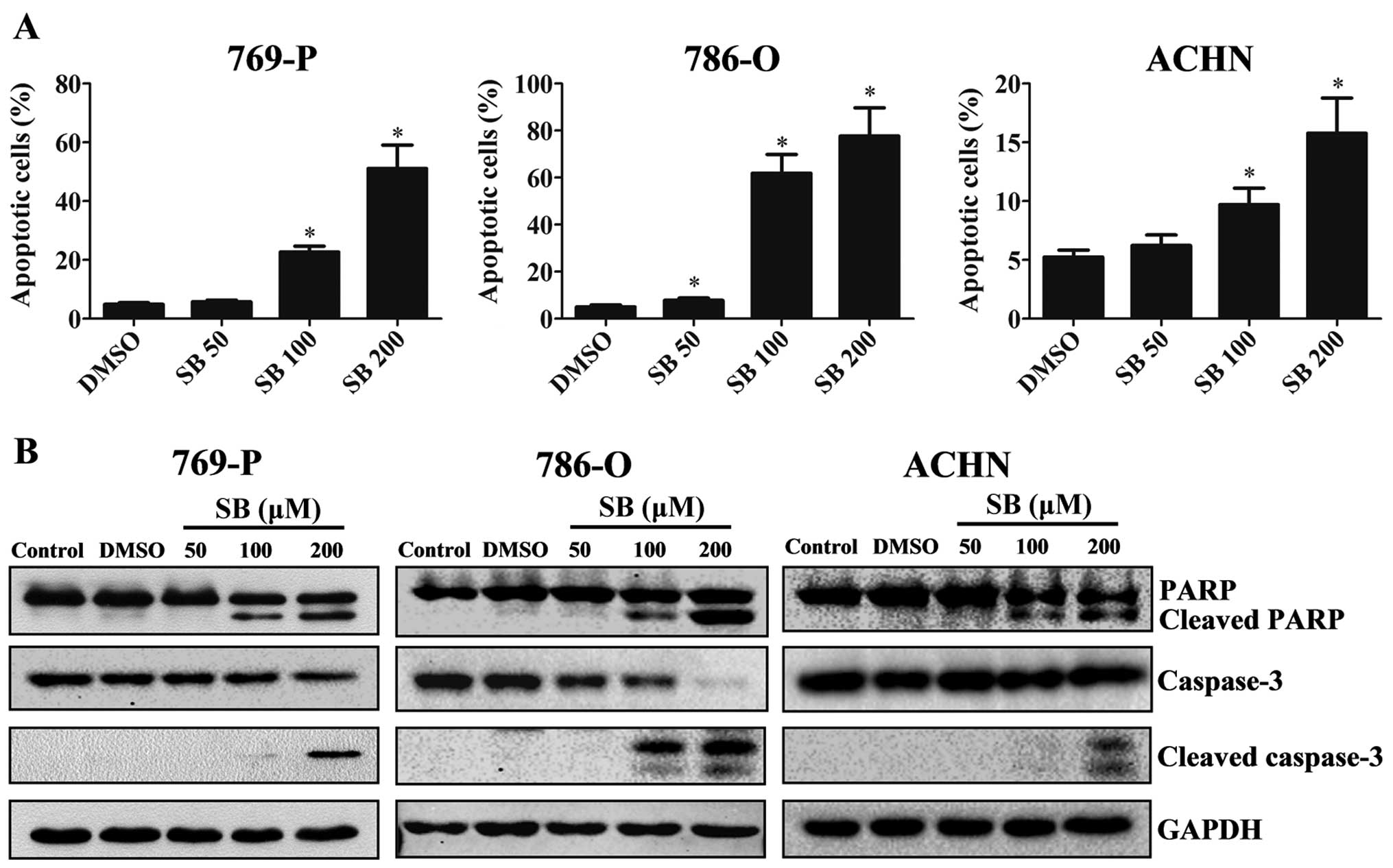
Silibinin induces the apoptosis of RCC
cells involving activation of caspase-3 and PARP
Since we observed the anti-proliferative effects of
silibinin on RCC, we next examined the apoptotic effects of
silibinin. RCC 769-P, 786-O and ACHN cells were treated with
silibinin (50, 100 and 200 µM) or DMSO (control) for 48 h.
Cells were then collected for flow cytometric assay and western
blot analysis. The percentages of apoptotic cells after treatment
with different dosages of silibinin (50, 100 and 200 µM) vs.
the control cells were 5.77/22.72/51.10 vs. 4.91% (769-P),
7.79/61.9/77.69 vs. 5.05% (786-O), 6.23/9.71/15.77 vs. 5.24%
(ACHN), respectively (P<0.05) (Fig.
2a). Western blot analysis data showed that cleaved subunits of
caspase-3 and PARP were increased upon silibinin treatment in a
dose-dependent manner (Fig. 2B),
indicating the activation of the caspase cascade by silibinin.
Taken together, our results demonstrated that silibinin induces RCC
cell apoptosis via activation of caspase cascade signaling.
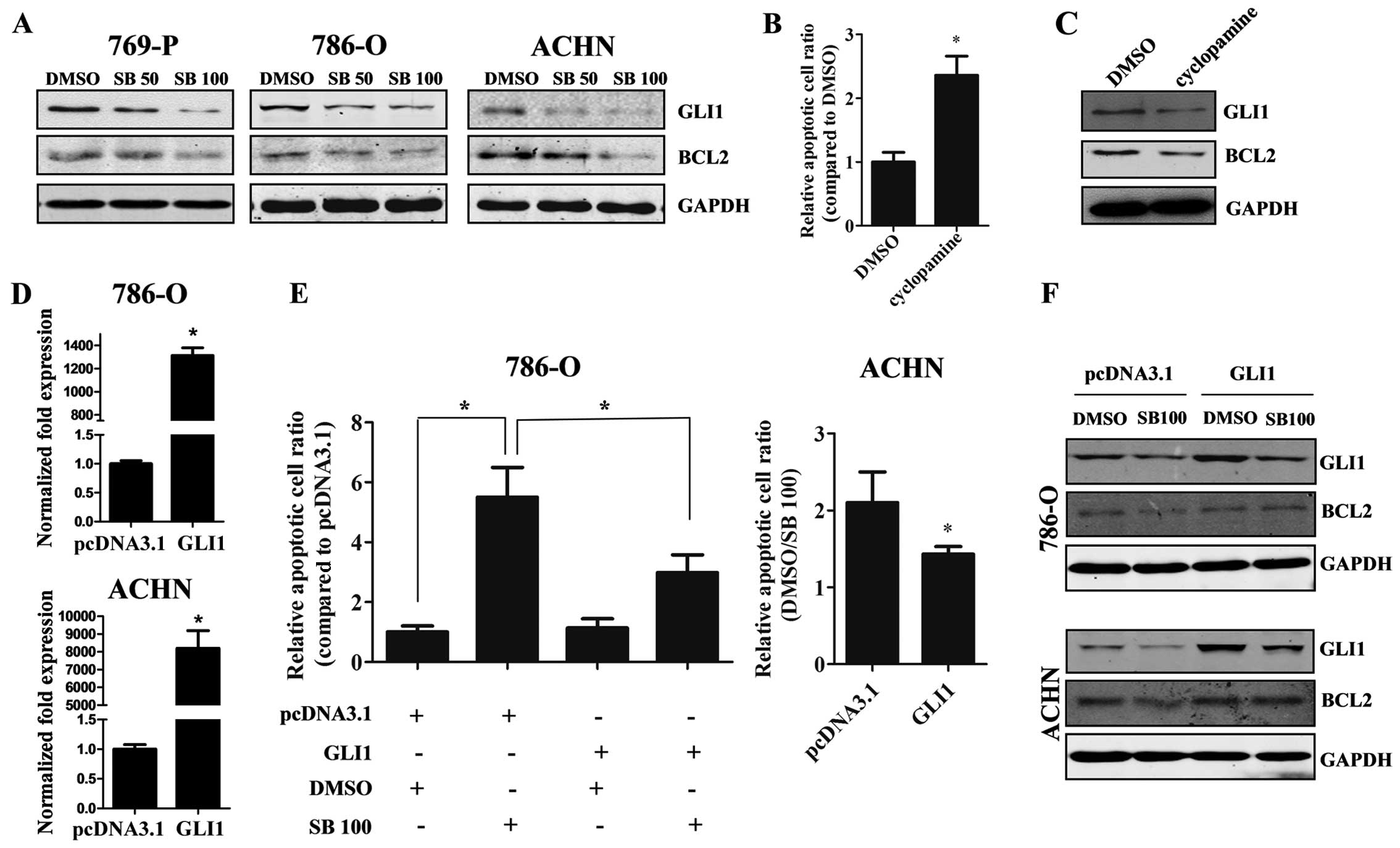
Silibinin induces apoptosis through
downregulation of GLI1 and BCL2
Since we demonstrated that silibinin treatment
inhibits cell proliferation, the underlying mechanism was
investigated. As shown in Fig. 3a,
silibinin markedly down-regulated GLI1 expression in all three RCC
cell lines in a dose-dependent manner, and BCL2, the downstream
molecule of GLI1, was also decreased. As known, GLI1 plays an
important role in cancer development and progression. In order to
detect the function of GLI1 in RCC, we used cyclopamine, a potent
Hh signaling inhibitor. The results showed that cyclopamine induced
the apoptosis of 769-P cells (Fig.
3B), while the levels of GLI1 and BCL2 were reduced (Fig. 3C). When the GLI1-overexpressing
plasmid was transfected into 786-O and ACHN cells, the apoptosis
and reduced BCL2 expression induced by silibinin was partially
reversed (Fig. 3D–F), suggesting
that the GLI1-BCL2 pathway participates in the silibinin-induced
apoptosis of RCC cells.
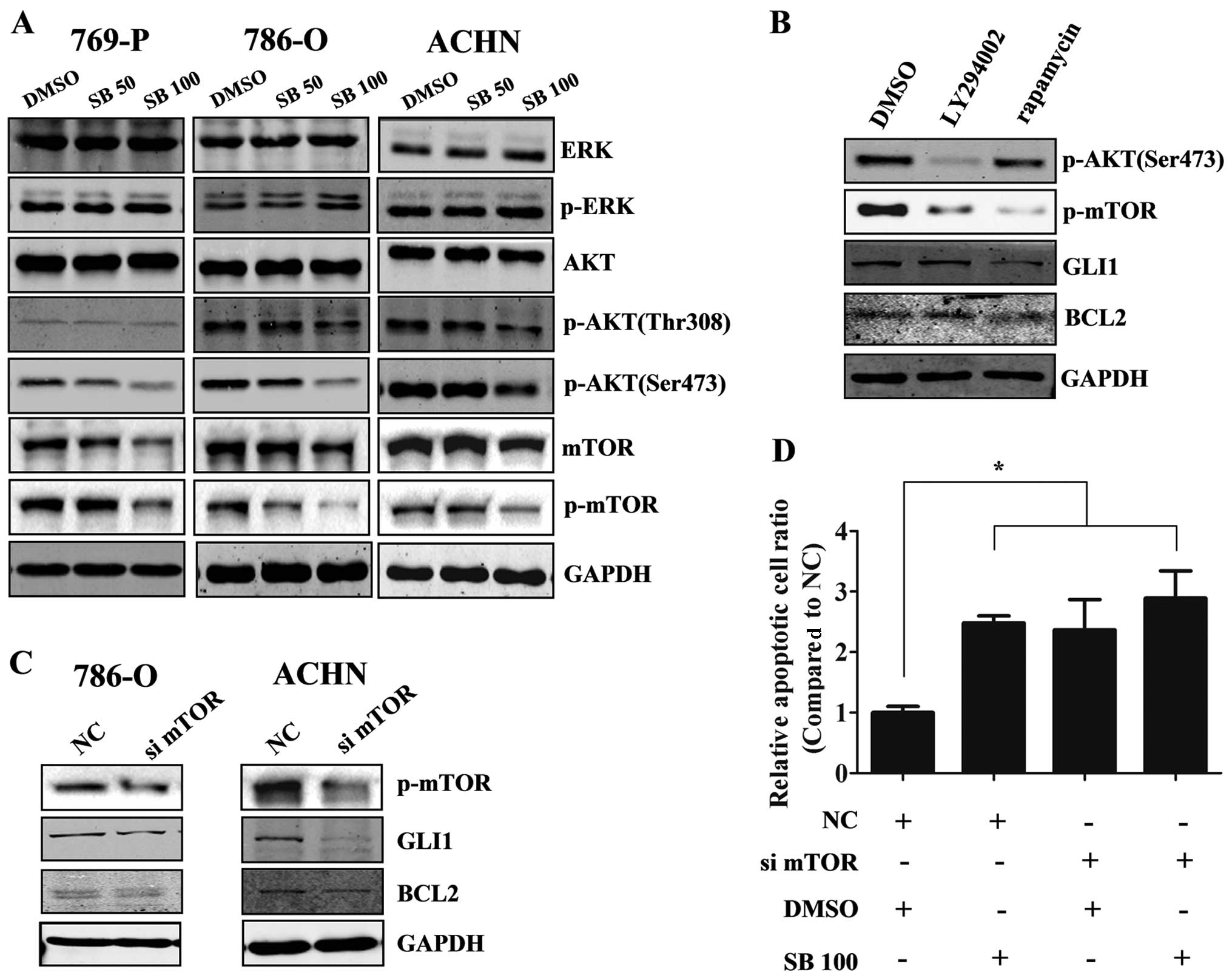
Downregulation of p-mTOR is involved in
the silibinin-induced GLI1 decrease
To further investigate the mechanisms of the
silibinin-induced decrease in GLI1, we detected the GLI1-associated
pathway which has been previously reported to regulate GLI1
expression. Western blot results showed that p-AKT, p-mTOR but not
p-ERK were downregulated after the treatment with silibinin
(Fig. 4a). Additionally, only the
p-mTOR inhibitor (rapamycin, 100 nM) but not the p-AKT inhibitor
(LY294002, 20 µM) could inhibit GLI1 and BCL2 expression in
the 786-O cells (Fig. 4B).
Furthermore, knockdown of mTOR by siRNA in the 786-O and ACHN cells
reduced GLI1 and BCL2 expression (Fig.
4C), indicating that the mTOR pathway regulated GLI1 expression
in the RCC cells. When knockdown of mTOR by siRNA was combined with
silibinin treatment or DMSO (control) in ACHN cells, we found this
combination did not increase the percentage of apoptotic cells
compared with silibinin alone or mTOR knockdown alone (Fig. 4D), which implied that the
silibinin-induced apoptosis of RCC cells was mediated by mTOR.
Taken together, our results indicate that silibinin inhibits GLI1
expression via reduction of mTOR activity.
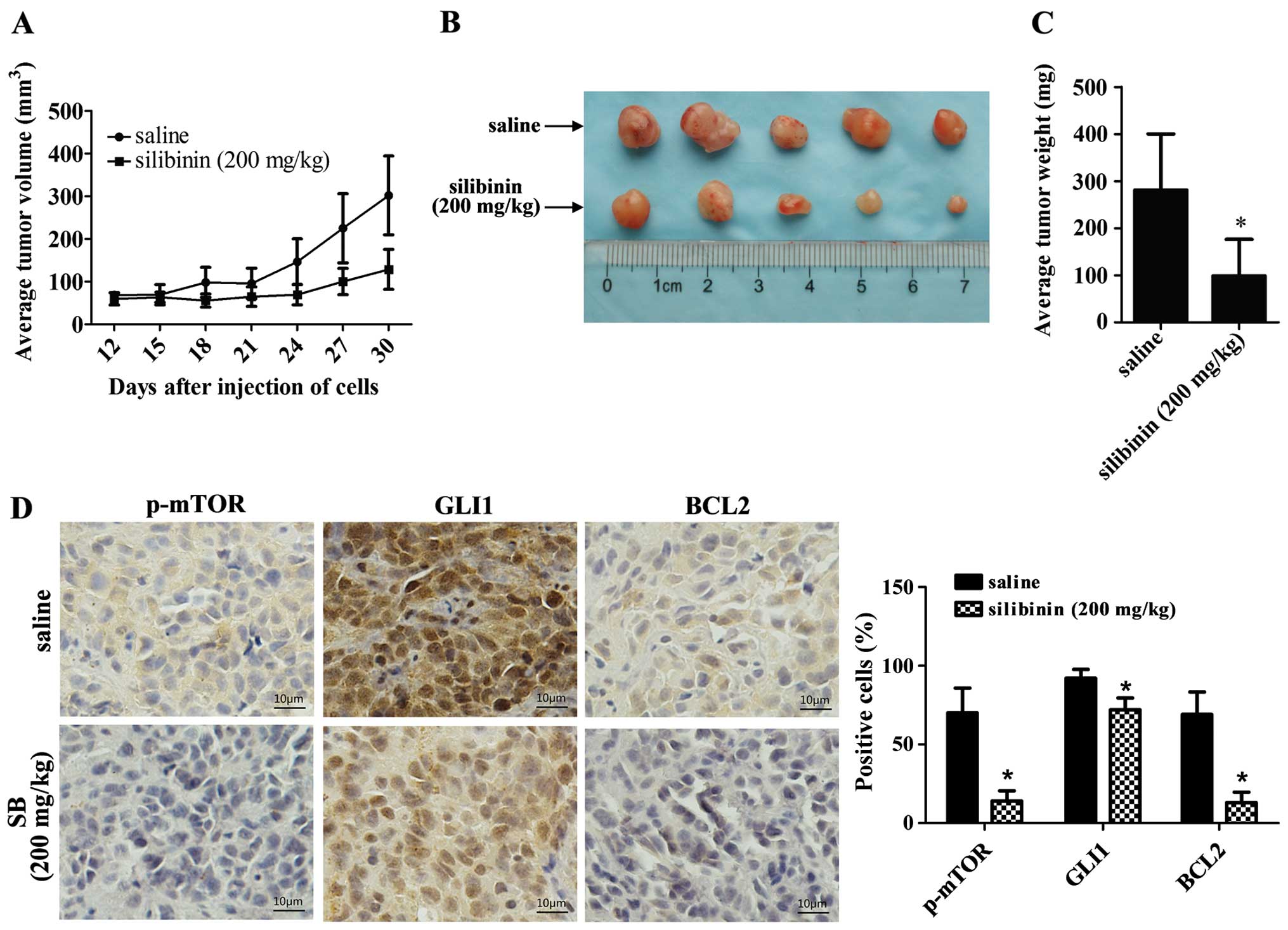
Silibinin inhibits the growth of RCC
xenografts in vivo
To further evaluate the anticancer effects of
silibinin in vivo, we chose 786-O cells, which have a high
potential for tumor formation, as a model system in vivo.
Silibinin (200 mg/kg) inhibited tumor growth compared with the
control group in vivo (Fig. 5a
and B). after 30 days, the average tumor weight of the
silibinin-treated group was 98.8±77.6 mg while the control group
was 281.22±118.87 mg (P<0.05) (Fig.
5C). Immunohistochemistry assay showed that the expression
levels of p-mTOR, GLI1 and BCL2 were decreased in the silibinin
(200 mg/kg) group compared with the saline group (control) in the
xenograft samples, which was consistent with the results observed
in vitro (Fig. 5D).
Discussion
There have been only a limited number of studies on
the apoptotic effects of silibinin on RCC cells. Our previous
studies showed the apoptotic and anti-metastatic effects of
silibinin on RCC cells, in which downregulated p-ERK was observed
after the treatment of silibinin (27,28).
However, in our present study, no significant change was detected
in p-ERK expression. Such disparate findings from our results could
be due to the difference in cell type used (we used RCC cell line
Caki-1 in our previous study, while 769-P, 786-O and OS-RC-2 cell
lines were used in our present study), implying that there may be
fundamental differences in the individual gene expression pattern
among various cell lines. Therefore, precautions should be taken
when extrapolating results from one to another study. More
recently, we reported the role of autophagy and the AMPK/mTOR
pathway in the silibinin-induced anti-metastatic effects on RCC
cells (29). as known, when cells
are subjected to adverse stress, such as nutrient starvation or
chemical agents, cancer cells may trigger an autophagic response
followed by degradation of unnecessary molecules or organelles to
prevent cells from death. On the other hand, treatment with many
anticancer reagents or ionizing radiation, has been shown to induce
autophagic cell death, directly leading to the inhibition of cancer
cell growth (30–32). Silibinin has been reported to induce
autophagic death in cervical and breast cancer (33,34).
Another study indicated that inhibition of autophagy with a
specific inhibitor enhanced cell death, suggesting a cytoprotective
function of autophagy in silibinin-treated cells (35). Thus, there is considerable interest
in elucidating the mechanisms of interplay between autophagy and
apoptosis induced by silibinin, which clearly requires further
research.
The Hh pathway was proven to play a crucial role in
pancreatic cancer tumorigenesis in early and late stages, and the
maintenance of Hh signaling is important for aberrant proliferation
and tumorigenesis (9). Our results
confirmed that GLI1 sustained the proliferation of RCC, and
inhibition of GLI1 by cyclopamine induced apoptosis by
downregulation of BCL2, which was consistent with previously
published results (12). We first
documented that silibinin effectively decreased the expression of
GLI1 in a dose-dependent manner and induced apoptosis by
downregulating the expression of BCL2 in RCC. Overexpression of
GLI1 could partially reverse the apoptosis induced by silibinin,
which implied that GLI1 is a regulator for the potential
therapeutic application of silibinin against mRCC.
As the downstream transcriptional factor of the Hh
pathway, GLI1 plays a key role in regulating cancer progression not
only in an SMO-dependent manner but also in an SMO-independent
manner. The canonical SMO-dependent pathway was found to be
activated by the binding of ligand HH to the membrane receptor PTC,
which triggered another membrane receptor SMO disassociated from
PTC and activated GLI1. However, Desch et al confirmed the
autonomous role of GLI in B-cell chronic lymphocytic leukemia (CLL)
cell apoptosis independent of SMO (36). The well known SMO-independent
pathway includes the akT pathway (37), MAPK/ERK pathway (38) and KRAS pathway (39). Wang et al found that
activation of the mTOR/S6K1 pathway could enhance GLI1
transcriptional activity and oncogenic function through
S6K1-mediated GLI1 phosphorylation at Ser84, which released GLI1
from its endogenous inhibitor SuFu (40). In order to explore the mechanisms
involved in the silibinin-induced GLI1 downregulation, we detected
p-ERK, p-akT and p-mTOR expression which have been reported to
regulate GLI1 expression after silibinin treatment in RCC. The
results showed that p-AKT and p-mTOR were downreg-ulated but p-eRk
had no significant change. Then we treated RCC cells with
inhibitors of PI3K (LY294002, 20 µM) and p-mTOR (rapamycin,
100 nM), followed by detection of GLI1 protein expression. However,
only the mTOR inhibitor decreased GLI1 expression in RCC.
Meanwhile, the results of the knockdown of mTOR by siRNA also
supported the conclusion, indicating that GLI1 is regulated by the
mTOR pathway in RCC. As known, mTOR has two existing complex forms
exerting various effects, mTORC1 (raptor) and mTORC2 (rictor).
mTORC1 is regulated by AKT whereas mTORC2 is the upstream of AKT.
Our results indicated that GLI1 might be potentially regulated by
mTORC2 but not mTORC1. However, the exact evidence remains to be
explored in further studies.
The activation of the mTOR pathway has been found in
RCC and is correlated with high grade and poor prognostic patient
features (41,42). Recently, the mTOR pathway has been
considered as one of the most important targets for mRCC therapy,
and mTOR antagonists rapamycin and three analogs of rapamycin
(temsirolimus, everolimus and deferolimus) have undergone clinical
evaluation as cancer therapeutics. It was reported that silibinin
could dephosphorylate mTOR and its effectors ribosomal protein S6
kinase (p70S6K) and eukaryotic initiation factor 4E-binding
protein-1 (4E-BP1) and suppress HIF-1α accumulation (43). Silibinin inhibited colon cancer
stem-like cell self-renewal and sphere formation by suppressing the
PP2Ac/AKT Ser473/mTOR pathway (44). Our data also supported that
silibinin could reduce the activation of the mTOR pathway in RCC.
However, the mechanisms that mediate the silibinin-regulated mTOR
pathway still need to be identified.
In conclusion, our in vitro and in
vivo results first demonstrated that silibinin-induced
apoptosis of RCC cells was mediated by the regulation of the
mTOR-GLI1-BCL2 pathway. This finding also implies that GLI1 is a
novel regulator mediated by silibinin for potential therapeutic
application in mRCC, providing a basis for future clinical trials
of silibinin for the treatment of patients with mRCC.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (NSFC no. 81072107 to Lei Li and NSFC
no. 81101936 to Jin Zeng) and China '863' program
(SS2014AA020607).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. Ca Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Motzer RJ, Bander NH and Nanus DM:
Renal-cell carcinoma. N Engl J Med. 335:865–875. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Johnsen JA and Hellsten S: Lymphatogenous
spread of renal cell carcinoma: An autopsy study. J Urol.
157:450–453. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fyfe G, Fisher RI, Rosenberg SA, Sznol M,
Parkinson DR and Louie AC: Results of treatment of 255 patients
with metastatic renal cell carcinoma who received high-dose
recombinant interleukin-2 therapy. J Clin Oncol. 13:688–696.
1995.PubMed/NCBI
|
|
5
|
Yang JC, Sherry RM, Steinberg SM, Topalian
SL, Schwartzentruber DJ, Hwu P, Seipp CA, Rogers-Freezer L, Morton
KE, White DE, et al: Randomized study of high-dose and low-dose
interleukin-2 in patients with metastatic renal cancer. J Clin
Oncol. 21:3127–3132. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McDermott DF, Regan MM, Clark JI, Flaherty
LE, Weiss GR, Logan TF, Kirkwood JM, Gordon MS, Sosman JA, Ernstoff
MS, et al: Randomized phase III trial of high-dose interleukin-2
versus subcutaneous interleukin-2 and interferon in patients with
metastatic renal cell carcinoma. J Clin Oncol. 23:133–141. 2005.
View Article : Google Scholar
|
|
7
|
Nüsslein-Volhard C and Wieschaus E:
Mutations affecting segment number and polarity in Drosophila.
Nature. 287:795–801. 1980. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ingham PW and McMahon AP: Hedgehog
signaling in animal development: Paradigms and principles. Genes
Dev. 15:3059–3087. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thayer SP, di Magliano MP, Heiser PW,
Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernández-del
Castillo C, Yajnik V, et al: Hedgehog is an early and late mediator
of pancreatic cancer tumorigenesis. Nature. 425:851–856. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sanchez P, Hernández AM, Stecca B, Kahler
AJ, DeGueme AM, Barrett A, Beyna M, Datta MW and Datta S:
Inhibition of prostate cancer proliferation by interference with
SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci USA.
101:12561–12566. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qualtrough D, Buda A, Gaffield W, Williams
AC and Paraskeva C: Hedgehog signalling in colorectal tumour cells:
Induction of apoptosis with cyclopamine treatment. Int J Cancer.
110:831–837. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Han Me, Lee YS, Baek SY, Kim BS, Kim JB
and Oh SO: Hedgehog signaling regulates the survival of gastric
cancer cells by regulating the expression of Bcl-2. Int J Mol Sci.
10:3033–3043. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hegde GV, Peterson KJ, Emanuel K, Mittal
AK, Joshi AD, Dickinson JD, Kollessery GJ, Bociek RG, Bierman P,
Vose JM, et al: Hedgehog-induced survival of B-cell chronic
lymphocytic leukemia cells in a stromal cell microenvironment: a
potential new therapeutic target. Mol Cancer Res. 6:1928–1936.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Von Hoff DD, LoRusso PM, Rudin CM, Reddy
JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, et
al: Inhibition of the hedgehog pathway in advanced basal-cell
carcinoma. N Engl J Med. 361:1164–1172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
LoRusso PM, Rudin CM, Reddy JC, Tibes R,
Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Chang I, Darbonne WC, et
al: Phase I trial of hedgehog pathway inhibitor vismodegib
(GDC-0449) in patients with refractory, locally advanced or
metastatic solid tumors. Clin Cancer Res. 17:2502–2511. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim DJ, Kim J, Spaunhurst K, Montoya J,
Khodosh R, Chandra K, Fu T, Gilliam A, Molgo M, Beachy PA, et al:
Open-label, exploratory phase II trial of oral itraconazole for the
treatment of basal cell carcinoma. J Clin Oncol. 32:745–751. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dormoy V, Danilin S, Lindner V, Thomas L,
Rothhut S, Coquard C, Helwig JJ, Jacqmin D, Lang H and Massfelder
T: The sonic hedgehog signaling pathway is reactivated in human
renal cell carcinoma and plays orchestral role in tumor growth. Mol
Cancer. 8:1232009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singh RP, Raina K, Deep G, Chan D and
Agarwal R: Silibinin suppresses growth of human prostate carcinoma
PC-3 orthotopic xenograft via activation of extracellular
signal-regulated kinase 1/2 and inhibition of signal transducers
and activators of transcription signaling. Clin Cancer Res.
15:613–621. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu KJ, Zeng J, Zhu GD, Zhang LL, Zhang D,
Li L, Fan JH, Wang XY and He DL: Silibinin inhibits prostate cancer
invasion, motility and migration by suppressing vimentin and MMP-2
expression. Acta Pharmacol Sin. 30:1162–1168. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu K, Ning Z, Zeng J, Fan J, Zhou J, Zhang
T, Zhang L, Chen Y, Gao Y, Wang B, et al: Silibinin inhibits
β-catenin/ZEB1 signaling and suppresses bladder cancer metastasis
via dual-blocking epithelial-mesenchymal transition and stemness.
Cell Signal. 25:2625–2633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zeng J, Sun Y, Wu K, Li L, Zhang G, Yang
Z, Wang Z, Zhang D, Xue Y, Chen Y, et al: Chemopreventive and
chemotherapeutic effects of intravesical silibinin against bladder
cancer by acting on mitochondria. Mol Cancer Ther. 10:104–116.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tyagi A, Singh RP, Ramasamy K, Raina K,
Redente EF, Dwyer-Nield LD, Radcliffe RA, Malkinson AM and Agarwal
R: Growth inhibition and regression of lung tumors by silibinin:
Modulation of angiogenesis by macrophage-associated cytokines and
nuclear factor-kappaB and signal transducers and activators of
transcription 3. Cancer Prev Res (Phila). 2:74–83. 2009. View Article : Google Scholar
|
|
23
|
Kaur M, Velmurugan B, Tyagi A, Deep G,
Katiyar S, Agarwal C and Agarwal R: Silibinin suppresses growth and
induces apoptotic death of human colorectal carcinoma LoVo cells in
culture and tumor xenograft. Mol Cancer Ther. 8:2366–2374. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Singh RP, Gu M and Agarwal R: Silibinin
inhibits colorectal cancer growth by inhibiting tumor cell
proliferation and angio-genesis. Cancer Res. 68:2043–2050. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li L, Zeng J, Gao Y and He D: Targeting
silibinin in the anti-proliferative pathway. Expert Opin Investig
Drugs. 19:243–255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheung CW, Taylor PJ, Kirkpatrick CM,
Vesey DA, Gobe GC, Winterford C, Nicol DL and Johnson DW:
Therapeutic value of orally administered silibinin in renal cell
carcinoma: Manipulation of insulin-like growth factor binding
protein-3 levels. BJU Int. 100:438–444. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li L, Gao Y, Zhang L, Zeng J, He D and Sun
Y: Silibinin inhibits cell growth and induces apoptosis by caspase
activation, down-regulating survivin and blocking EGFR-ERK
activation in renal cell carcinoma. Cancer Lett. 272:61–69. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liang L, Li L, Zeng J, Gao Y, Chen YL,
Wang ZQ, Wang XY, Chang LS and He D: Inhibitory effect of silibinin
on EGFR signal-induced renal cell carcinoma progression via
suppression of the EGFR/MMP-9 signaling pathway. Oncol Rep.
28:999–1005. 2012.PubMed/NCBI
|
|
29
|
Li F, Ma Z, Guan Z, Chen Y, Wu K, Guo P,
Wang X, He D and Zeng J: autophagy induction by silibinin
positively contributes to its anti-metastatic capacity via
AMPK/mTOR pathway in renal cell carcinoma. Int J Mol Sci.
16:8415–8429. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Green DR, Galluzzi L and Kroemer G: Cell
biology. Metabolic control of cell death Science.
345:12502562014.
|
|
31
|
Green DR and Levine B: To be or not to be?
How selective autophagy and cell death govern cell fate. Cell.
157:65–75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Navarro-Yepes J, Burns M, Anandhan A,
Khalimonchuk O, del Razo LM, Quintanilla-Vega B, Pappa A,
Panayiotidis MI and Franco R: Oxidative stress, redox signaling,
and autophagy: Cell death versus survival. Antioxid Redox Signal.
21:66–85. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fan S, Li L, Chen S, Yu Y, Qi M, Tashiro
S, Onodera S and Ikejima T: Silibinin induced-autophagic and
apoptotic death is associated with an increase in reactive oxygen
and nitrogen species in HeLa cells. Free Radic Res. 45:1307–1324.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zheng N, Zhang P, Huang H, Liu W, Hayashi
T, Zang L, Zhang Y, Liu L, Xia M, Tashiro SI, Onodera S and Ikejima
T: ERalpha down-regulation plays a key role in silibinin-induced
autophagy and apoptosis in human breast cancer MCF-7 cells. J
Pharmacol Sci. May 12–2015.epub ahead of print. View Article : Google Scholar
|
|
35
|
Kauntz H, Bousserouel S, Gossé F and Raul
F: Silibinin triggers apoptotic signaling pathways and autophagic
survival response in human colon adenocarcinoma cells and their
derived metastatic cells. Apoptosis. 16:1042–1053. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Desch P, Asslaber D, Kern D, Schnidar H,
Mangelberger D, Alinger B, Stoecher M, Hofbauer SW, Neureiter D,
Tinhofer I, et al: Inhibition of GLI, but not Smoothened, induces
apoptosis in chronic lymphocytic leukemia cells. Oncogene.
29:4885–4895. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stecca B, Mas C, Clement V, Zbinden M,
Correa R, Piguet V and Beermann F: Melanomas require HEDGEHOG-GLI
signaling regulated by interactions between GLI1 and the
RAS-MEK/AKT pathways. Proc Natl Acad Sci USA. 104:5895–5900. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Seto M, Ohta M, Asaoka Y, Ikenoue T, Tada
M, Miyabayashi K, Mohri D, Tanaka Y, Ijichi H, Tateishi K, et al:
Regulation of the hedgehog signaling by the mitogen-activated
protein kinase cascade in gastric cancer. Mol Carcinog. 48:703–712.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nolan-Stevaux O, Lau J, Truitt ML, Chu GC,
Hebrok M, Fernández-Zapico Me and Hanahan D: GLI1 is regulated
through Smoothened-independent mechanisms in neoplastic pancreatic
ducts and mediates PDAC cell survival and transformation. Genes
Dev. 23:24–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG,
Lang JY, Li CW, Hsu JL, Miller SA, Wang X, et al: The crosstalk of
mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 21:374–387. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Robb VA, Karbowniczek M, Klein-Szanto AJ
and Henske EP: activation of the mTOR signaling pathway in renal
clear cell carcinoma. J Urol. 177:346–352. 2007. View Article : Google Scholar
|
|
42
|
Pantuck AJ, Seligson DB, Klatte T, Yu H,
Leppert JT, Moore L, O'Toole T, Gibbons J, Belldegrun AS and Figlin
RA: Prognostic relevance of the mTOR pathway in renal cell
carcinoma: implications for molecular patient selection for
targeted therapy. Cancer. 109:2257–2267. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
García-Maceira P and Mateo J: Silibinin
inhibits hypoxia-inducible factor-1alpha and mTOR/p70S6K/4E-BP1
signalling pathway in human cervical and hepatoma cancer cells:
Implications for anticancer therapy. Oncogene. 28:313–324. 2009.
View Article : Google Scholar
|
|
44
|
Wang JY, Chang CC, Chiang CC, Chen WM and
Hung SC: Silibinin suppresses the maintenance of colorectal cancer
stem-like cells by inhibiting PP2A/AKT/mTOR pathways. J Cell
Biochem. 113:1733–1743. 2012.PubMed/NCBI
|