Introduction
Neuroblastoma (NB), one of the most common tumors in
childhood, accounts for ~10% of all pediatric cancers and 15% of
childhood cancer-related mortality (1). The survival outcomes of NB remain
unsatisfactory, although a variety of surgical techniques have been
developed in the past few decades. One of the clinical hallmarks of
NB is multidrug resistance (2), and
many patients, particularly those with malignant NB, often develop
chemoresistance (3). It is well
established that tumor cells are able to resist chemotherapeutic
agents through a variety of mechanisms, which includes enhancing
drug metabolism, altering the accumulation of medicine.
High mobility group box 1 (HMGB1), a conserved
non-histone nuclear protein, binds DNA and promotes the assembly of
proteins with a specific DNA target site (4). In addition to its role in
transcription, HMGB1 also functions in the cytoplasm as an
extracellular signaling protein during tumor progression. HMGB1 is
closely associated with each of the hallmarks of cancer, including
cell proliferation, ability to develop angiogenesis, evasion of
apoptosis, tissue invasion and metastasis and represents a
potential target in the therapy of various types of cancers
(5,6). It has been reported that HMGB1 is
highly expressed in hepatocellular carcinoma (7), colorectal cancer (8), lymphoma (9) and breast cancer (10). Increased expression of HMGB1 is
correlated with progression and poor prognosis in human
nasopharyngeal carcinoma (11) and
colorectal cancer (12). In
addition, targeting HMGB1 by RNA interference was found to inhibit
ovarian cancer growth and metastasis in vitro (13).
It has been widely reported that HMGB1 plays a role
in facilitating autophagy (14,15),
since it can disrupt the interaction between Beclin-1 and its
negative regulator Bcl-2 by competitively binding to Beclin-1
(16). Moreover, recent studies
suggest that HMGB1-mediated autophagy promotes chemoresistance in
osteosarcoma, lung adenocarcinoma and ovarian cancer (4,17,18).
These studies found that the level of HMGB1 protein increased after
anticancer agent treatment, and HMGB1 protein contributed to
inducing autophagy to evade apoptosis. Therefore, HMGB1 is a newly
identified gene associated with cancer growth and metastasis, as
well as tumor chemoresistance. Moreover, HMGB1-mediated autophagy
is considered as a potential marker of therapeutic effect and may
be used to predict clinical outcome. However, the mechanism and
significance of HMGB1-mediated autophagy in NB remain largely
unknown.
In the present study, we found that HMGB1 expression
levels, particularly in the cytoplasm, increased rapidly in
response to anticancer agents including doxorubicin (Dox),
cisplatin (Cis) and etoposide (eto). RNA interference-mediated
knockdown of HMGB1 restored the chemosensitivity of SH-SY5Y cells.
Overexpression of HMGB1 promoted cell growth and migration in
vitro and increased chemotherapy resistance. Furthermore, the
results revealed that HMGB1-overexpressing SH-SY5Y cells acquired
resistance to multidrug treatment through regulation of autophagy,
an intracellular self-defense mechanism known to confer drug
resistance. We found that HMGB1 promoted cell growth and migration
in vitro and increased chemotherapy resistance by
facilitating autophagic progression. Therefore, HMGB1 is a critical
factor in the development of chemoresistance and it offers a novel
target for improving the efficacy of NB therapy.
Materials and methods
Cell culture and regents
The NB SH-SY5Y cell line used in the experiments was
purchased from the Institute of Biochemistry and Cell Biology
(Shanghai, China). The cells were cultured in Dulbecco's modified
eagle's medium (DMEM) (Gibco, Life Technologies) supplemented with
10% fetal bovine serum (FBS; Gibco, Uruguay), 100 µg/ml of
penicillin and 100 µg/ml of streptomycin. They were cultured
in a humidified atmosphere containing 5% CO2 at 37°C.
Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA,
USA). 7′-Dichlorofluorescein diacetate (DCFH-DA), Cell Counting
Kit-8 (CCK-8) and the ECL-plus kit were purchased from Beyotime
(China); M-MLV reverse transcriptase was purchased from promega
(Madison, WI, USA); all antibodies were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). 3-Methyladenine (3-MA) was
purchased from Sigma (St. Louis, MO, USA).
GFP-LC3 transfection
The green fluorescent protein-LC3 (GFP-LC3) plasmid
was purchased from GeneChem (China). Cells were seeded at a density
of 9×105/ml on glass coverslips placed into 24-well
tissue culture plates (Corning Glass Works, Corning, NY, USA). On
the following day, the cells were transfected at 50–80% confluence
and transfected with the GFP-LC3 plasmid according to the
manufacturer's instructions. After 4 h, the medium was replaced by
DMEM containing 10% FBS, and the cells were left for another 24 h.
The stable cells were selected according to our previous study
(19).
CCK-8 assay for cell proliferation
Cells were plated in 96-well culture plates
(5×103 cells/well). At the indicated time, viable cell
numbers were determined by a cell proliferation assay using CCK-8.
The absorbance of optical densities at each time point was detected
by a microplate spectrophotometer at 450 nm.
Overexpression of HMGB1
The HMGB1 gene was amplified by polymerase chain
reaction (PCR) and was inserted into the Agel site of the
pGC-FU-3FLAG vector plasmid (Addgene, Cambridge, MA, USA). For
stable overexpression of HMGB1, HeK293T cells were plated in 75
cm2 culture flasks and transfected with 10 µg
lentivirus-HMGB1 vector (lenti-HMGB1) or lentivirus-GFP vectors
(lenti-GFP). The medium was changed the next day, and the viral
supernatant was harvested 48 h later. All medium containing viruses
were collected and passed through 0.45-µm syringe filters.
SH-SY5Y cells were incubated with the lentivirus supernatant for 24
h and selected with 2 µg/ml puromycin (Sigma) according to
the manufacturer's instructions.
Knockdown of HMGB1, Beclin-1 and
Atg5
Transfection with HMGB1-siRNA, Beclin-1-siRNA and
Atg5-siRNA (Sigma) was carried out by the Lipofectamine 2000
transfection reagent according to the manufacturer's
instructions.
Analysis of apoptosis by flow
cytometry
The incidence of apoptosis in NB cells was detected
using the Annexin V-FITC/PI apoptosis detection kit (BD Pharmingen,
USA) as previously described (20).
Apoptotic cells, including those staining positively for Annexin
V-FITC and negatively for PI and those that were double-positive,
were counted and represented as a percentage of the total cell
count.
Western blotting assay
The cells were extracted at the indicated time using
lysis buffer. Protease and phosphatase with whole-cell extracts
were prepared in RIPA buffer. Cell extracts were boiled for 10 min
in loading buffer and then equal amounts of cell extracts were
separated on 6–15% SDS-PAGE gels. Separated protein bands were
transferred to polyvinylidene fluoride (PVDF) membranes, and the
membranes were blocked in 5% skim milk powder. The primary
antibodies against HMGB1, Beclin-1, Atg5, cyclin E, cyclin B1 and
β-actin were diluted according to the instructions concerning the
antibodies and incubated overnight at 4°C. Subsequently,
horseradish peroxidase-linked secondary antibodies were incubated
at room temperature for 4 h at a dilution ratio of 1:1,000
according to the kit's instructions. The membranes were washed with
TBST for three times (5 min each time), and the immunoreactive
bands were visualized using the ECL-plus kit. The relative protein
level was normalized to β-actin concentration.
Autophagolysosome detection by
transmission electron microscopy
At the indicated time, the cells were fixed in 0.2%
glutaraldehyde in PBS (pH 7.4) for 2 h at room temperature,
post-fixed in 1% osmium tetroxide in water for 1 h, and then
stained in 2% uranyl acetate in water for 1 h in the dark. After
dehydration in an ascending series of ethanol, the samples were
embedded in Durcopan ACM for 6 h, and cut into 80-nm sections.
These sections were stained with uranyl acetate and lead citrate
and examined with a transmission electron microscope (Philips CM,
The Netherlands).
Measurements of intracellular ROS
To determine ROS generation within
H2O2-treated cells, flow cytometry was
performed. Cells were exposed to H2O2 for
different hours and then stained with 5 µg/ml of DCFH-DA for
30 min and subjected to flow cytometry and analyzed by CellQuest
software (Becton-Dickinson, San Jose, CA, USA) according to a
previously described method (21).
Wound-healing assay
A wound-healing assay was also performed to confirm
the influence of HMGB1 on SH-SY5Y cell migration. When the cells
transfected with lenti-HMGB1 or the lentivirus were grown to
confluency, a scratch in the cell monolayer was made with a cell
scratch spatula. After the cells were incubated under standard
conditions for 24 and 48 h, images of the scratches were captured
using a digital camera system coupled with a microscope.
Transwell migration assays
The invasion assays were performed using 24-well
Transwell chambers (8 µm; Corning). For the invasion assay,
tumor cells were resuspended in serum-free DMEM, and
2×105 cells were seeded into the upper chambers. DMEM
(0.5 ml) containing 10% FBS was added to the bottom chambers.
Following a 24-h incubation, cells on the upper surface of the
membrane were scrubbed off, and the migrated cells were fixed with
75% ethanol, stained with 0.1% crystal violet and counted under a
light microscope.
Statistical analysis
SPSS 13.0 was used for statistical analysis. One-way
analysis of variance (ANOVA) was used to analyze the differences
between groups. The LSD method of multiple comparisons was used
when the probability for ANOVA was statistically significant.
Statistical significance was set at P<0.05.
Results
Anticancer agents promote HMGB1
expression, and knock-down of HMGB1 increases sensitivity to
chemotherapy in NB cells
First, we assayed the effects of the anticancer
agents, Dox, Cis and Eto on the expression of HMGB1. These
anticancer agents significantly enhanced the expression of HMGB1 in
the SH-SY5Y cells (Fig. 1A).
Moreover, this effect was dose-dependent in the case of Dox
(Fig. 1B). In addition,
immunofluorescence indicated that in the absence of treatment,
HMGB1 was mainly located in the nucleus, with very low levels in
the cytoplasm. Treatment of anticancer agents markedly enhanced
total levels of HMGB1 in both the nucleus and cytoplasm (Fig. 1C).
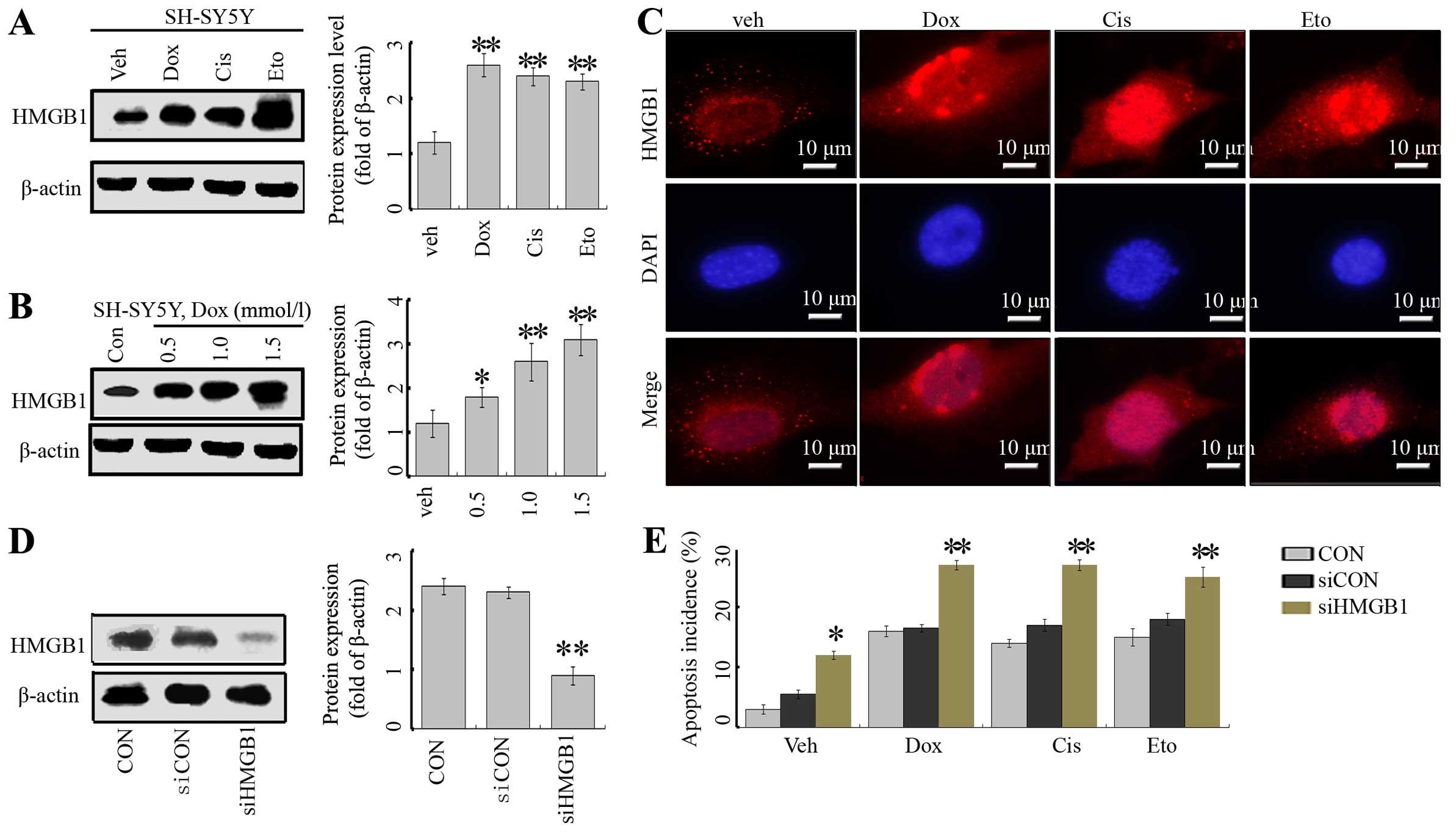 | Figure 1Anticancer agents induce HMGB1
expression in neuroblastoma cells. (A) SH-SY5Y cells were treated
with Dox (1 µM), Cis (20 µM), Eto (10 µM) or
vehicle (distilled water) for 24 h, and the HMGB1 protein level was
analyzed by western blotting (WB) (vs. untreated group). (B) The
cells were treated with Dox at concentrations of 0.5, 1 and 1.5
mmol/l, respectively, and the HMGB1 level was tested by WB assays.
(C) SY-SH5Y cells were treated with anticancer agents, and the
HMGB1 protein was detected by IF assays. (D) SY-SH5Y cells were
transfected with siCON or siHMGB1, and the level of HMGB1 protein
was suppressed in the siHMGB1 group. The cells without transfection
served as the control (CON). **P<0.01 vs. CON. (e)
Flow cytometric analysis of apoptosis incidence. Cells in the
siHMGB1, siCON and CON groups were treated with Dox (1 µM),
Cis (20 µM), Eto (10 µM) or vehicle (distilled water)
for 24 h, and the apoptosis was assessed by flow cytometry with
Annexin V/FITC staining. The results are the representative of
three identical experiments and the bars are the mean ± SD.
*P<0.05 vs. control, **P<0.01 vs.
control. |
To explore the potential role of HMGB1 in the
regulation of cell death in NB cells, a target-specific siRNA
against HMGB1 (siHMGB1) was transfected into SH-SY5Y cells. The
transfection inhibited HMGB1 expression, evidenced by a decrease in
the HMGB1 protein level (Fig. 1D).
The inhibition of HMGB1 rendered cells more sensitive to Dox-, Cis-
and Eto-induced cell injury, by assessment of the incidence of
apoptosis (Fig. 1E).
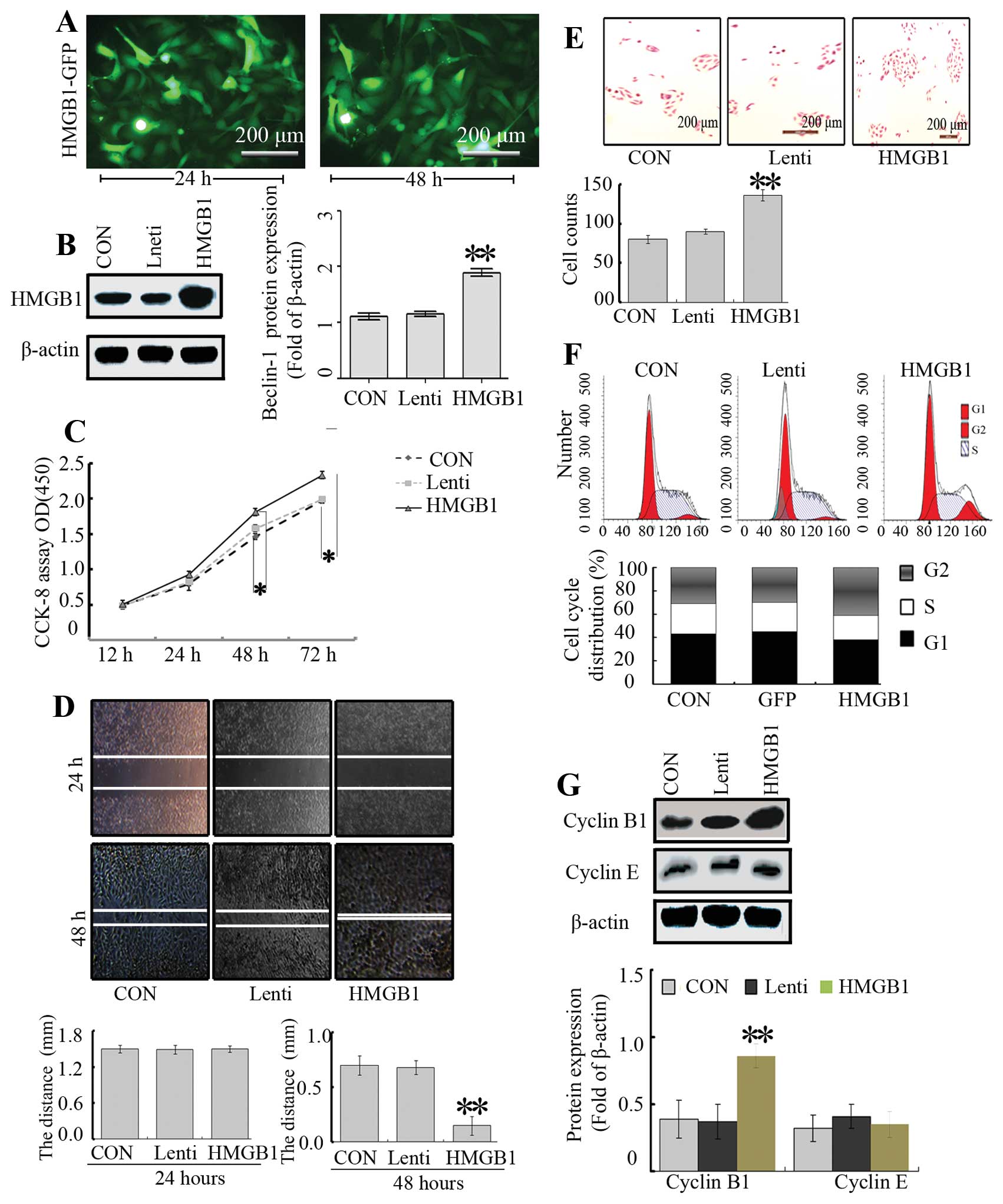
Effect of HMGB1 on NB cell proliferation,
invasion, metastasis and cell cycle distribution
To further explore the function of HMGB1 and its
mechanism, the HMGB1 gene was overexpressed by a lentivirus in the
SH-SY5Y cells. A lenti-virus vector containing the HMGB1 gene
(lenti-HMGB1) was transfected into SH-SY5Y cells, and the infection
efficiency was >90% as detected by fluorescence microscopy
(Fig. 2A). As expected, the HMGB1
expression was markedly enhanced after transfection at 48 h
(Fig. 2B). In order to test the
effect of HMGB1 overexpression on cell growth, we investigated the
proliferative activity of the cells by CCK-8. As shown in Fig. 2C, overexpression of HMGB1
intensified the SH-SY5Y cell growth, compared with the growth noted
in the Lenti and CON groups.
To determine the effect of HMGB1 on NB cell invasion
and migration, Transwell and wound-healing assays were carried out.
The migratory ability of the cells in the HMGB1 group was markedly
higher when compared with that in the CON and Lenti groups
(Fig. 2D). However, there were no
significant differences between the Lenti and CON groups.
Furthermore, a Transwell assay was performed to determine the
ability of cells to invade a matrix barrier, and the representative
micrographs of Transwell filters are presented in Fig. 2E. The invasive cell count
demonstrated that invasive potential was significantly increased in
the HMGB1 group relative to the CON and Lenti groups.
Next, to understand the effects of HMGB1 on the cell
cycle distribution of SH-SY5Y NB cells, flow cytometry was carried
out. Flow cytometry revealed that the proportion of cells in the
G2/M phase in the HMGB1 group was higher than that in the CON and
Lenti groups, while the percentage of cells in the S phase was not
altered (P<0.01, Fig. 2F).
Subsequently, cyclin B1 and cyclin E are responsible for cell cycle
progression in the G2 phase (22)
and thus were assessed by western blotting. The results showed that
overexpression of HMGB1 led to an increased level of cyclin B1, but
no significant changes in the levels of cyclin E were noted
(Fig. 2G), suggesting that HMGB1
modulates the cell cycle through regulation of cyclin B1.
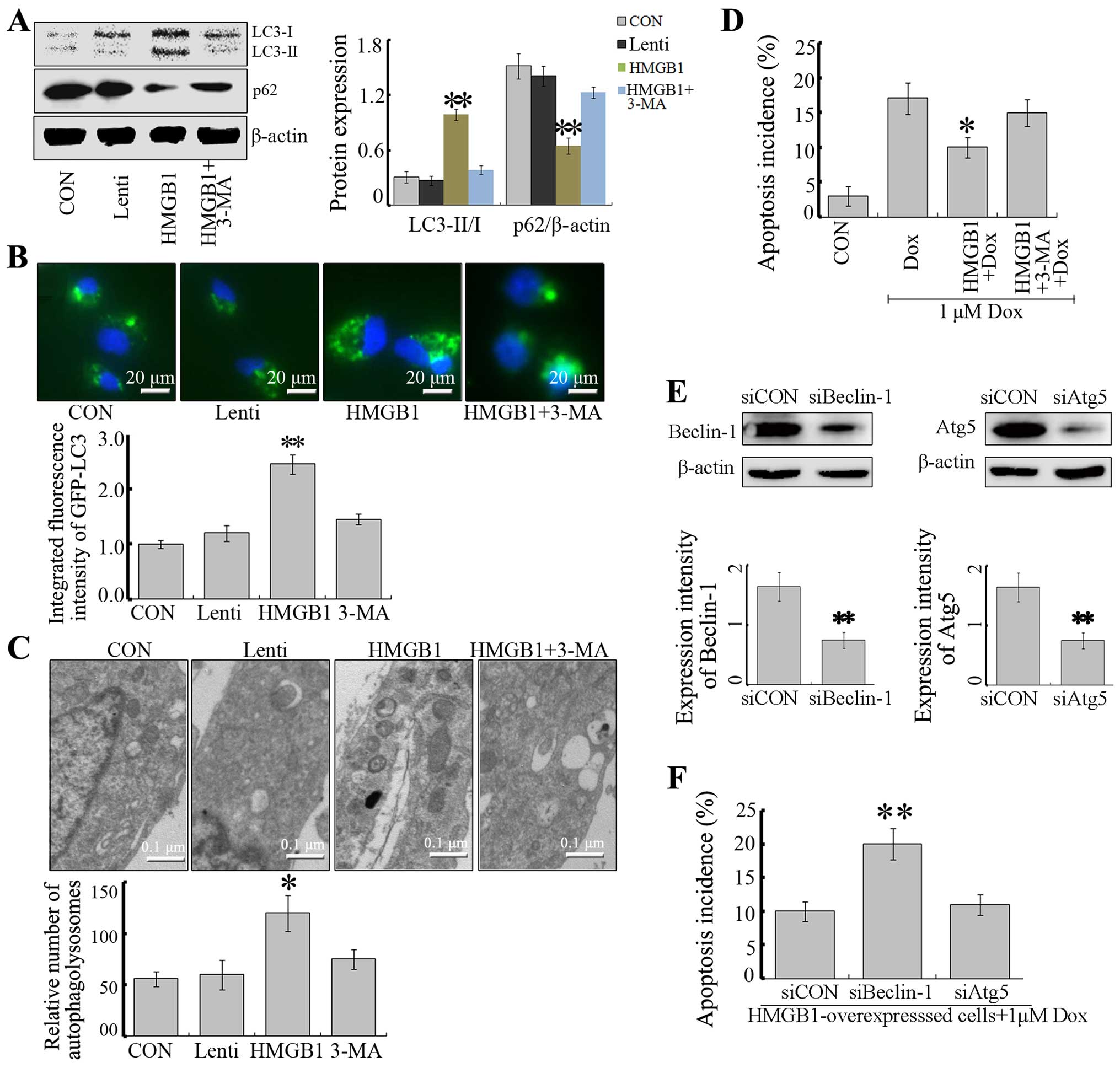
HMGB1 activates autophagy and reduces the
sensitivity to anticancer agents in vitro
Previous studies have reported that HMGB1-mediated
autophagy is a significant contributor to chemotherapy resistance
in several types of malignant tumors (4,17,18).
To investigate whether HMGB1 is a direct activator of autophagy
that protects SH-SY5Y cells from apoptosis, we further evaluated
the LC3-I to LC3-II conversion, p62 and GFP-LC3 puncta by
fluorescent analysis. Overexpression of HMGB1 increased the
appearance of LC3-II and the degradation of p62, promoting the
formation by GFP-LC3 (Fig. 3A and
B). However, this elevated flux of autophagy was abolished with
3-MA, an autophagy inhibitor (Fig.
3A). Consistently, fluorescence micrographs showed that the
puncta of GFP-LC3 in HMGB1-overexpressing NB cells could be partly
attenuated by 3-MA (Fig. 3A).
Moreover, ultrastructure analysis revealed that
HMGB1-overexpressing cells exhibited more autophagosomes compared
with this number in the control cells in the NB cells, and 3-MA
reduced the effect of HMGB1 (Fig.
3C). These data suggest that HMGB1 is a positive regulator of
autophagy in NB cells.
To clarify the role of HMGB1-mediated autophagy in
NB cells following chemotherapy, we analyzed the responses of
HMGB1-overexpressing NB cells to Dox treatment. Dox induced
apoptotic cell death to a great extent while overexpression of
HMGB1 partly reversed the effect of Dox (Fig. 3D). Next, to investigate the role of
HMGB1-mediated autophagy, autophagy was suppressed by 3-MA, an
autophagy inhibitor by inhibiting class III PI3K (23), and RNA interfering technique
targeted at Beclin-1 or Atg5 (Fig.
3E). As a result, treatment with 3-MA or Beclin-1 siRNA
reversed HMGB1-induced protection against Dox. Nevertheless,
blockade of autophagy by Atg5 siRNA failed to reverse HMGB1-induced
protection (Fig. 3F).
HMGB1 decreases Dox-induced oxidative
stress and cell death
Previous studies have demonstrated that Dox
treatment leads to an increase in intracellular reactive oxygen
species (ROS), oxidative stress injury, and cell death (24–26).
To detect the impact of HMGB1 on Dox-induced oxidative stress,
DCFH-DA probes were used to assess H2O2
levels. H2O2 generation was greatly increased
by Dox in the NB cells (Fig. 4A).
Importantly, HMGB1-overexpressing NB cells were less sensitive to
Dox, evidenced by a decrease in H2O2
generation following Dox treatment (Fig. 4A). Furthermore, HMGB1-inhibited
oxidative stress was partly reversed by the application of 3-MA or
Beclin-1 siRNA (Fig. 4B), but not
by Atg5 siRNA. Our data suggest that HMGB1 reduced oxidative stress
induced by Dox through regulation of Beclin-1-mediated
autophagy.
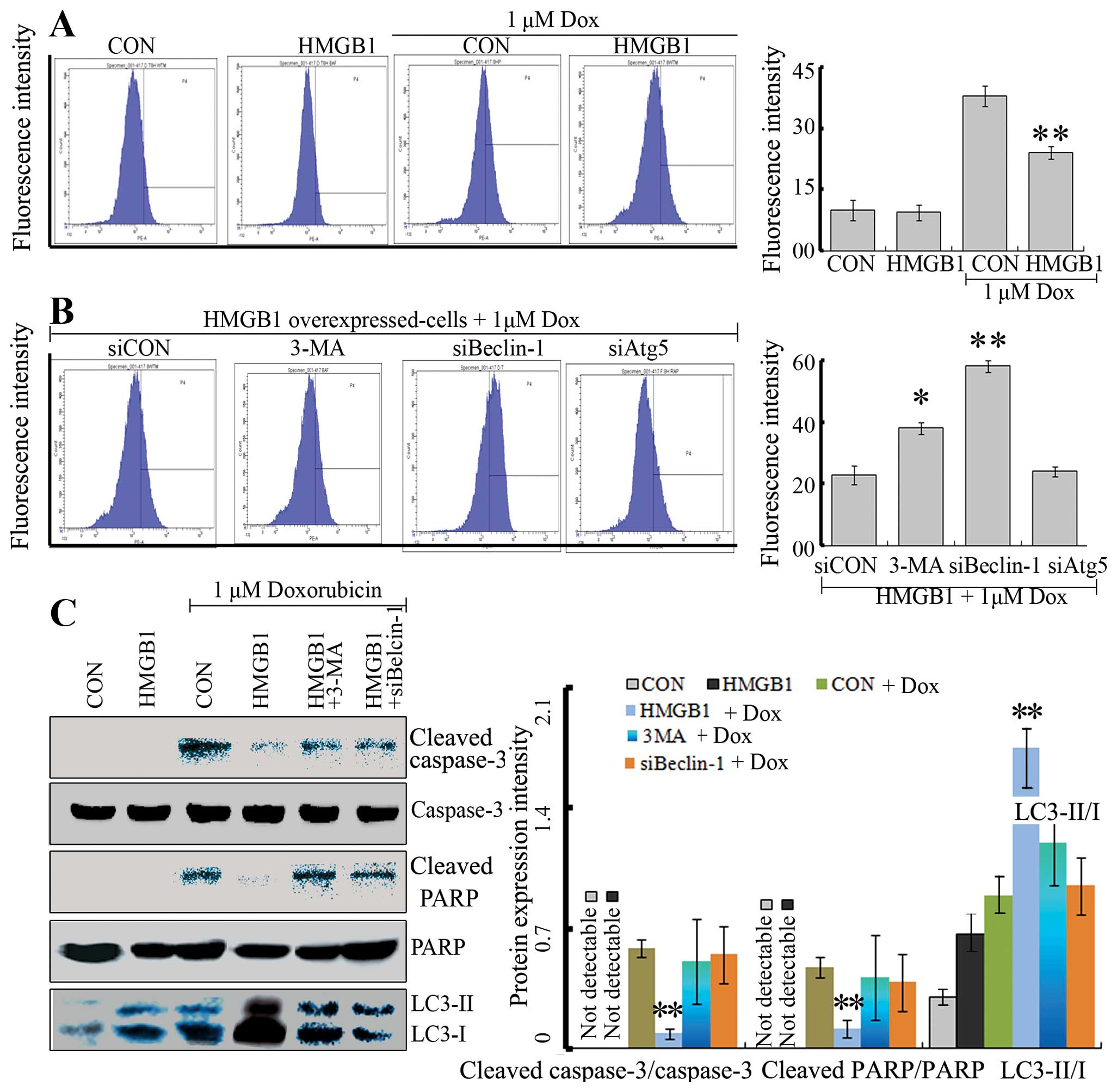 | Figure 4HMGB1 decreases Dox-induced oxidative
stress and cell death. (A) Flow cytometric assay of Dox-induced ROS
in SY-SH5Y cells transfected with HMGB1 or without. SY-SH5Y cells
(CON) and HMGB1-overexpressing cells were treated with Dox (1
µM) or vehicle for 24 h. Subsequently, the ROS level was
detected by flow cytometry. HMGB1 did not alter the basal ROS level
while it reduced the increased ROS level induced by Dox;
**P<0.05 vs. cells treated with cells. (B) Flow
cytometry for Dox-induced ROS in HMGB1-overexpressing cells
following inhibition of autophagy by pharmacological inhibitor
(3-MA) and gene interference (siBelcin-1 or siAtg5). The
HMGB1-overexpressing cells were treated with 5 mM 3-MA,
co-transfected with siBeclin-1 or siAtg5, and were then treated
with Dox (1 µM) for 24 h. Dox-induced ROS in
HMGB1-overexpressing cells was promoted by siBeclin-1 or 3-MA,
rather than Atg5; **P<0.05 vs. siCON. (C) Dox
treatment induced expression of cell death markers, and
HMGB1-mediated autophagy reduced the cell death. Cells as described
in A and B were treated with Dox (1 µM) or vehicle for 24 h
and then cleaved PARP, PARP, cleaved caspase-3, caspase-3 and
LC3-II/I protein levels were assessed by western blotting. The
cleaved PARP, cleaved caspase-3 and LC3-II expression levels were
normalized against PARP, caspase-3 and LC3-I, respectively. Dox
treatment led to an increase in cleaved PARP and cleaved caspase-3.
HMGB1 partly reversed the increase in cleaved PARP and cleaved
caspase-3 induced by Dox treatment. However, the protective effect
of HMGB1 against Dox was reversed when autophagy was inhibited by
pharmacological inhibitor (3-MA) ors gene interference
(siBeclin-1); **P<0.05 vs. CON cells treated with
Dox. |
In addition, cleaved PARP and cleaved caspase-3
facilitate cellular disassembly and serve as markers of cells
undergoing apoptosis. Thus, the levels of cleaved PARP and cleaved
caspase-3 were assessed by western blotting. Our results showed
that HMGB1 reduced cleaved PARP and cleaved caspase-3 induced by
Dox treatment. Moreover, both 3-MA and Beclin-1 siRNA effectively
reversed the effect of HMGB1. Consistently, these data demonstrated
that HMGB1 exerted a protective effect against oxidative
stress-mediated apoptosis by regulating autophagy.
Discussion
Neuroblastoma (NB) is one of the predominant tumors
that occurs mainly in children. Although a large number of studies
from basic research on oncogenes have been carried out, the
prognosis of patients, particularly for these with advanced NB,
remains poor. Most patients develop chemoresistance and metastatic
dissemination (27). The mechanisms
involved in drug resistance have not been clarified. Researchers
found that HMGB1 is expressed in brain cells and HMGB1 subcellular
localization changes during retinoic acid-induced differentiation
of P19 NB cells (28). Moreover,
HMGB1 protein is released by NB cells upon different stresses, such
as carbon dioxide, hypoxia and low pH (29–31).
Therefore, we investigated the role of HMGB1-mediated autophagy in
NB. We observed that anticancer agents promoted HMGB1 expression,
promoted cytosolic HMGB1 expression and the elevation of autophagic
activity, suggesting that cytosolic localization of HMGB1 may
correlate with autophagy induction. Consistently, cytosolic
translocation of HMGB1 as an origin of autophagy has been
demonstrated in other cell types under various cytotoxic stresses
(4,17,18).
Our results showed that the HMGB1 expression in NB cells was
increased, and knockdown of HMGB1 rendered them more sensitive to
anticancer agents.
In order to clarify the role of HMGB1 in NB cells,
the present study using a gain-of-function experiment that showed
that overexpression of HMGB1 significantly promoted the
proliferative activity and invasive potential of NB cells,
indicating that HMGB1 may play an important role in the development
and progression of NB, and may represent a potential therapeutic
target for the treatment of cancer. Consistent with previous
findings, overexpression of HMGB1 was associated with altered
hallmarks of autophagy, including LC3-II/I and p62 and we confirmed
that HMGB1 serves as a positive regulator of autophagy and possibly
mediates the resistance of anticancer agents. To confirm our
hypothesis, the apoptosis in HMGB1-overexpressing cells was induced
by Dox in the present study for its common clinical application
(32). HMGB1 reduced Dox-induced
oxidative stress injury and cell death. However, inhibition of
autophagy with 3-MA or knock-down of Beclin-1 reversed the effect
of HMGB1 and restored the cytotoxicity of Dox treatment, suggesting
that cytosolic HMGB1 caused autophagic activation, which resulted
in resistance to Dox. Consistent with our results, Mohan et
al showed that combination of LC3 short hairpin RNA plasmid
transfection inhibited autophagy and increased apoptosis induced by
rapamycin in human malignant NB SK-N-BE2 cells (33). These data suggest that HMGB1 is a
pro-survival protein, promoting cancer growth and development in NB
cells.
Next, we further investigated the mechanism of
HMGB1-mediated autophagy involved in NB cell death induced by Dox,
since autophagy is able to promote or inhibit apoptosis under
different stressors (19,34,35).
Autophagy in HMGB1-overexpressing cells was inhibited by a
pharmacological inhibitor (3-MA), siRNA Beclin-1 or siRNA Atg5,
respectively, and then the apoptosis and oxidative stress were
assessed after Dox treatment. As expected, apoptosis was increased
in the HMGB1-overexpressing cells treated with 3-MA or siRNA
Beclin-1 exposed to Dox, which is consistent with previous research
in several tumor cell lines (36–38),
that found decreased protein expression of Beclin-1. However,
knock-down of Atg5 failed to reverse the protective effect of HMGB1
against Dox treatment. These data suggest that HMGB1 exerts a
protective effect against oxidative stress-mediated apoptosis by
regulating Beclin-1-mediated autophagy.
To the best of our knowledge, this is the first
study to investigate the role and clinical significance of
HMGB1-mediated autophagy in NB. Yet, the use of NB cells from only
one cell line provides limited evidence. Further research using
more cell lines, xenograft models and primary tumors in vivo
are warranted to confirm our hypothesis. In conclusion, our
investigation revealed that HMGB1 promotes proliferative activity,
invasion and metastasis. Moreover, HMGB1-mediated autophagy exerts
a protective effect against oxidative stress-mediated apoptosis by
regulating Beclin-1-mediated autophagy.
References
|
1
|
Biedler JL: Drug resistance: Genotype
versus phenotype - thirty-second G. H. A. Clowes Memorial Award
Lecture. Cancer Res. 54:666–678. 1994.PubMed/NCBI
|
|
2
|
Michaelis M, Klassert D, Barth S, Suhan T,
Breitling R, Mayer B, Hinsch N, Doerr HW, Cinatl J and Cinatl J Jr:
Chemoresistance acquisition induces a global shift of expression of
aniogenesis-associated genes and increased pro-angogenic activity
in neuroblastoma cells. Mol Cancer. 8:802009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Furfaro AL, Piras S, Passalacqua M,
Domenicotti C, Parodi A, Fenoglio D, Pronzato MA, Marinari UM,
Moretta L, Traverso N, et al: HO-1 up-regulation: A key point in
high-risk neuroblastoma resistance to bortezomib. Biochim Biophys
Acta. 1842:613–622. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Y, Cheng Y, Ren X, Zhang L, Yap KL,
Wu H, Patel R, Liu D, Qin ZH, Shih IM, et al: NAC1 modulates
sensitivity of ovarian cancer cells to cisplatin by altering the
HMGB1-mediated autophagic response. Oncogene. 31:1055–1064. 2012.
View Article : Google Scholar :
|
|
5
|
Yang S, Xu L, Yang T and Wang F:
High-mobility group box-1 and its role in angiogenesis. J Leukoc
Biol. 95:563–574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tang D, Kang R, Cheh CW, Livesey KM, Liang
X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et
al: HMGB1 release and redox regulates autophagy and apoptosis in
cancer cells. Oncogene. 29:5299–5310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang L, Han J, Wu H, Liang X, Zhang J, Li
J, Xie L, Xie Y, Sheng X and Yu J: The association of HMGB1
expression with clinicopathological significance and prognosis in
hepatocellular carcinoma: A meta-analysis and literature review.
PLoS One. 9:e1106262014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Süren D, Yıldırım M, Demirpençe Ö, Kaya V,
Alikanoğlu AS, Bülbüller N, Yıldız M and Sezer C: The role of high
mobility group box 1 (HMGB1) in colorectal cancer. Med Sci Monit.
20:530–537. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meyer A, Staratschek-Jox A, Springwald A,
Wenk H, Wolf J, Wickenhauser C and Bullerdiek J: Non-Hodgkin
lymphoma expressing high levels of the danger-signalling protein
HMGB1. Leuk Lymphoma. 49:1184–1189. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brezniceanu ML, Völp K, Bösser S, Solbach
C, Lichter P, Joos S and Zörnig M: HMGB1 inhibits cell death in
yeast and mammalian cells and is abundantly expressed in human
breast carcinoma. FASEB J. 17:1295–1297. 2003.PubMed/NCBI
|
|
11
|
Wu D, Ding Y, Wang S, Zhang Q and Liu L:
Increased expression of high mobility group box 1 (HMGB1) is
associated with progression and poor prognosis in human
nasopharyngeal carcinoma. J Pathol. 216:167–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ueda M, Takahashi Y, Shinden Y, Sakimura
S, Hirata H, Uchi R, Takano Y, Kurashige J, Iguchi T, Eguchi H, et
al: Prognostic significance of high mobility group box 1 (HMGB1)
expression in patients with colorectal cancer. Anticancer Res.
34:5357–5362. 2014.PubMed/NCBI
|
|
13
|
Chen J, Liu X, Zhang J and Zhao Y:
Targeting HMGB1 inhibits ovarian cancer growth and metastasis by
lentivirus-mediated RNA interference. J Cell Physiol.
227:3629–3638. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fu LL, Cheng Y and Liu B: Beclin-1:
Autophagic regulator and therapeutic target in cancer. Int J
Biochem Cell Biol. 45:921–924. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang D, Kang R, Livesey KM, Cheh CW,
Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ III,
et al: Endogenous HMGB1 regulates autophagy. J Cell Biol.
190:881–892. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang D, Kang R, Livesey KM, Kroemer G,
Billiar TR, Van Houten B, Zeh HJ III and Lotze MT: High-mobility
group box 1 is essential for mitochondrial quality control. Cell
Metab. 13:701–711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R,
Vernon P, Cao L and Tang D: HMGB1 promotes drug resistance in
osteosarcoma. Cancer Res. 72:230–238. 2012. View Article : Google Scholar
|
|
18
|
Pan B, Chen D, Huang J, Wang R, Feng B,
Song H and Chen L: HMGB1-mediated autophagy promotes docetaxel
resistance in human lung adenocarcinoma. Mol Cancer. 13:1652014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang YH, Chen K, Li B, Chen JW, Zheng XF,
Wang YR, Jiang SD and Jiang LS: Estradiol inhibits osteoblast
apoptosis via promotion of autophagy through the ER-ERK-mTOR
pathway. Apoptosis. 18:1363–1375. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang L, Zhang H, Qian J, Wang K and Zhu J:
Interleukin-10 blocks in vitro replication of human cytomegalovirus
by inhibiting the virus-induced autophagy in MRC5 cells. Biochem
Biophys Res Commun. 448:448–453. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang YH, Li B, Zheng XF, Chen JW, Chen K,
Jiang SD and Jiang LS: Oxidative damage to osteoblasts can be
alleviated by early autophagy through the endoplasmic reticulum
stress pathway - implications for the treatment of osteoporosis.
Free Radic Biol Med. 77:10–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Murai T, Nakagawa Y, Maeda H and Terada K:
Altered regulation of cell cycle machinery involved in
interleukin-1-induced G1 and G2 phase growth
arrest of A375S2 human melanoma cells. J Biol Chem. 276:6797–6806.
2001. View Article : Google Scholar
|
|
23
|
Petiot A, Ogier-Denis E, Blommaart EF,
Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol
3′-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem. 275:992–998. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moreira AC, Branco AF, Sampaio SF,
Cunha-Oliveira T, Martins TR, Holy J, Oliveira PJ and Sardão VA:
Mitochondrial apoptosis-inducing factor is involved in
doxorubicin-induced toxicity on H9c2 cardiomyoblasts. Biochim
Biophys Acta. 1842:2468–2478. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Singla S, Kumar NR and Kaur J: In vivo
studies on the protective effect of propolis on doxorubicin-induced
toxicity in liver of male rats. Toxicol Int. 21:191–195. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shokoohinia Y, Hosseinzadeh L, Moieni-Arya
M, Mostafaie A and Mohammadi-Motlagh HR: Osthole attenuates
doxorubicin-induced apoptosis in PC12 cells through inhibition of
mitochondrial dysfunction and ROS production. Biomed Res Int.
2014:1568482014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tonini GP and Pistoia V: Molecularly
guided therapy of neuroblastoma: A review of different approaches.
Curr pharm Des. 12:2303–2317. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guazzi S, Strangio A, Franzi AT and
Bianchi ME: HMGB1, an architectural chromatin protein and
extracellular signalling factor, has a spatially and temporally
restricted expression pattern in mouse brain. Gene Expr Patterns.
3:29–33. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Faraco G, Fossati S, Bianchi ME, Patrone
M, Pedrazzi M, Sparatore B, Moroni F and Chiarugi A: High mobility
group box 1 protein is released by neural cells upon different
stresses and worsens ischemic neurodegeneration in vitro and in
vivo. J Neurochem. 103:590–603. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reismann M, Wehrmann F, Schukfeh N,
Kuebler JF, Ure B and Glüer S: Carbon dioxide, hypoxia and low pH
lead to overexpression of c-myc and HMGB-1 oncogenes in
neuroblastoma cells. Eur J Pediatr Surg. 19:224–227. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pedrazzi M, Averna M, Sparatore B, Patrone
M, Salamino F, Marcoli M, Maura G, Cervetto C, Frattaroli D,
Pontremoli S, et al: Potentiation of NMDA receptor-dependent cell
responses by extracellular high mobility group box 1 protein. PLoS
One. 7:e445182012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wittig JC, Bickels J, Priebat D, Jelinek
J, Kellar-Graney K, Shmookler B and Malawer MM: Osteosarcoma: A
multi-disciplinary approach to diagnosis and treatment. Am Fam
Physician. 65:1123–1132. 2002.PubMed/NCBI
|
|
33
|
Mohan N, Chakrabarti M, Banik NL and Ray
SK: Combination of LC3 shRNA plasmid transfection and genistein
treatment inhibited autophagy and increased apoptosis in malignant
neuroblastoma in cell culture and animal models. PLoS One.
8:e789582013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lockshin RA and Zakeri Z: Apoptosis,
autophagy, and more. Int J Biochem Cell Biol. 36:2405–2419. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao Z, Tao L, Shen C, Liu B, Yang Z and
Tao H: Silencing of Barkor/ATG14 sensitizes osteosarcoma cells to
cisplatin-induced apoptosis. Int J Mol Med. 33:271–276. 2014.
|
|
37
|
Lanvers-Kaminsky C, Winter B, Koling S,
Frodermann B, Braun Y, Schaefer KL, Diallo R, Koenemann S, Wai D,
Willich N, et al: Doxorubicin modulates telomerase activity in
Ewing's sarcoma in vitro and in vivo. Oncol Rep. 14:751–758.
2005.PubMed/NCBI
|
|
38
|
Zhou Y, Sun K, Ma Y, Yang H, Zhang Y, Kong
X and Wei L: Autophagy inhibits chemotherapy-induced apoptosis
through downregulating Bad and Bim in hepatocellular carcinoma
cells. Sci Rep. 4:53822014.PubMed/NCBI
|