Introduction
Ovarian cancer is a common cancer of the female
reproductive organs, and is associated with the highest death rate
among all gynecological cancers (1). Cisplatin chemotherapy and adjuvant
therapy are common in ovarian cancer, and drug resistance is a
major cause of death (1–3). Cisplatin resistance is mostly a
secondary effect, and its mechanism is unclear. Cisplatin
resistance may be associated with the altered regulation of
multiple signaling pathways, including downregulation of apoptotic
signals and activation of pro-survival signals (3–5).
Recent studies have shown that cisplatin can induce endoplasmic
reticulum (ER) stress-associated apoptosis, and that ER stress
tolerance may be involved in cisplatin resistance (6–9).
A variety of physiological and pathological
conditions, including viral infection, hypoxia, oxidative damage
and antitumor therapy, can induce ER stress (10,11).
ER stress can activate ER stress-related proapoptotic molecules
such as growth arrest and DNA damage-inducible transcript 3 (DDIT3)
(also known as CHOP) and caspase-12, and induce the expression and
activation of pro-survival molecules such as GADD34 and 78 kDa
glucose-regulated protein (Grp-78). The balance between these
processes determines cellular fate, i.e., adaptation or apoptosis
(11–13). ER-mediated apoptosis involves at
least two mechanisms: the unfolded protein response (UPR) and
calcium signaling (12).
Calcium ions can function as mitogenic or
proapoptotic messengers, depending on their intracellular location
and cytoplasmic concentration (14,15).
The storage, release and uptake of all non-muscle cell calcium ions
are subject to ER regulation. Disrupted ER calcium homeostasis can
induce cellular apoptosis. There is a crosstalk between the UPR and
ER calcium signaling (16).
Excessive ER stress may synergize with mitochondrial cytochrome
c release, leading to caspase activation and apoptosis
(17). Close contact between the ER
and mitochondria membranes enables the exchange of lipid, calcium
ions and glycosylated proteins (18–20).
ER inositol trisphosphate receptor (IP3R) channel opening has a
destabilizing role on mitochondrial calcium balance, and ER
Ca2+ channel activity regulates cellular susceptibility
to ER stress (21,22). However, it is unclear whether the ER
stress-mediated apoptosis induced by cisplatin regulation of the
UPR and calcium signaling are involved in ovarian cancer drug
resistance.
In the present study, we found that cisplatin
resistance in ovarian carcinoma is linked to ER stress tolerance.
The intensity of ER stress induced by cisplatin was weaker in
cisplatin-resistant SKOV3/DDP ovarian cancer cells than in the
parental cisplatin-sensitive SKOV3 cells. Both ER stress-mediated
apoptosis and mitochondrial pathway-mediated apoptosis were induced
in the cisplatin-sensitive SKOV3 cells, but not in the
cisplatin-resistant SKOV3/DDP cells. Cisplatin-induced
Ca2+ flow from the ER to mitochondria led to
mitochondrial calcium overload, which amplified proapoptotic
signaling in the cisplatin-sensitive SKOV3 cells. Tolerance to
cisplatin-induced ER stress involved the preservation of ER and
mitochondria calcium homeostasis, which maintained cell survival
and led to cisplatin resistance in ovarian cancer cells.
Materials and methods
Cell culture
The cisplatin-sensitive human ovarian cancer cell
line SKOV3 and the cisplatin-resistant clone SKOV3/DDP were
obtained from the Chinese Academy of Medical Sciences and the
Peking Union Medical College, respectively. Cells were cultured at
37°C with 5% CO2 in RPMI-1640 medium (Gibco, Carlsbad,
CA, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen,
Carlsbad, CA, USA). SKOV3/DDP cells were maintained in the same
medium containing 1 µg/ml cisplatin (Sigma, St. Louis, MO,
USA) to maintain the multidrug-resistant phenotype.
Cell viability assays
Cell viability was determined by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Exponentially growing cells were seeded into 96-well culture
plates in 100 µl medium at a density of 1×104
cells/well. After 16–24 h, varying concentrations of cisplatin were
added to quadruplicate wells and cells were incubated for 24 or 48
h. For the MTT assays (Beyotime, Shanghai, China), 20
µl/well MTT [5 mg/ml in phosphate-buffered saline (PBS)] was
added to the cells and incubated for 4 h. Dimethylsulfoxide (150
µl/well; Beijing Chemical Industry, Beijing, China) was then
added, the plates were shaken at room temperature for 10 min, and
the absorbance was measured at a 570-nm wavelength using a
microplate reader (BioTek Instruments, Winooski, VT, USA).
Immunofluorescence staining and confocal
laser microscopy
Cells were seeded onto coverslips into 24-well
plates at a density of 5×104 cells/well and allowed to
recover overnight. After treatment with 6 µg/ml cisplatin
for 0 or 24 h, the cells were fixed with 4% paraformaldehyde,
stained with the Hoechst 33342 (2 µg/ml; Sigma) nuclear
stain for 30 min, and washed in PBS; chromatin condensation was
examined by an Olympus FV1000 confocal laser microscope (Olympus,
Tokyo, Japan). Expression of Grp78, protein disulfide isomerase
(PDI) and active caspase-3, and PDI colocalization with
voltage-dependent anion-selective channel protein 1 (VDAC1), IP3R
and mitochondria were examined by indirect immunofluorescence. For
this, the cells were cultured on coverslips overnight, treated with
6 µg/ml cisplatin for different time periods, and then
rinsed three times with PBS. The cells were then fixed with 4%
paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100
for 5 min, blocked with 5% bovine serum albumin, and incubated with
a primary antibody (anti-Grp78, anti-PDI, anti-active caspase-3,
anti-VDAC1, anti-IP3R; all used at 1:100 dilution; anti-Grp78,
anti-PDI and anti-active caspase-3 were obtained from Santa Cruz
Biotechnology, Santa Cruz, CA, USA; anti-VDAC1 and anti-IP3R were
obtained from Abcam, Hong Kong; and MitoTracker Red was obtained
from Invitrogen) overnight at 4°C. The next day, coverslips were
incubated with the Alexa Fluor 488/546-conjugated secondary
antibody (1:400 dilution; Molecular Probes, Eugene, OR, USA) for 1
h, and stained with Hoechst 33342 (2 µg/ml) for 2 min,
followed by three washes in PBS. After mounting onto slides, the
cells were examined using an Olympus FV1000 confocal laser
microscope.
Flow cytometric analysis
The Muse™ Annexin V Dead Cell kit (EMD Millipore
Corporation, Hayward, CA, USA) was used to monitor cell death.
Exponentially-growing SKOV3 and SKOV3/DDP cells were seeded into
6-well culture plates at a density of 2×105 cells/well.
Following exposure to experimental conditions, the cells were
trypsinized and resuspended in RPMI-1640 medium with 10% FBS at a
concentration of 1×106 cells/ml. Cells were incubated
with Annexin V and Dead Cell reagent in a darkroom at room
temperature for 20 min. Finally, the samples were measured using
the Muse™ Cell Analyzer (EMD Millipore Corporation). All
experiments were performed in triplicate.
Western blot analysis
Whole cell protein extracts were prepared using cell
lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM
Na2EDTA, 1 mM EDTA, 1% Triton, 2.5 mM sodium
pyrophosphate, 1 mM β-glycerophosphate, 1 mM
Na3VO4, 1 mM NaF, 1 µg/ml leupeptin
and 1 mM PMSF) for western blotting. The protein concentration was
quantified using a Bradford protein assay kit (Bio-Rad, Hercules,
CA, USA). For cytoplasmic protein extraction, the cells were
harvested, washed with ice-cold PBS and sedimented for 5 min at 600
× g at 4°C. Cells were then incubated in cell lysis buffer (150 mM
NaCl, 1 mM EDTA, 10 mM HEPES, 1 mM PMSF and 0.6% NP-40). Cell
lysates were sonicated, incubated for 15 min on ice, and then
clarified at 700 × g for 10 min at 4°C. The supernatant was
centrifuged at 14,000 × g for another 30 min at 4°C; cytoplasmic
proteins were present in the supernatant. For western blot
analysis, lysate proteins (30–50 µg) were resolved by 8, 10
or 15% SDS-polyacrylamide gel electrophoresis and transferred onto
Immobilon-P membranes (EMD Millipore, Billerica, MA, USA).
Membranes were then blocked with 5% non-fat dry milk in buffer (10
mM Tris-HCl, pH 7.6, 100 mM NaCl and 0.1% Tween-20) for 1 h at room
temperature and incubated with the relevant primary antibody
overnight at 4°C. Anti-Grp78 and anti-PDI antibodies (used at 1:200
dilution) were obtained from Santa Cruz Biotechnology. Anti-ATF4,
DDIT3/CHOP, anti-cytochrome c, anti-caspase-3 and
anti-caspase-4 (used at 1:1,000 dilution) and CTR1 were obtained
from Abcam (Cambridge, UK). Anti-β-actin antibody (1:1,000
dilution) was obtained from ProteinTech Group (Chicago, IL, USA).
Membranes were then incubated with horseradish
peroxidase-conjugated secondary antibody (Thermo Fisher Scientific,
Waltham, MA, USA) at 1:2,000 dilution for 1 h at room temperature.
Immunodetection was performed using enhanced chemiluminescence
reagents (Thermo Scientific, Rockford, IL, USA) and images were
captured using a Syngene Bio Imaging System (Synoptics, Cambridge,
UK). Specific proteins were quantified by densitometry using
Quantity One software (Bio-Rad Laboratories), normalized to actin,
and presented as the mean ± SD of three independent
experiments.
Calcium concentration analysis
The Ca2+-sensitive fluorescent dyes
Fluo-4/AM (Molecular Probes) and Rhod-2/AM (AAT Bioquest,
Sunnyvale, CA, USA) were used to measure the Ca2+
concentration according to the manufacturer's instructions. Before
exposure to different experimental conditions, the cells were
incubated with Fluo-4/AM or Rhod-2/AM for 30 min at 37°C. Cell
samples were then analyzed by confocal laser microscopy. All
experiments were performed in triplicate.
JC-1 staining
The mitochondrial membrane potential was examined by
JC-1 staining kit (Beyotime Institute of Biotechnology, China).
After cisplatin treatment, the cells were incubated with a JC-1
working solution at 37°C in the dark for 20 min and then observed
by confocal microscopy. In healthy cells, where mitochondrial
membranes remain depolarized, JC-1 forms complexes of J aggregates,
which are seen as punctate red fluorescence at the 590-nm emission
wavelength; however, in apoptotic cells, JC-1 remains in the
monomeric form, and is seen as diffused green fluorescence at the
530-nm emission wavelength.
Statistical analysis
Experiments were performed at least three times, and
data are presented as means ± SD. Data analysis was performed using
one-way ANOVA. Tukey's post-hoc test was used to determine the
statistical significance in all pairwise comparisons of interest.
P<0.05 was considered to represent a statistically significant
difference.
Results
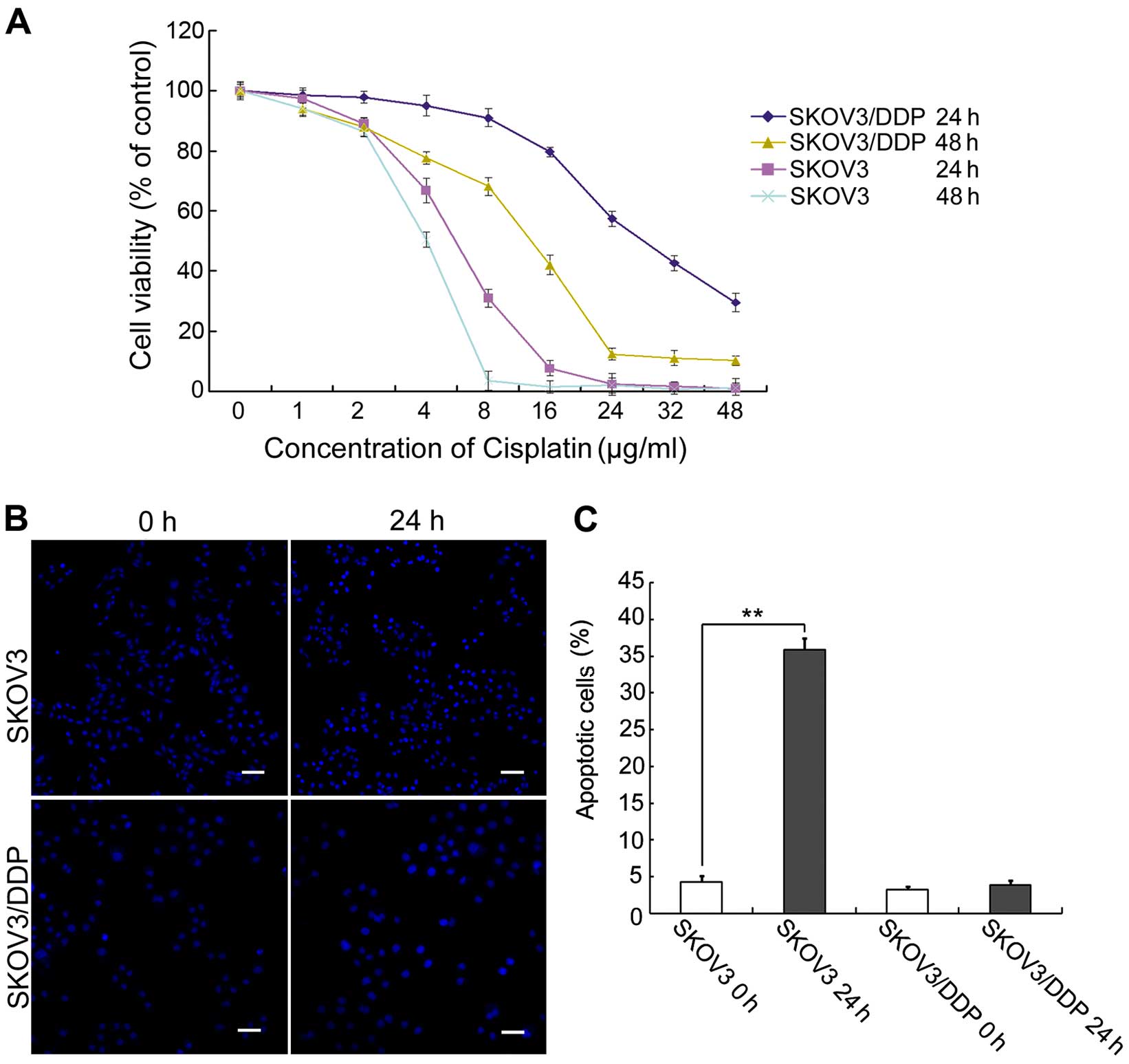
Cisplatin inhibits proliferation and
induces apoptosis in the ovarian cancer cells
We compared cisplatin sensitivity in
cisplatin-sensitive SKOV3 cells and cisplatin-resistant SKOV3/DDP
cells. Both cell types were treated with increasing doses of
cisplatin for 24 or 48 h, and then growth inhibition was examined
using MTT assays. We found that cisplatin inhibited the
proliferation and/or survival of both ovarian cancer cell lines
(Fig. 1A). This result confirmed
that cisplatin-sensitive SKOV3 cells were more sensitive to
cisplatin than cisplatin-resistant SKOV3/DDP cells.
Based on the results of the MTT assay and previous
studies (9), both cell lines were
treated with 6 µg/ml cisplatin for 0 or 24 h, and apoptotic
chromatin condensation was examined by Hoechst 33342 staining
(Fig. 1B). Cisplatin-induced
apoptotic chromatin condensation was clearly observed in the SKOV3
cells, but not in the SKOV3/DDP cells. We also determined the
apoptotic ratio bya quantitative analysis. Cisplatin increased the
apoptotic ratio from 4.3% (at 0 h) to 36.4% (at 24 h) in the SKOV3
cells. In contrast, there were no obvious changes in the SKOV3/DDP
cells over this period (Fig.
1C).
These results indicate that cisplatin can
efficiently induce apoptosis in SKOV3 cells, but not in SKOV3/DDP
cells.
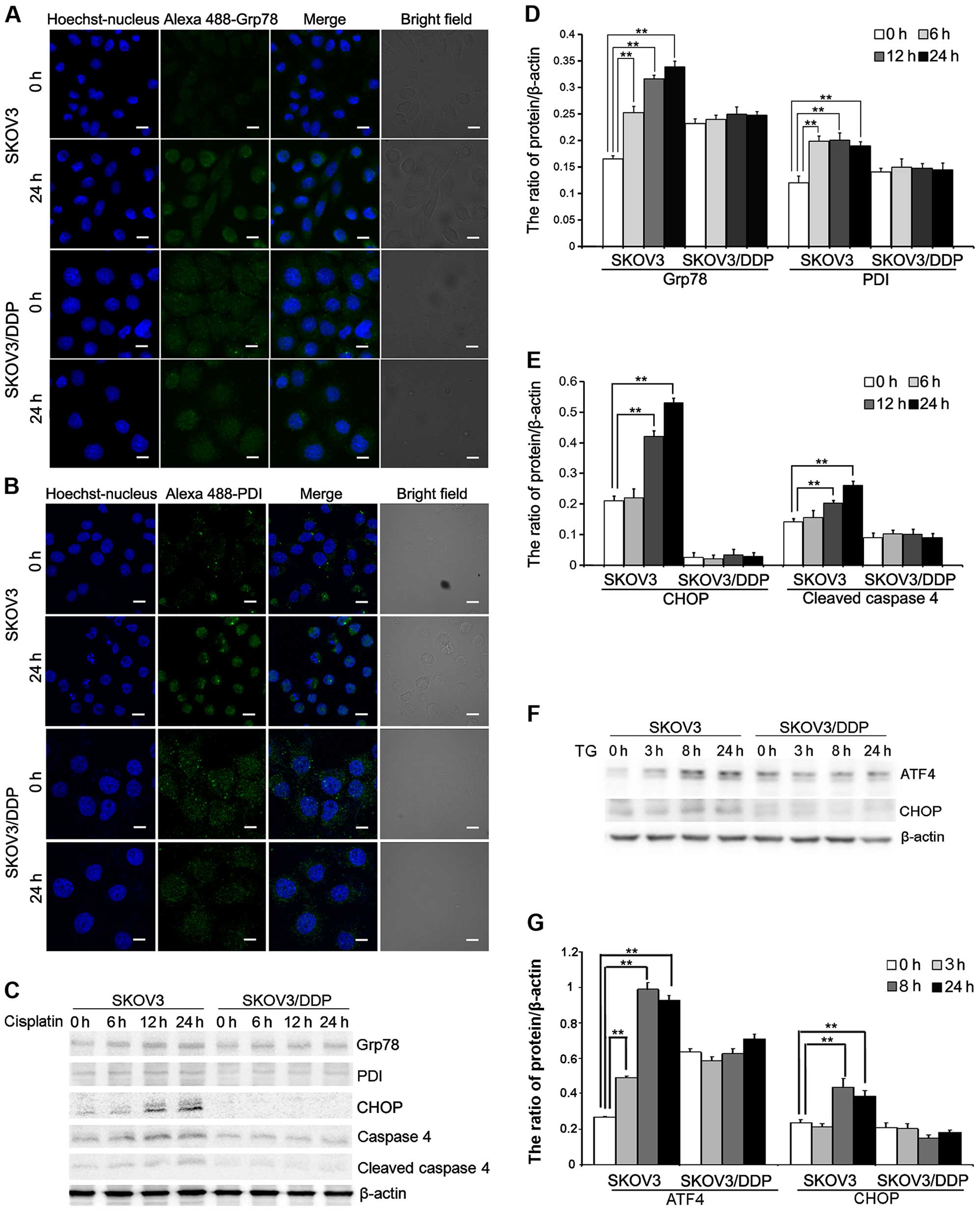
Cisplatin induces ER stress in the SKOV3
cells but not in the SKOV3/DDP cells
To further evaluate the mechanism of
cisplatin-induced apoptosis in ovarian cancer cells, we assessed ER
stress protein expression in cells treated with cisplatin.
Cisplatin treatment induced the expression of Grp78 and PDI, two ER
stress marker proteins that accumulate after ER stress. Confocal
microscopy showed that Grp78 and PDI accumulation occurred after 24
h in the SKOV3 cells treated with cisplatin (Fig. 2A and B). Western blotting showed
that Grp78 and PDI were upregulated after 24 h in the SKOV3 cells
(Fig. 2C and D). We also detected
upregulation of DDIT3/CHOP, which is involved in ER stress-induced
cell death. These results showed that DDIT3/CHOP is induced in the
cisplatin-treated SKOV3 cells (Fig. 2C
and E). Caspase-4 is an ER-resident caspase that is processed
in response to ER stress (8,9,23).
Caspase-4 activation, reflected as caspase-4 cleavage, was
significantly increased in the SKOV3 cells after cisplatin
treatment (Fig. 2C and E).
Similarly, we determined that the expression of ATF4 and
DDIT3/CHOP, two ER-stress mediators, was significantly upregulated
in the SKOV3 cells treated with ER stress inducer, thapsigargin
(TG) (Fig. 2F and G).
These results demonstrated that cisplatin can induce
ER stress-mediated apoptosis in SKOV3 cells but not in SKOV3/DDP
cells.
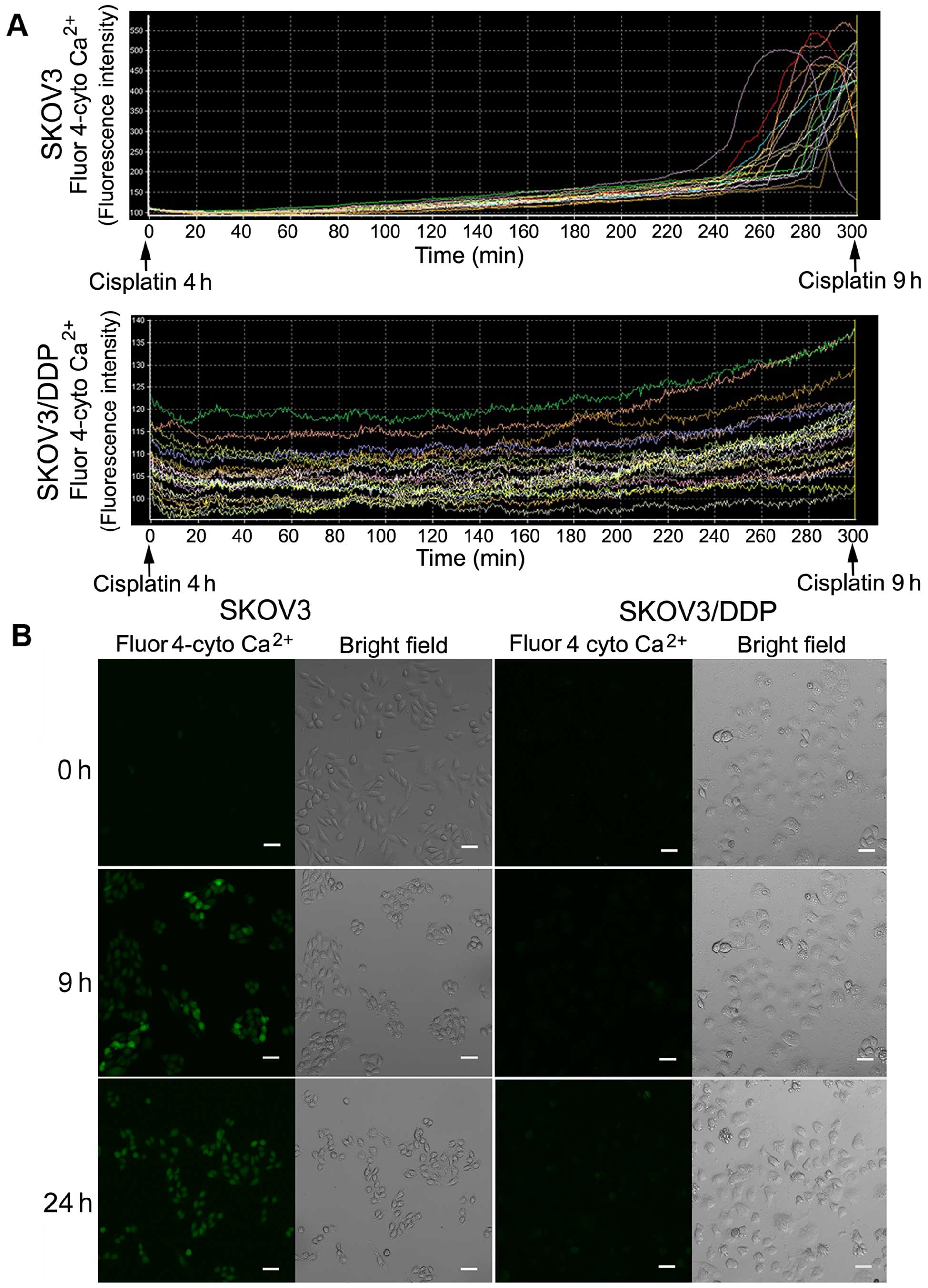
Cisplatin induces cytosolic
Ca2+ influx in SKOV3 cells but not in SKOV3/DDP
cells
The ER is the major intracellular calcium
(Ca2+) storage organelle and is critically involved in
Ca2+ homeostasis. Sustained ER stress causes
Ca2+ release to the cytosol, leading to calcium
overload. Thus, we investigated cytosolic Ca2+ changes
using a calcium-sensitive fluorescent probe and confocal
microscopy. We observed a sharp increase in cytosolic
Ca2+ in the SKOV3 cells after ~9 h of cisplatin
treatment, but no obvious changes in the SKOV3/DDP cells (Fig. 3A).
Confocal microscopy images of both cell lines
treated with cisplatin at 0, 9 and 24 h showed a clear increase in
green fluorescence after 9 and 24 h in the SKOV3 cells (Fig. 3B).
These results demonstrated that cisplatin induces
calcium overload in SKOV3 cells, but not in SKOV3/DDP cells.
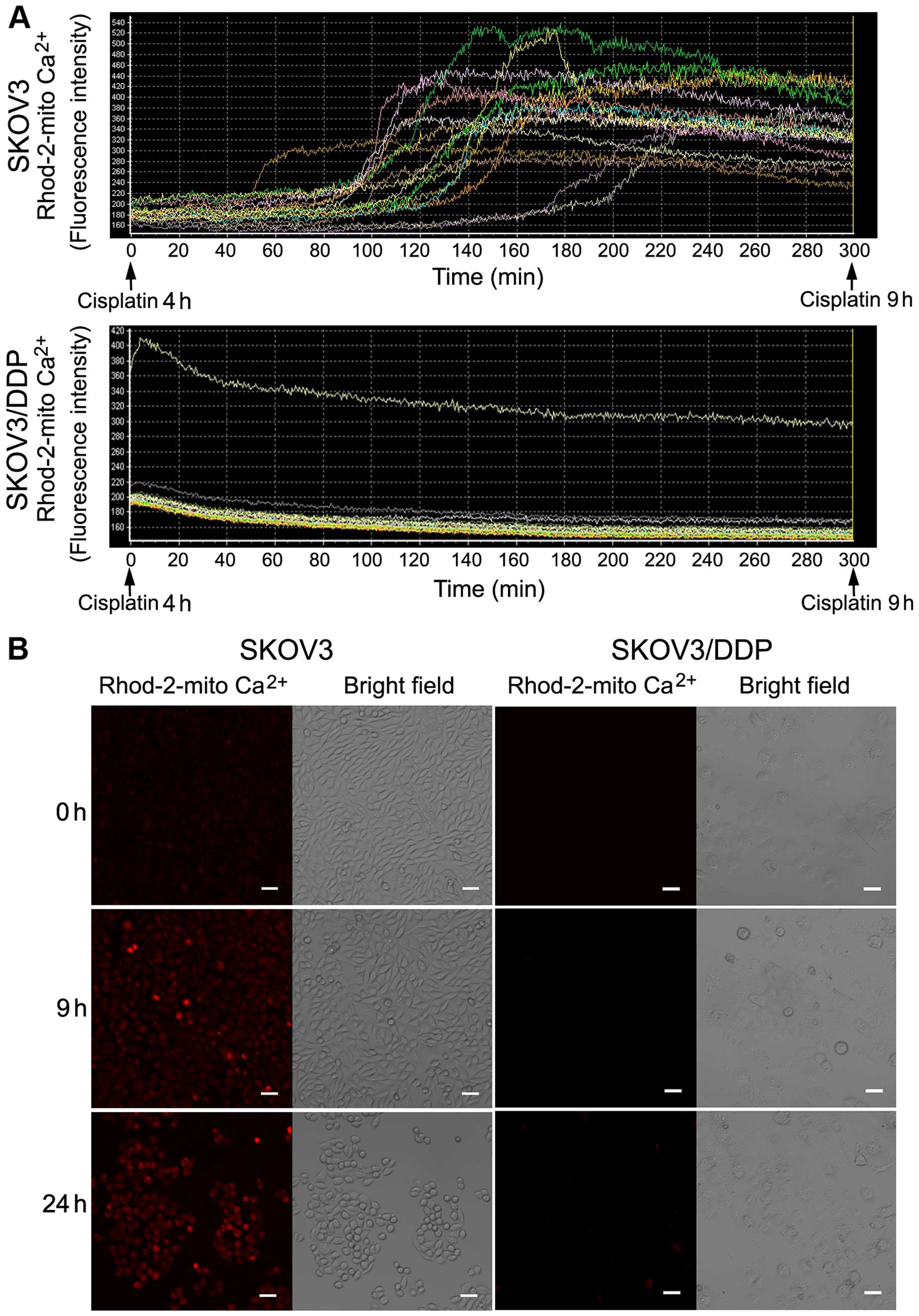
Cisplatin induces continuous
Ca2+ flux into the mitochondria of SKOV3 cells
High cytoplasmic Ca2+ concentrations lead
to increased mitochondrial Ca2+ uptake, which damage the
mitochondria, leading to apoptosis. We measured mitochondrial
Ca2+ changes using a calcium-sensitive fluorescent probe
and confocal microscopy. There was a sharp increase in
mitochondrial Ca2+ in the SKOV3 cells after cisplatin
treatment for ~6 h, but a reduction in mitochondrial calcium in
SKOV3/DDP cells (Fig. 4A).
Confocal microscopy images of both cell lines
treated with cisplatin for 0, 9 and 24 h showed a clear increase in
red fluorescence after 9 and 24 h in the SKOV3 cells (Fig. 4B).
These results demonstrated that cisplatin can
increase mitochondrial Ca2+ in SKOV3 cells, but not in
SKOV3/DDP cells.
Mitochondrial Ca2+ overload
leads to mitochondrial-mediated apoptosis in the SKOV3 cells
Mitochondrial calcium overload can damage the
mitochondrial structure and induce apoptosis via the mitochondrial
pathway. Therefore, our next goal was to determine the effect of
mitochondrial Ca2+ overload on SKOV3 cells.
First, we used
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolyL-carbocyanine
chloride (JC-1) to measure the mitochondrial membrane potential.
The mitochondrial membrane potential started to decrease after
cisplatin treatment for 12 h in the SKOV3 cells, but not in the
SKOV3/DDP cells (Fig. 5A).
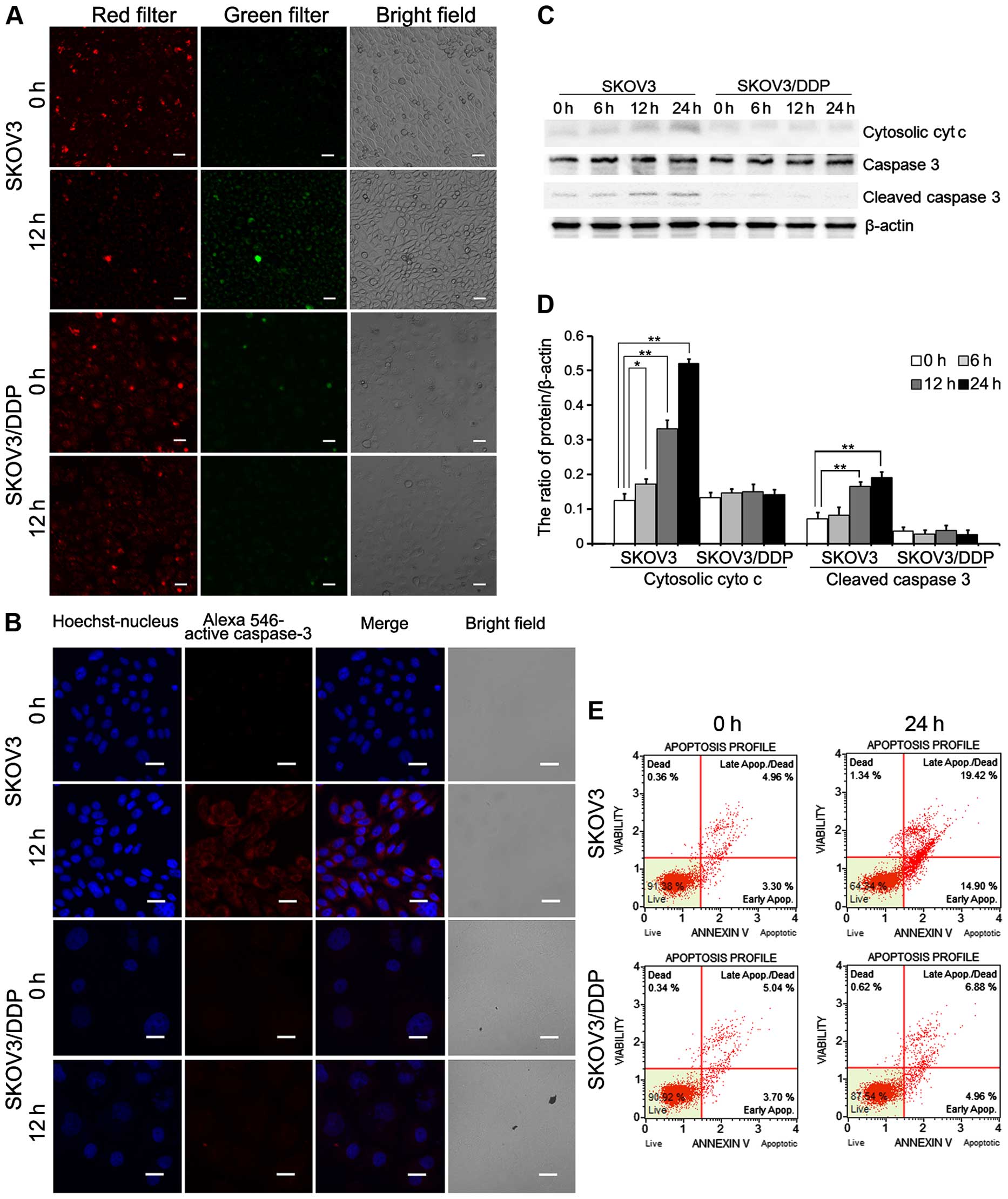
Confocal microscopy showed caspase-3 activation in
both the SKOV3 and SKOV3/DDP cell lines treated with cisplatin.
Cisplatin treatment increased caspase-3 activation in the SKOV3
cells relative to the SKOV3/DDP cells (Fig. 5B). As shown in Fig. 5C and D, cytosolic cytochrome
c and cleaved caspase-3 were upregulated in the SKOV3 cells
compared with the SKOV3/DDP cells. Analysis of apoptosis by flow
cytometry revealed a higher apoptosis rate in the SKOV3 cells
treated with cisplatin (34.32%) compared with the rate in the
SKOV3/DDP cells exposed to cisplatin (11.84%; Fig. 5E).
These results demonstrate that cisplatin-induced
mitochondrial Ca2+ overload leads to
mitochondrial-mediated apoptosis in SKOV3 cells.
Cisplatin-induced increase in
mitochondrial-associated membrane structures leads to mitochondrial
calcium overload in the SKOV3 cells
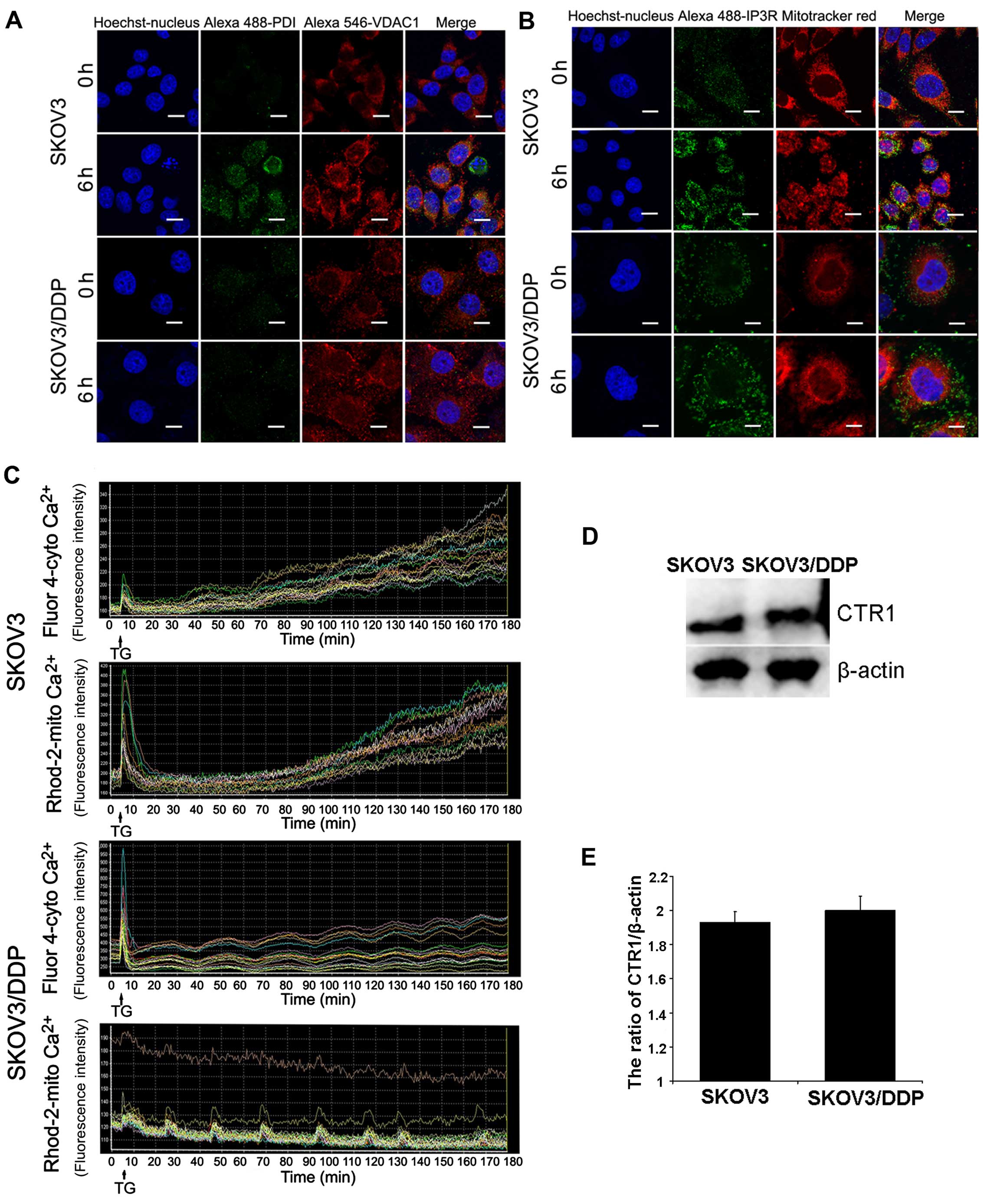
Mitochondrial-associated membranes (MAMs) linking ER
to the mitochondria contain calcium channels. Our results showed
that mitochondrial calcium overload (6 h) occurred before cytosolic
calcium overload (9 h). Therefore, we used ER and mitochondrial
markers to observe structural changes in the MAMs after cisplatin
treatment in both cell lines. In addition, confocal microscopy
showed that ER-mitochondria contact areas increased in the
cisplatin-treated SKOV3 cells (Fig. 6A
and B).
We also test whether the ER stress inducer TG
induced the same response in both cell lines. Using
calcium-sensitive fluorescent probes, we found that the cytoplasmic
Ca2+ concentration increased further after TG treatment;
a parallel increase in mitochondrial Ca2+ concentration
occurred in the SKOV3 cells, but not in the SKOV3/DDP cells
(Fig. 6C). To investigate the role
of CTR1 drug-importer on the acquisition of tolerance to cisplatin,
we also detected CTR1 in the SKOV3 and SKOV3/DDP cells. According
to Fig. 6D and E, expression of
CTR1 in both cell lines exhibited no statistical differences.
These results demonstrated that SKOV3/DDP cells are
more resistant to ER stress compared with SKOV3 cells.
Discussion
Chemotherapy resistance is a main cause of death in
ovarian cancer patients (3,24); identifying the resistance mechanism
is therefore very important. Cisplatin is a classic chemotherapy
reagent; drug resistance is mostly acquired and the mechanism is
unclear. Cisplatin resistance may be caused by adaptive changes
affecting multiple signaling molecules in tumor cells, for example,
downregulation of apoptotic signaling and activation of
pro-survival signaling. The ER is reported to be a cytoplasmic
target of cisplatin (8,9). Cisplatin can induce ER
stress-associated apoptosis; therefore, ER stress tolerance may be
involved in cisplatin resistance (6).
We used SKOV3/DDP cells as an in vitro model
of cisplatin resistance. Morphometric analysis of cell apoptosis
and analysis of cell viability after cisplatin treatment showed
that the sensitivity of SKOV3/DDP cells to cisplatin was
significantly lower than that of the parental SKOV3 cell line.
ER is the main storage organelle for intracellular
Ca2+, and the site of protein synthesis, folding,
modification and transport (25,26).
New proteins in the ER need to be modified in various ways,
including glycosylation and disulfide bond formation, for correct
folding, maturation and stabilization (26). These processes need the assistance
of several resident molecular chaperones and
Ca2+-binding proteins, including glucose-regulated
proteins (such as Grp78 or BiP), calreticulin and cadherin, and
several protein-folding enzymes, such as protein disulfide
isomerase (PDI) (26,27). A variety of physiological and
pathological conditions can interfere with the ER protein-folding
process, leading to the deposition and aggregation of unfolded
proteins in the ER, and resulting in ER stress that triggers the
UPR (28). The primary function of
the UPR is to reduce the burden of ER proteins by temporarily
closing down protein synthesis and altering a complex gene
transcriptional program to increase protein folding. If this
transcriptional program cannot restore ER homeostasis, sustained ER
stress induces cell death, known as ER-mediated apoptosis (10,12,29).
The main apoptotic effector molecule triggered by ER stress is
DDIT3/CHOP (12,29). DDIT3/CHOP switches on the expression
of a series of genes, leading to apoptosis. Caspase-4 is an
ER-resident caspase that is processed in response to ER stress; it
is required for ER stress-induced apoptosis (similar to caspase-12
in murine cells) (8,9,23,29,30).
Active caspase-4 enters the cytoplasm, where it activates other
caspase family members to complete apoptosis (30,31).
In addition, ER stress-induced calcium signaling can also mediate
cellular apoptosis (12,29).
The present study showed that cisplatin treatment
induced Grp78 and PDI expression in the SKOV3 cells, but not in the
SKOV3/DDP cells. In addition, DDIT3/CHOP expression was increased
and caspase-4 was activated in the SKOV3 cells. Moreover, when both
SKOV3 and SKOV3/DDP cells were treated with TG, the expression
levels of ATF4 and DDIT3/CHOP were upregulated in the SKOV3 cells.
These results indicated that cisplatin can trigger ER stress in the
SKOV3 cells, leading to ER stress-mediated apoptosis. In contrast,
SKOV3/DDP cells were ER stress tolerant and did not undergo ER
stress-mediated apoptosis.
Excessive ER stress synergizes with mitochondria and
cytochrome c to activate caspases and induce apoptosis
(16,32,33).
The ER stress inducer TG induces ER stress, which triggers
cytochrome c release accompanied by elevated intracellular
calcium (16). ER-mitochondria
contacts represent many points of close contact between ER and
mitochondria that can mediate the intermembrane exchange of lipids,
calcium ions and glycosylated proteins (19,20).
ER-mitochondria contacts form the basis for two-way organellar
communication that regulates mitochondrial energy and lipid
metabolisms, Ca2+ signaling, and cell death during many
physiological and pathological processes (18–20).
ER-mitochondria contacts can mediate phospholipid and
Ca2+ transport between the ER and mitochondria. This
Ca2+ shuttling provides a convenient way to prevent
pancytoplasmic non-specific upregulation of Ca2+
(17,21). The ER can release calcium ions,
leading to calcium uptake by adjacent mitochondria to stimulate
cell necrosis/apoptosis (18).
Mitochondrial calcium overload can lead to reduced membrane
potential, which causes cytochrome c release and induces
cell apoptosis (17,34).
Our experiments showed that the cytoplasmic
Ca2+ concentration in SKOV3 cells increased gradually
with prolonged cisplatin treatment: a significant increase was
observed at ~9 h, and it continued with longer exposure periods. In
contrast, the mitochondrial Ca2+ concentration increased
after ~6 h and was then was maintained at the higher level. We
further showed that in SKOV3 cells, cisplatin treatment for 6 h led
to decreased mitochondrial membrane potential and cytochrome
c release, followed by caspase-3 activation. By labeling the
ER and mitochondria, we found that cisplatin treatment clearly
resulted in increased numbers of ER-mitochondria contact
structures, which probably explains the rapid calcium influx from
the ER into mitochondria after ER stress induction. These results
suggest that when ER stress intensifies in SKOV3 cells, the number
of connection structures between the ER and mitochondria increases,
causing the ER to release large amounts of Ca2+ through
the MAMs to the mitochondria; this happens before Ca2+
influx into the cytoplasm. In contrast, SKOV3/DDP cells are ER
stress tolerant and do not exhibit this phenomenon; therefore, they
survive.
We used the ER stress inducer TG to test this
mechanism. In cisplatin-sensitive SKOV3 ovarian cancer cells,
cytoplasmic Ca2+ levels rose rapidly to a peak, and then
continued to rise, while the mitochondrial calcium concentration
peaked sharply and then rose continuously. In contrast, in
SKOV3/DDP cells, the cytoplasmic calcium level transiently
increased and then fluctuated at a lower level; more importantly,
the mitochondrial Ca2+ concentration did not increase
significantly, although it showed cyclical fluctuations. These
results indicated that cisplatin-resistant SKOV3/DDP cells have a
strong tolerance to ER stress. The ER stress inducer TG did not
induce cytosolic and mitochondrial calcium overload in this cell
line.
To the best of our knowledge, CTR1 has been found to
regulate platinum drug uptake, such as cisplatin (35,36).
However, there was no significant difference in the expression of
CTR1 between SKOV3 and SKOV3/DDP cells. This indicates that CTR1
plays no role in cisplatin-resistant SKOV3/DDP cells.
In conclusion, cisplatin-resistant SKOV3/DDP cells
were used to study the role of ER stress in ovarian cancer
cisplatin resistance. We found that cisplatin treatment did not
induce ER stress in SKOV3/DDP cells, and that apoptosis signaling
downstream of the UPR (related to ER stress) was not activated.
Cytosolic or mitochondrial calcium overload did not occur in
response to ER stress. The ER stress inducer TG also failed to
induce calcium overload. This suggests that SKOV3/DDP cells are
resistant to cisplatin-induced ER stress, and can thus avoid ER
stress-mediated apoptosis to maintain cell survival, resulting in
cisplatin resistance. These data provide new insights into the
mechanism of cisplatin resistance in ovarian cancer.
Acknowledgments
This study was supported by the National Nature and
Science Foundation of China (NSFC81372793, 81272876 and 81202552),
and the Department of Education of Jilin Province Project (no.
2013361). We thank Liwen Bianji (Edanz Group China) for editing the
English in this manuscript.
References
|
1
|
Mei L, Chen H, Wei DM, Fang F, Liu GJ, Xie
HY, Wang X, Zou J, Han X and Feng D: Maintenance chemotherapy for
ovarian cancer. Cochrane Database Syst Rev.
6:CD0074142013.PubMed/NCBI
|
|
2
|
Bookman MA: First-line chemotherapy in
epithelial ovarian cancer. Clin Obstet Gynecol. 55:96–113. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davis A, Tinker AV and Friedlander M:
'Platinum resistant' ovarian cancer: What is it, who to treat and
how to measure benefit? Gynecol Oncol. 133:624–631. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ali AY, Farrand L, Kim JY, Byun S, Suh JY,
Lee HJ and Tsang BK: Molecular determinants of ovarian cancer
chemoresistance: New insights into an old conundrum. Ann NY Acad
Sci. 1271:58–67. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar
|
|
6
|
Xu Y, Wang C and Li Z: A new strategy of
promoting cisplatin chemotherapeutic efficiency by targeting
endoplasmic reticulum stress. Mol Clin Oncol. 2:3–7.
2014.PubMed/NCBI
|
|
7
|
Xu Y, Li D, Zeng L, Wang C, Zhang L, Wang
Y, Yu Y, Liu S and Li Z: Proteasome inhibitor lactacystin enhances
cisplatin cytotoxicity by increasing endoplasmic reticulum
stress-associated apoptosis in HeLa cells. Mol Med Rep. 11:189–195.
2015.
|
|
8
|
Xu Y, Yu H, Qin H, Kang J, Yu C, Zhong J,
Su J, Li H and Sun L: Inhibition of autophagy enhances cisplatin
cytotoxicity through endoplasmic reticulum stress in human cervical
cancer cells. Cancer Lett. 314:232–243. 2012. View Article : Google Scholar
|
|
9
|
Yu H, Su J, Xu Y, Kang J, Li H, Zhang L,
Yi H, Xiang X, Liu F and Sun L: p62/SQSTM1 involved in cisplatin
resistance in human ovarian cancer cells by clearing ubiquitinated
proteins. Eur J Cancer. 47:1585–1594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Groenendyk J and Michalak M: Endoplasmic
reticulum quality control and apoptosis. Acta Biochim Pol.
52:381–395. 2005.PubMed/NCBI
|
|
11
|
Yadav RK, Chae SW, Kim HR and Chae HJ:
Endoplasmic reticulum stress and cancer. J Cancer Prev. 19:75–88.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sano R and Reed JC: ER stress-induced cell
death mechanisms. Biochim Biophys Acta. 1833:3460–3470. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Szegezdi E, Fitzgerald U and Samali A:
Caspase-12 and ER-stress-mediated apoptosis: The story so far. Ann
NY Acad Sci. 1010:186–194. 2003. View Article : Google Scholar
|
|
14
|
Hajnóczky G, Davies E and Madesh M:
Calcium signaling and apoptosis. Biochem Biophys Res Commun.
304:445–454. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Santella L, Ercolano E and Nusco GA: The
cell cycle: A new entry in the field of Ca2+ signaling.
Cell Mol Life Sci. 62:2405–2413. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deniaud A, Sharaf el dein O, Maillier E,
Poncet D, Kroemer G, Lemaire C and Brenner C: Endoplasmic reticulum
stress induces calcium-dependent permeability transition,
mitochondrial outer membrane permeabilization and apoptosis.
Oncogene. 27:285–299. 2008. View Article : Google Scholar
|
|
17
|
Pinton P, Giorgi C, Siviero R, Zecchini E
and Rizzuto R: Calcium and apoptosis: ER-mitochondria
Ca2+ transfer in the control of apoptosis. Oncogene.
27:6407–6418. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Grimm S: The ER-mitochondria interface:
The social network of cell death. Biochim Biophys Acta.
1823:327–334. 2012. View Article : Google Scholar
|
|
19
|
Raturi A and Simmen T: Where the
endoplasmic reticulum and the mitochondrion tie the knot: The
mitochondria-associated membrane (MAM). Biochim Biophys Acta.
1833:213–224. 2013. View Article : Google Scholar
|
|
20
|
Rowland AA and Voeltz GK: Endoplasmic
reticulum-mitochondria contacts: Function of the junction. Nat Rev
Mol Cell Biol. 13:607–625. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rizzuto R, Marchi S, Bonora M, Aguiari P,
Bononi A, De Stefani D, Giorgi C, Leo S, Rimessi A, Siviero R, et
al: Ca2+ transfer from the ER to mitochondria: When, how
and why. Biochim Biophys Acta. 1787:1342–1351. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Luciani DS, Gwiazda KS, Yang TL, Kalynyak
TB, Bychkivska Y, Frey MH, Jeffrey KD, Sampaio AV, Underhill TM and
Johnson JD: Roles of IP3R and RyR Ca2+ channels in
endoplasmic reticulum stress and beta-cell death. Diabetes.
58:422–432. 2009. View Article : Google Scholar :
|
|
23
|
Liu N, Xu Y, Sun JT, Su J, Xiang XY, Yi
HW, Zhang ZC and Sun LK: The BH3 mimetic S1 induces endoplasmic
reticulum stress-associated apoptosis in cisplatin-resistant human
ovarian cancer cells although it activates autophagy. Oncol Rep.
30:2677–2684. 2013.PubMed/NCBI
|
|
24
|
Galluzzi L, Vitale I, Michels J, Brenner
C, Szabadkai G, Harel-Bellan A, Castedo M and Kroemer G: Systems
biology of cisplatin resistance: Past, present and future. Cell
Death Dis. 5:e12572014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ali Khan H and Mutus B: Protein disulfide
isomerase a multifunctional protein with multiple physiological
roles. Front Chem. 2:702014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Halperin L, Jung J and Michalak M: The
many functions of the endoplasmic reticulum chaperones and folding
enzymes. IUBMB Life. 66:318–326. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koenig PA and Ploegh HL: Protein quality
control in the endoplasmic reticulum. F1000Prime Rep. 6:492014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chaudhari N, Talwar P, Parimisetty A,
Lefebvre d'Hellencourt C and Ravanan P: A molecular web:
Endoplasmic reticulum stress, inflammation, and oxidative stress.
Front Cell Neurosci. 8:2132014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Urra H, Dufey E, Lisbona F, Rojas-Rivera D
and Hetz C: When ER stress reaches a dead end. Biochim Biophys
Acta. 1833:3507–3517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hitomi J, Katayama T, Eguchi Y, Kudo T,
Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K,
et al: Involvement of caspase-4 in endoplasmic reticulum
stress-induced apoptosis and Abeta-induced cell death. J Cell Biol.
165:347–356. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Oda T, Kosuge Y, Arakawa M, Ishige K and
Ito Y: Distinct mechanism of cell death is responsible for
tunicamycin-induced ER stress in SK-N-SH and SH-SY5Y cells.
Neurosci Res. 60:29–39. 2008. View Article : Google Scholar
|
|
32
|
Dou G, Sreekumar PG, Spee C, He S, Ryan
SJ, Kannan R and Hinton DR: Deficiency of αB crystallin augments ER
stress-induced apoptosis by enhancing mitochondrial dysfunction.
Free Radic Biol Med. 53:1111–1122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rodriguez D, Rojas-Rivera D and Hetz C:
Integrating stress signals at the endoplasmic reticulum: The BCL-2
protein family rheostat. Biochim Biophys Acta. 1813:564–574. 2011.
View Article : Google Scholar
|
|
34
|
Chaudhuri D and Clapham DE: Outstanding
questions regarding the permeation, selectivity, and regulation of
the mitochondrial calcium uniporter. Biochem Biophys Res Commun.
449:367–369. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fung KL, Tepede AK, Pluchino KM, Pouliot
LM, Pixley JN, Hall MD and Gottesman MM: Uptake of compounds that
selectively kill multidrug-resistant cells: The copper transporter
SLC31A1 (CTR1) increases cellular accumulation of the
thiosemicarbazone NSC73306. Mol Pharm. 11:2692–2702. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Matsumoto S, Tanaka T, Kurokawa H, Matsuno
K, Hayashida Y and Takahashi T: Effect of copper and role of the
copper transporters ATP7A and CTR1 in intracellular accumulation of
cisplatin. Anticancer Res. 27:2209–2216. 2007.PubMed/NCBI
|