Introduction
It is an accepted concept of the cancer paradigm
that tumorigenesis results from an imbalance between cell
proliferation and cell death. It is also becoming clearer that in
response to cellular stress, cells may utilize multiple mechanisms
that ultimately determine cell death or survival. Cancer cells are
exposed to a wide range of stress sources, such as nutrient
deprivation and hypoxia, as well as cytotoxic chemotherapy and
radiotherapy. Cancer cells utilize several mechanisms to withstand
the cell death signals that are generated upon exposure to stress.
Cancer cells undergoing certain forms of stress, such as starvation
or hypoxia, can also promote survival by modulating their
intracellular metabolism (1); for
instance activation of the metabolic autophagy pathway. Autophagy
or 'cell death occurring with autophagy' is an evolutionarily
conserved process aimed at maintaining cellular homeostasis by
lysosomal degradation of excessive, damaged and/or aged proteins
and organelles. To date, at least three distinct autophagic
pathways are known: macroautophagy, microautophagy and
chaperone-mediated autophagy. Microautophagy is the sequestration
of cytosolic components directly by the lysosomal membrane, whereas
chaperone-mediated autophagy involves translocation of targeted
proteins directly into the lysosome via a chaperone. Macroautophagy
is the most commonly studied form of autophagy in mammalian cells
and is characterized by the sequestration of proteins and
organelles, such as mitochondria, within an autophagosome (2). Microtubule-associated protein 1 light
chain (LC3) is known to be associated with the autophagosomal
membrane. The LC3 precursor (pro-LC3) is proteolytically cleaved by
Atg4 protease, resulting in the formation of LC3B-I. At the onset
of autophagy, LC3B-I is activated and conjugated to an amino group
of phosphatidylethanol-amine generating LC3B-II, and then recruited
to autophagosomes (3). An increase
in LC3 was shown to be directly correlated with the number of
autophagosomes and LC3B-II is considered to be the most specific
marker of the autophagic process (4).
Autophagy is dramatically increased under stressful
(nutrient-deprived) conditions and hypoxia in both normal and
cancer cells. Under these conditions, it is becoming evident that
autophagy protects cells, by providing an alternative energy source
and by eliminating dysfunctional organelles or proteins (2). Its role in tumorigenesis is more
controversial and both the presence and the absence of autophagy
have been implicated (5). Autophagy
is known to be associated with the poor outcome of patients with
several types of cancers, and its effectiveness as a prognostic
marker in colorectal cancer was demonstrated by several studies
(6,7).
Colorectal cancer (CRC) is the result of
interactions between environmental and genetic factors. Mutations
in human DNA mismatch repair (MMR) genes such as MLH1,
MSH2 and MSH6 are associated with the development of
both hereditary and sporadic CRCs (8). The importance of genetic stabilization
by MMR is illustrated by the fact that defects in the human MMR
pathway confer a strong predisposition to human hereditary
non-polyposis colorectal cancer (HNPCC) and may contribute to the
development of 15–20% of human sporadic CRCs (9). Moreover the DNA MMR system is of
clinical importance since mismatch repair-deficient tumor cells are
resistant to several classes of commonly used chemotherapy drugs
such as the methylating agent temozolomide, the platinum-based drug
cisplatin and the antimetabolite 6-thioguanine (6-GT) (10–12).
The inhibition of autophagy has two potential
therapeutic targets in CRC. CRC cells have been shown to have
functional autophagic machinery that is upregulated during nutrient
deprivation, providing an alternative nutrient source and acting as
a survival mechanism. Inhibition of this pathway led to marked
apoptotic death in CRC cells in vitro (13). Moreover, modulation of autophagy is
of interest in overcoming 5-FU resistance. 5-FU-based combination
chemotherapy remains the most common drug regimen used in both
adjuvant and palliative settings. 5-FU, an anti-metabolite
chemotherapy, exerts its cytotoxic effects through both the
inhibition of thymidylate synthetase and the incorporation of
macromolecules into RNA and DNA, leading to activation of p53 and
ultimately apoptosis (14).
In vitro experiments (15,16)
have shown that the inhibition of autophagy increases 5-FU-induced
apoptosis. There are two trials (NCT01206530, NCT01006369)
investigating the addition of chloroquine to 5-FU-based
chemotherapy and bevacizumab. As well as assessing efficacy and
side-effects, these trials are also aiming to identify surrogate
biomarkers for autophagy detection in patient tissue samples and
for in vivo molecular imaging (17). This highlights the need for robust
biomarkers that reflect functional autophagy and the role of
individualized autophagy profiles that can then be used to
determine whether to inhibit or induce autophagy.
In the present study, we evaluated the expression of
LC3B-II in samples of human colorectal microadenomas and carcinomas
compared to normal mucosa. Furthermore, the expression pattern of
LC3B-II was assessed in carcinomas classified as DNA microsatellite
stable (MSS) and unstable (MSI). Thus, immunofluorescence
techniques coupled with confocal microscopy and immunoblot
experiments were performed. The results clearly showed a
significant increase in the expression of the autophagic key factor
in microadenomas and carcinomas with respect to normal mucosa. In
MSS carcinomas the level of LC3B-II expression was higher than that
in the MSI carcinomas. To our knowledge, this is the first study
evaluating the role of autophagy in human colorectal preneoplastic
lesions such as microadenomas, and in colorectal carcinomas
according to the presence or absence of DNA microsatellite
instability; in other words in the presence or deficiency of the
DNA mismatch repair pathway.
Materials and methods
Ethics statement
All patients enrolled in the present study, which
underwent colonoscopy or surgical resection for colorectal cancer
at the University Hospital of Modena, were asked to provide
informed written consent, and the study protocol was specifically
approved by the Comitato Etico Provinciale di Modena.
Study population
Forty samples of normal colorectal mucosa (NM) were
collected from 20 patients during colonoscopy. All samples were
frozen at −80°C. All patients had normal colonoscopy results.
Thirty microadenomas (MAs) were also identified in 15 patients, and
removed after surgery for colorectal cancer on surgical specimens,
after staining of the mucosa with a 0.1% methylene-blue solution in
saline, and observation under a dissecting microscope (31). The average multiplicity of the MA
examined was 79 (range, 30–120), and all showed low grade
dysplasia. All MAs for each patient were frozen at −80°C. Finally,
30 samples of colorectal cancer (CRC) were collected from 20
patients operated on for colon or rectal cancer; all samples were
frozen at −80°C, as described above.
Moreover, we studied histological sections retrieved
from pathological archives of 49 carcinomas (CRC). In all 49
samples of carcinoma, the presence of instability at DNA
microsatellites was determined (23 were microsatellite stable, MSS,
and 26 microsatellite unstable, MSI).
Western blot analysis
One sample frozen at −80°C for each subject was used
for western blot analysis. Whole cell lysates were obtained from 20
samples of NM, 15 MA, and 15 CRC, extracted with hypotonic buffer
(50 mM Tris-Cl, pH 7.8, containing 1% Nonidet P-40, 140 mM NaCl,
0.1% SDS and Na-deoxycholate, 1 mM Na3VO4,
and freshly added protease inhibitor cocktail). Lysates were then
cleared by centrifugation for 15 min in a refrigerated centrifuge,
max speed, and immediately boiled in SDS sample buffer. An amount
of 40 mg of protein extracted from each sample (NM, MA, and CRC)
was electrophoresed on SDS-PAGE and transferred to nitrocellulose
membranes. The membranes were blocked with 3% dry milk and 2% BSA
in PBS-T, and incubated with the following antibody, diluted at
1:1,000 overnight at 4°C under agitation: [rabbit anti-human MAPLC3
(Santa Cruz Biotechnology)]. After washing, the membranes were
incubated with a secondary HPR-conjugated goat anti-mouse IgG
antibody (1:5,000) for 30 min at room temperature. Immunoreactive
proteins were detected with ECL (Amersham). Anti-mouse-β-tubulin
(Sigma) was used as loading control. Densitometric analysis was
performed using a Kodak Image Station 440cf system (Kodak,
Rochester, NY, USA), and semi-quantitative analysis was performed
with NIH Image J software. For each sample and marker, the band
intensities were normalized to β-tubulin, and the results are
expressed as the normalized treatment to the control ratio.
Microsatellite instability
The DNA microsatellite status of all tumors was
evaluated using four fluorescent-labeled mono-nucleotide markers,
BAT25, BAT26, NR24 and CAT25. These quasi-monomorphic markers were
selected after in-depth review of the literature for their very
high sensitivity and specificity in identifying mismatch
repair-deficient tumors (18–20).
Using this mononucleotide marker panel, a tumor was defined as
MSI-positive when showing instability with at least three
markers.
DNA from the tumor tissues was amplified in a
10-µl volume containing 30–50 ng of DNA, 0.15 pmol of
dye-labeled forward and unlabeled reverse primers, 2 mM
concentration of each deoxynucleotide triphosphate, 1.5 mM
MgCl2, 50 mM KCl, 10 mM Tris (pH 8.3) and 0.6 units of
Taq polymerase. Thermocycling conditions were: 94°C for 4
min, followed by 11 touchdown cycles, each with a denaturing step
at 94°C for 30 sec, an extension step at 72°C for 15 sec and a 75
sec annealing step that decreased 1°C/cycle (beginning at 65°C in
the first cycle and decreasing to 55°C in the 11th cycle). The 11th
cycle was then repeated 26 times for a total of 37 cycles of PCR;
finally an extension step of 4 min at 72°C followed by storage at
4°C. PCR products were prepared for analysis by pooling 2 µl
of dye-D2 reaction, 1 µl of dye-D3 and 0.5 µl of
dye-D4 reaction; 40 µl of deionized formamide and 0.5
µl of CEQ DNA size standard-400 were added to 0.7 µl
of the each mixture. All samples were run on a CEQ 8000 sequencer
and analyzed using a CEQ 8000 Fragment Analysis system (Beckman
Coulter) (21).
Evaluation of immunofluorescence by
confocal microscopy
One sample frozen at −80°C for each subject was used
for immunofluorescence analysis to evaluate the expression of
LC3B-II, vimentin, myeloperoxidase, CD-20 and CD-3 proteins. Twenty
samples of NM, 15 MAs and 15 CRCs were fixed in 4% paraformaldehyde
in PBS, cryoprotected in 15% sucrose in PBS, and frozen in
isopentane cooled in liquid nitrogen. Horizontal cryosections of
the samples were cut (10 µm thick), and hematoxylin and
eosin (H&E) staining was performed on sections for control
tissue integrity and histology. Moreover, the 49 samples of MSS and
MSI carcinomas, embedded in paraffin, were used for
immunofluorescence analysis as described above. Before
immunohistochemistry, routine histology of all tissue samples was
carried out after H&E staining of the sections. Slides were
dried overnight at 37°C, dewaxed in two changes of fresh xylene,
and rehydrated in a descending alcohol series. Antigen retrieval
involved treatment with a protease (Pronase 1:20; DakoCytomation)
for 7 min at 37°C. After treatment with 3% BSA in PBS for 30 min at
room temperature, the cryostatic and paraffin sections were
incubated with the primary antibodies: rabbit anti-human MAPLC3
(Santa Cruz Biotechnology); mouse anti-human CD-3, mouse anti-human
CD-20, mouse anti-vimentin and mouse anti-myeloperoxidase (Dako),
diluted 1:25 in PBS containing 3% BSA for 1 h at room temperature.
After washing in PBS, the samples were incubated for 1 h at room
temperature with the secondary antibodies diluted 1:20 in PBS
containing 3% BSA [sheep anti-mouse FITC-conjugated, goat
anti-rabbit TRITC-conjugated (Sigma)]. After washing in PBS and in
H2O, the samples were counterstained with 1 mg/ml DAPI
in H2O and then mounted with anti-fading medium (0.21 M
DABCO and 90% glycerol in 0.02 M Tris, pH 8.0). Negative control
samples were not incubated with the primary antibody. The confocal
imaging was performed on a Leica TCS SP2 AOBS confocal laser
scanning microscope. Excitation and detection of the samples were
carried out in sequential mode to avoid overlapping of signals.
Sections were scanned with laser intensity, confocal aperture, gain
and black level setting kept constant for all samples. Optical
sections were obtained at increments of 0.3 mm in the z-axis and
were digitized with a scanning mode format of 512×512 or
1,024×1,024 pixels and 256 grey levels. The confocal serial
sections were processed with the Leica LCS software to obtain
three-dimensional projections. Image rendering was performed by
Adobe Photoshop software. The original green fluorescent confocal
images were converted to grey-scale and median filtering was
performed. An intensity value ranging from 0 (black) to 255 (white)
was assigned to each pixel. Background fluorescence was subtracted
and immunofluorescence intensity (IF) was calculated as the average
for each selected area. The IF of the selected areas, linearly
correlated with the number of pixels, was quantitatively analyzed
using the standard imaging analysis software of an NIS-Elements
system. Each sample was assigned a code number and the score,
referred to as immunofluorescence intensity score (IFIS), was
determined by an observer who was blinded to the tissue groups
during the analysis (22).
Statistical analysis
All quantitative data for NM, MA, and CRC are
reported as mean ± SD. The average expression difference in the
various groups of colorectal lesions, was tested for statistical
significance using Kruskal-Wallis analysis, followed by
Student-Newman-Keuls test. A value of P<0.05 was chosen to
indicate a statistically significant difference.
Results
Patterns of LC3B-II autophagic activity
in the adenoma-carcinoma sequence
In order to evaluate the expression pattern of
LC3B-II protein in the samples of normal colorectal mucosa (NM),
microadenoma (MA) and colorectal cancer (CRC), immunofluorescence
experiments coupled with confocal analysis were performed.
Moreover, this technique allowed us to define the accurate
distribution pattern of LC3B-II protein in very thick samples, and
its immunostaining quantification.
Cells expressing LC3B-II protein showed two distinct
autophagic patterns: a diffuse finely and granular reactivity
dispersed in the cytoplasm, or a rounded densely stained material,
probably enclosed within a cytoplasmic vacuole that accumulated
prevalently around the nucleus.
The diffuse granular pattern, often similar to small
clumps, was noted in many cancerous epithelial cells of all
sections studied, although to a different extent and staining
intensity (Fig. 1D).
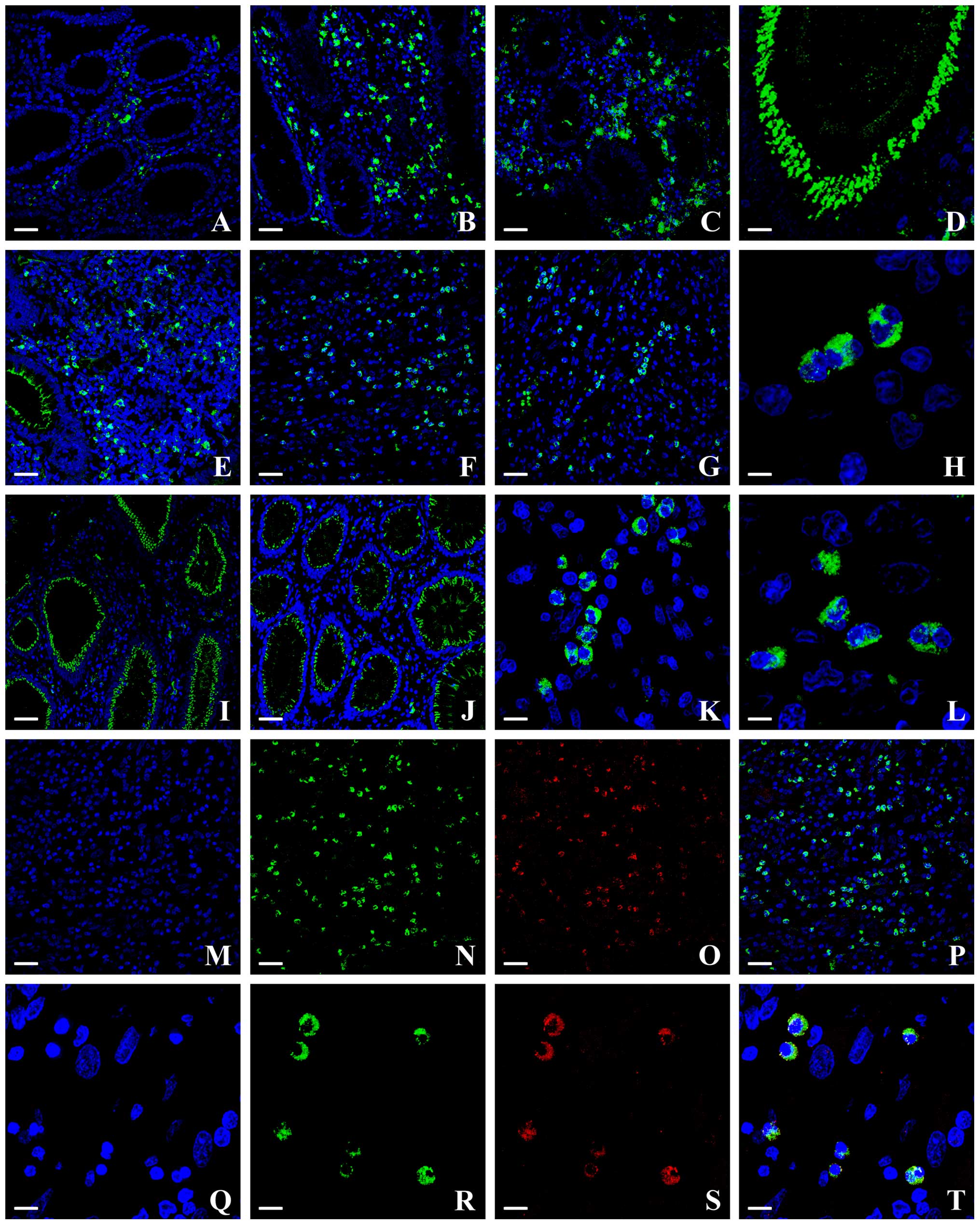 | Figure 1(A–L) Confocal analysis of colonic
mucosa cryosections labeled by DAPI (blue) and anti-LC3B-II
(green). (A) Normal colorectal mucosa: LC3B-II protein is localized
mainly in stromal cells at the cytoplasmic level; the amount of
immunostaining is low. (B) Microadenoma: the marked increase in
LC3B-II staining with respect to normal mucosa is evident; the
staining patterns of LC3B-II protein varies from a diffuse mode to
a granular one in the cytoplasm of stromal cells. (C) Colorectal
cancer: the immunostaining is significantly enhanced with respect
to microadenomas in stromal cells. Many cancerous epithelial cells
show marked staining with a diffuse granular pattern (D). Positive
cells tended to gather at the edge of the neoplastic tissue (E), in
the stroma inside the tumor (F), and also among the neoplastic
cells at the margins of the lesions (G). Magnification of stromal
cancer cells, the dense rounded autophagic vacuoles were well
recognizable (H). (I and J) Confocal analysis of DNA microsatellite
stable (MSS) and unstable (MSI) carcinomas. In the epithelium the
LC3B-II protein was distributed in a diffuse/granular staining
pattern throughout the cytoplasm, accumulated predominantly at the
apical region of cells towards the lumen of colonic crypts. The
pattern is more evident in MSS tumors (I) than in MSI ones (J). (K
and L) Magnification of stromal cells, bright tiny or large
aggregates (autophagosomes), are well recognizable (K, MSS; L,
MSI). (M–P) Samples of MSS carcinoma stained with DAPI (M, blue),
LC3B-II (N, green), vimentin (O, red) and merged (P). In the
stromal compartment the presence is evident of many labeled cells
with LC3B-II and vimentin, whose co-immunostaining (yellow in P)
corresponds to 40%. (Q-T) Samples of MSS carcinoma stained with
DAPI (Q, blue), LC3B-II (R, green), CD-20 (S, red) and merged (T).
Only 10% of T lymphocytes/natural killer cells expressed LC3B-II as
revealed by co-immunostaining (yellow in T). Scale bars: A, B, C,
E, F, G, I, J, M, N, O and P, 50 µm; D, 25 µm; H and
L, 5 µm; K, Q, R, S and T, 10 µm. |
The dense rounded autophagic vacuoles were well
recognizable in stromal cancer cells; such structures varied in
size and density, but usually formed coarse, rather than fine,
granules (Fig. 1H).
The staining patterns of LC3B-II protein that varied
from a diffuse mode to a granular one in the cytoplasm of stromal
cells, appeared consistent and unchanged in the same samples,
without appreciable variations in intensity and localization among
the cells. In all NM samples a few scattered areas in the stromal
compartment exhibited a moderate LC3B-II-reactivity, although the
epithelial cells were not marked (Fig.
1A).
An intense staining was evident in MA, in stromal
cells surrounding colonic crypts, but no appreciable LC3B-II
staining of epithelial cells was found (Fig. 1B).
LC3B-II-positive cells were observed throughout the
resected tissue in all CRC lesions and they were particularly
numerous within the central tumor stroma (Fig. 1C). Concerning the stromal
compartment, the samples of CRC were characterized by a higher
number of LC3B-II-positive cells with respect to the MAs (Fig. 1B and C); in a subset of carcinoma
samples, positive cells were not evenly distributed in the tissue,
but tended to gather in some areas, usually at the edge of the
neoplastic tissue (Fig. 1E), not
only in the stroma inside the tumor (Fig. 1F) but also among the neoplastic
cells at the margins of the lesions (Fig. 1G).
Moreover, interestingly, a consistent epithelial
positivity was observed in CRC: the protein aggregated in small
clumps distributed in the cytoplasm at the lateral and basal
portions of the epithelial cells (Fig.
1D and E).
The semi-quantitative evaluation of immunostaining
intensity reported as immunofluorescence intensity score (IFIS)
(Table I) showed that the level of
LC3B-II protein in both epithelial and stromal compartments had an
increasing trend from NM to CRC, with statistical significance of
the different expression among the groups.
 | Table IImmunofluorescence intensity score
(mean ± SD) of LC3B-II protein in samples of normal colorectal
mucosa, microadenoma, colorectal cancer. |
Table I
Immunofluorescence intensity score
(mean ± SD) of LC3B-II protein in samples of normal colorectal
mucosa, microadenoma, colorectal cancer.
| NM | MA | CRC |
|---|
| IFIS | 37±5.0 | 74±4.0 | 107±9.0 |
LC3B-II expression pattern in DNA
microsatellite stable (MSS) and unstable (MSI) carcinomas
Tumor samples from patients registered in the
Colorectal Cancer Registry of Modena, evaluated for DNA
microsatellite instability, were analyzed with immunofluorescence
techniques, and semi-quantitative evaluation of immunofluorescence
staining intensity was performed.
Many stromal cells showed an intense LC3B-II
immuno-reactivity in the cytoplasm; bright tiny or large
aggregates, as expected for autophagosomes, as well as a more
diffuse cytoplasmic staining were well recognizable (Fig. 1K and L). The positive stromal cells
appeared deeply located, inside the tumor, both in stable and
unstable tumor samples.
In the epithelium, LC3B-II protein was distributed
homogenously in a diffuse/granular staining pattern throughout the
cytoplasm, accumulated predominantly at the apical region of cell
towards the lumen of colonic crypts (Fig. 1I and J). These features were more
evident in MSS tumors (Fig. 1I)
than in MSI ones (Fig. 1J),
although positive staining was still detectable in the cells of the
surface epithelium and upper glands.
Concerning the semi-quantitative analysis of
immunofluorescence intensity, there were substantial differences
between stable and unstable tumors. Indeed the IFIS of MSS tumors
was 110±11 (SD), whereas IFIS of MSI tumors was 89±11 (SD); the
difference was statistically significant (Table II). In the lamina propria of both
stable and unstable tumor samples, some infiltrating mononuclear
cells were strongly positive for LC3B-II in the cytoplasm.
| Table IIImmunofluorescence intensity score
(mean ± SD) of LC3B-II protein in samples of DNA microsatellite
stable (MSS) and unstable (MSI) colorectal carcinoma. |
Table II
Immunofluorescence intensity score
(mean ± SD) of LC3B-II protein in samples of DNA microsatellite
stable (MSS) and unstable (MSI) colorectal carcinoma.
In order to clarify the role of the stromal
compartment in the synthesis of LC3B-II, we tried to identify the
type of stromal cells mostly involved in this process. For this
purpose, double immunofluorescence analyses were performed with the
LC3B-II antibody together with several antibodies specific for
different stromal cells, i.e., anti-vimentin, specific for
fibroblast-like cells, anti-myeloperoxidase, specific for
neutrophils, anti-CD-20 and anti-CD-3 which recognize T and B
lymphocytes, respectively. Interestingly, the staining profiles
observed showed the highest amount of LC3B-II-positive cells, ~40%,
in fibroblast-like cells and 10% T lymphocytes/natural killer
cells, as revealed by co-immunostaining with LC3B-II and the
antibodies specific for these different cell types (Fig. 1M–T). Very few LC3B-II-positive
neutrophils were observed in the tumor stroma. No match with
LC3B-II and CD-3 was found. Those percentages were almost the same
in the two different types of samples, i.e., stable and unstable
tumors.
Western blot analysis
To confirm the findings of the morphological
evaluation as described above, cell lysates of NM, MA, and CRC,
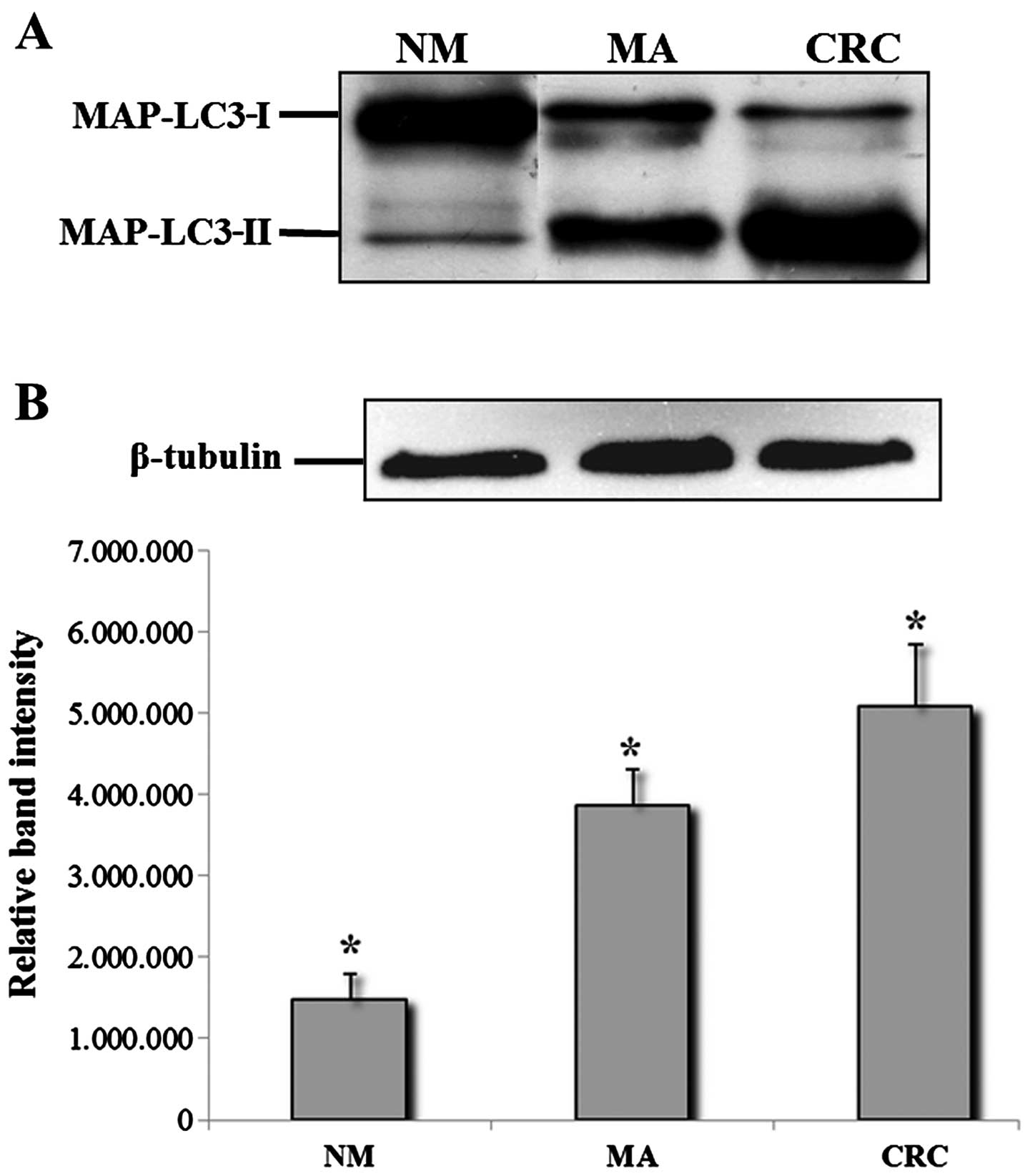
were subjected to western blot analysis. As shown in Fig. 2A, protein bands immunopositive for
LC3-I and LC3-II forms were clearly evident in a representative
sample of MA and CRC. Samples of NM generally yielded only a faint
band for LC3-I and LC3-II. All tumor tissues showed stronger bands
for LC3, especially for LC3-II, compared with the MAs.
Densitometric analysis and normalization (with equal amounts of
protein loading) of the immunoreactivity signals from protein
extracts of NM, MA and CRC showed that the level of LC3-I and
LC3-II proteins had an increasing trend from NM to CRC, with
statistical significance of the different expression levels
(Fig. 2B).
Discussion
To underline the novelty of our study, the following
main findings of the present investigation are summarized as
follows. i) The staining patterns of LC3B-II protein varied both in
morphological and quantitative aspects during the neoplastic
progression of human intestinal mucosa. The results clearly outline
that there is a striking increase in the intensity and density of
staining for LC3B-II protein in the normal
mucosa-microadenoma-carcinoma sequence. Immunofluorescence analyses
showed that LC3B-II was expressed in normal mucosa, although at low
levels, whereas it was significantly increased in microadenoma and
in cancer. ii) The results obtained with immunofluorescence
analyses were confirmed by western blotting data, with no
difference according to the various techniques used. iii) In DNA
microsatellite stable and unstable tumors, a strong positive
staining was detectable both in epithelial and stromal
compartments, and there were substantial differences between stable
and unstable tumors in the semi-quantitative analysis of
immunofluorescence. In MSS carcinomas, the level of LC3B-II
expression was higher than that in MSI carcinomas, and
LC3B-II-positive cells were mostly (40%) fibroblast-like cells.
The first point to emphasize is that the aim of this
study was to deepen the knowledge of the relationship between
autophagy and human CRC progression from the early steps of this
process. To our knowledge, this is the only study in which the
LC3B-II expression pattern in DNA microsatellite stable and
unstable CRCs was investigated.
Emerging evidence has shown that tumorigenesis and
the progression of human cancers are affected by disturbances in
the molecular machinery regulating autophagy (5,23).
However, the role of autophagy in cancer development and
progression remains controversial and may be regarded as a
double-edged sword. Recycling old and damaged cellular material
induced by stress and toxic injuries, in addition to providing an
alternative energy source, could be seen as assisting cancer cell
survival. On the other hand, when autophagy is pushed beyond a
crucial 'switch' point, it can lead to cell death which, if
harnessed, would be a useful mechanism for eradication of tumor
cells. Autophagy is considered a form of programmed cell death
(type II) responsible for eliminating damaged and/or harmful cells
such as cancer cells treated with anticancer reagents. Several
studies citing the pros and cons of autophagy in the cancer context
may be found in the scientific literature. Autophagy can be
involved in either the promotion or inhibition of cancer cell
survival (24,25). Experimental studies have indicated
that in the case of CRC cell lines, that were resistant to nutrient
deprivation culture conditions, autophagy may provide an
alternative source of energy through degradation of organelles
(13). Degraded material would
either be coverted into energy or biomass production, and the
energy aspect would certainly benefit CRC cells which are under
hypoxia when the blood supply is limited in the central portion of
the tumor mass. Interestingly, we found that the expression levels
of LC3B-II were increased in precancerous lesions (i.e.,
microadenomas), with respect to normal mucosa. The increment was
statistically significant and it was even more evident in the
carcinoma samples. These data are in keeping with the results
reported in our previous study, in which we demonstrated a gradual
increase in hypoxia-inducible factor-1α (HIF-1α) protein expression
in microadenomas, adenomas and carcinomas, showing a strong
relation to dysplasia (26). HIF-1α
is a key transcription factor that accumulates under hypoxic
conditions (27). The importance of
the association between hypoxia/HIF-1α and autophagy has previously
been demonstrated (28,29). This relationship seems to indicate
that hypoxia, an early event in tumor growth, may trigger autophagy
during malignant transformation (30). Moreover, we showed a marked
downregulation of apoptosis in human colorectal microadenomas with
respect to normal mucosa (31). It
is an accepted feature of the cancer paradigm that tumorigenesis
results from an imbalance between cell proliferation and cell
death. It is also becoming clearer that in response to cellular
stress, cells may utilize multiple mechanisms that ultimately
determine cell death or survival. Apoptosis and autophagy are
regulated by common upstream signals, share common components, and
there is also a functional relationship between autophagy and
apoptosis (32). Regarding cancer
development and treatment, the two processes commonly occur in the
same cells; in some cases inhibition of apoptosis causes autophagy
(33), while, in other cases,
inhibition of autophagy triggers apoptosis (34). Taken together our findings indicate
that autophagy can be viewed as a survival pathway to promote
cancer development, especially given the fact that the biomarker of
autophagy is strongly detectable from the early stage of CRC
progression. Thus targeting autophagy may be a useful strategy for
cancer treatment. These observation are in keeping with those of
Yoshioka et al (35).
Moreover, autophagy is known to be associated with
poor outcome in several types of cancers and several studies have
also demonstrated its effectiveness as a prognostic marker in CRC
(6,7,36,37).
Autophagy is reported to be upregulated and associated with drug
resistance in various treatment modalities for CRC (15). This process can prevent or promote
colorectal carcinogenesis as well as modulate the response to
various treatments (38,39). Therapeutic research and clinical
trials are currently underway to target both these properties
(40). Another interesting finding
of our study is the analysis of LC3B-II in samples of DNA
microsatellite stable and unstable carcinomas. The DNA mismatch
repair system plays a critical role in mutation avoidance and tumor
suppression. The importance of genetic stabilization by MMR is
illustrated by the fact that defects in the human MMR pathway
confer a strong predisposition to human hereditary non-polyposis
colorectal cancer (HNPCC) and may contribute to the development of
15–20% of human sporadic CRCs (41). The MSI phenotype is characterized by
a very specific clinicopathological profile. Colorectal tumors with
MSI are more often localized in the right colon and are associated
with high histological grade, mucinous component,
tumor-infiltrating lymphocytes, more necrosis and the presence of a
Crohn's like host response (42).
Thus, it was interesting to see whether these histological findings
had an impact on patient outcome. A better prognosis of MSI tumors
has been reported in studies describing this genetic instability in
CRC (43,44), as in further retrospective studies
(45–49). However, many of the studies were of
small sample size and included stage I–IV cancers and/or patients
treated by adjuvant or palliative chemotherapy. Studies with a
larger population restricted to stages II and III and randomized
versus no treatment are crucial to demonstrate the prognostic
significance of the altered MMR system in resected CRC. However,
research recently showed that most (90%) MSI tumors were part of
the low risk group among stage II colon cancer patients classified
by the prognostic ColoPrint® gene expression signature
in a series of 114 stage II tumors (50). Moreover, MSI remained a strong
independent prognostic marker in a multivariate analysis including
the other well-known prognostic Oncotype® gene
expression signature performed in 1,436 stage II colon cancers from
the QUASAR study (51,52). Mechanisms underlying the favorable
prognosis of MSI CRC remain widely unknown. The presence of a local
antitumor cytotoxic immune response due to the high content of
tumor-infiltrating lymphocytes that characterizes these tumors may
partially explain their better clinical outcome (53). This immune response also probably
explains the higher number of lymph nodes found in resected samples
of MSI stages I and II CRC compared to their MSS counterpart
(54). This study showed a
significant difference in the semi-quantitative analysis of LC3-II
protein expression pattern, i.e., the higher intensity of
fluorescence in MSS carcinomas with respect to MSI carcinomas in
the epithelium of colon cancer cells. Another point of the present
study was the identification of the types of stromal cells
producing LC3-II proteins in MSS and MSI carcinomas, which has
never been previously reported. LC3-II was expressed by stromal
fibroblasts in a high percentage and by T lymphocytes/natural
killer cells in smaller amounts. Several studies using mouse models
of genetically altered fibroblasts demonstrated a direct
involvement of resident fibroblasts in the onset of cancer
(55); moreover, both genetic and
cell-biology studies indicate that tumor growth is not just
determined by malignant cancer cells themselves, but also by the
tumor stroma (56). The expression
of autophagy-related proteins in the stroma can be explained by the
reverse Warburg effect - the interaction between tumor and stromal
metabolism in breast cancer. According to this hypothesis, reactive
oxygen species from breast cancer cells induce stromal glycolysis,
mitochondrial dysfunction, and increased autophagy. Ketone bodies
and lactate resulting from stromal cell glycolysis enter the tumor
cell, which generates ATP through oxidative phosphorylation.
Therefore, according to this theory, the expression of LC3B-II we
observed in the stroma may reflect increased stromal autophagic
activity. Thus, the cells interacting with the tumor cells are
probably cancer-associated fibroblasts (CAFs) (57–59).
CAFs represent the major cellular component of the desmoplastic
stroma of solid cancers and their metastases (60).
In clinical practice, two molecular features are
currently used in order to select the best therapeutic approach in
CRC patients. The first is the DNA microsatellite status and second
is the KRAS status. KRAS is mutated in a substantial part of all
CRCs, with a particular increased prevalence in MSSs, while when we
consider only MSIs this prevalence drops. This is of great interest
for our study, since we demonstrated that autophagy is
significantly higher in MSSs than in MSIs. Thus, a clinical
evaluation of the expression of autophagy in CRC patients could
play a role in selecting potential responders or non-responders to
autophagy inhibitory drugs. In conclusion, we elucidated the
expression pattern of an autophagy-related protein in the
adenoma-carcinoma sequence of the human large bowel, investigating
the relationship between the autophagic process and the DNA
microsatellite status. Our data suggest that autophagy is activated
in human CRC cells and may represent a pro-survival mechanism by
enhancing the ability of cancer cells to adapt to apoptotic stimuli
and hypoxic conditions during cancer progression.
Acknowledgments
The present study was supported by funds of the
Associazione per la Ricerca sui Tumori Intestinali (ARTI), which
also provided support to G.M. The authors wish to thank the Centro
Interdipartimentale Grandi Strumenti (CIGS) of the University of
Modena and Reggio Emilia, for software, instrument availability and
assistance.
References
|
1
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mizushima N, Ohsumi Y and Yoshimori T:
Autophagosome formation in mammalian cells. Cell Struct Funct.
27:421–429. 2002. View Article : Google Scholar
|
|
3
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Giatromanolaki A, Koukourakis MI, Harris
AL, Polychronidis A, Gatter KC and Sivridis E: Prognostic relevance
of light chain 3 (LC3A) autophagy patterns in colorectal
adenocarcinomas. J Clin Pathol. 63:867–872. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park JM, Huang S, Wu TT, Foster NR and
Sinicrope FA: Prognostic impact of beclin 1, p62/sequestosome 1 and
LC3 protein expression in colon carcinomas from patients receiving
5-fluorouracil as adjuvant chemotherapy. Cancer Biol Ther.
14:100–107. 2013. View Article : Google Scholar :
|
|
8
|
Buermeyer AB, Deschênes SM, Baker SM and
Liskay RM: Mammalian DNA mismatch repair. Annu Rev Genet.
33:533–564. 1999. View Article : Google Scholar
|
|
9
|
Jacob S and Praz F: DNA mismatch repair
defects: Role in colorectal carcinogenesis. Biochimie. 84:27–47.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Modrich P and Lahue R: Mismatch repair in
replication fidelity, genetic recombination, and cancer biology.
Annu Rev Biochem. 65:101–133. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fink D, Aebi S and Howell SB: The role of
DNA mismatch repair in drug resistance. Clin Cancer Res. 4:1–6.
1998.PubMed/NCBI
|
|
12
|
Stojic L, Brun R and Jiricny J: Mismatch
repair and DNA damage signalling. DNA Repair (Amst). 3:1091–1101.
2004. View Article : Google Scholar
|
|
13
|
Sato K, Tsuchihara K, Fujii S, Sugiyama M,
Goya T, Atomi Y, Ueno T, Ochiai A and Esumi H: Autophagy is
activated in colorectal cancer cells and contributes to the
tolerance to nutrient deprivation. Cancer Res. 67:9677–9684. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Hou N, Faried A, Tsutsumi S,
Takeuchi T and Kuwano H: Inhibition of autophagy by 3-MA enhances
the effect of 5-FU-induced apoptosis in colon cancer cells. Ann
Surg Oncol. 16:761–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li J, Hou N, Faried A, Tsutsumi S and
Kuwano H: Inhibition of autophagy augments 5-fluorouracil
chemotherapy in human colon cancer in vitro and in vivo model. Eur
J Cancer. 46:1900–1909. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suraweera N, Duval A, Reperant M, Vaury C,
Furlan D, Leroy K, Seruca R, Iacopetta B and Hamelin R: Evaluation
of tumor microsatellite instability using five quasimonomorphic
mono-nucleotide repeats and pentaplex PCR. Gastroenterology.
123:1804–1811. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xicola RM, Llor X, Pons E, Castells A,
Alenda C, Piñol V, Andreu M, Castellví-Bel S, Payá A, Jover R, et
al Gastrointestinal Oncology Group of the Spanish
Gastroenterological Association: Performance of different
microsatellite marker panels for detection of mismatch
repair-deficient colorectal tumors. J Natl Cancer Inst. 99:244–252.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deschoolmeester V, Baay M, Wuyts W, Van
Marck E, Van Damme N, Vermeulen P, Lukaszuk K, Lardon F and
Vermorken JB: Detection of microsatellite instability in colorectal
cancer using an alternative multiplex assay of quasi-monomorphic
mononucleotide markers. J Mol Diagn. 10:154–159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pedroni M, Roncari B, Maffei S, Losi L,
Scarselli A, Di Gregorio C, Marino M, Roncucci L, Benatti P, Ponti
G, et al: A mononucleotide markers panel to identify hMLH1/hMSH2
germline mutations. Dis Markers. 23:179–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mariani F, Sena P, Pedroni M, Benatti P,
Manni P, Di Gregorio C, Manenti A, Palumbo C, de Leon MP and
Roncucci L: Th inducing POZ-Kruppel Factor (ThPOK) is a key
regulator of the immune response since the early steps of
colorectal carcinogenesis. PLoS One. 8:e544882013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang J: Beclin 1 bridges autophagy,
apoptosis and differentiation. Autophagy. 4:947–948. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mariani F, Sena P, Marzona L, Riccio M,
Fano R, Manni P, Gregorio CD, Pezzi A, Leon MP, Monni S, et al:
Cyclooxygenase-2 and hypoxia-inducible factor-1alpha protein
expression is related to inflammation, and up-regulated since the
early steps of colorectal carcinogenesis. Cancer Lett. 279:221–229.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Beasley NJ, Wykoff CC, Watson PH, Leek R,
Turley H, Gatter K, Pastorek J, Cox GJ, Ratcliffe P and Harris AL:
Carbonic anhydrase IX, an endogenous hypoxia marker, expression in
head and neck squamous cell carcinoma and its relationship to
hypoxia, necrosis, and microvessel density. Cancer Res.
61:5262–5267. 2001.PubMed/NCBI
|
|
28
|
Mizushima N: Methods for monitoring
autophagy. Int J Biochem Cell Biol. 36:2491–2502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jin S and White E: Role of autophagy in
cancer: Management of metabolic stress. Autophagy. 3:28–31. 2007.
View Article : Google Scholar
|
|
30
|
Giatromanolaki A, Koukourakis MI,
Koutsopoulos AV, Harris AL, Gatter KC and Sivridis E: Autophagy and
hypoxia in colonic adenomas related to aggressive features.
Colorectal Dis. 15:e223–e230. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sena P, Roncucci L, Marzona L, Mariani F,
Maffei S, Manenti A and De Pol A: Altered expression of apoptosis
biomarkers in human colorectal microadenomas. Cancer Epidemiol
Biomarkers Prev. 19:351–357. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Inbal B, Bialik S, Sabanay I, Shani G and
Kimchi A: DAP kinase and DRP-1 mediate membrane blebbing and the
formation of autophagic vesicles during programmed cell death. J
Cell Biol. 157:455–468. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Amaravadi RK, Yu D, Lum JJ, Bui T,
Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoshioka A, Miyata H, Doki Y, Yamasaki M,
Sohma I, Gotoh K, Takiguchi S, Fujiwara Y, Uchiyama Y and Monden M:
LC3, an autophagosome marker, is highly expressed in
gastrointestinal cancers. Int J Oncol. 33:461–468. 2008.PubMed/NCBI
|
|
36
|
Guo GF, Jiang WQ, Zhang B, Cai YC, Xu RH,
Chen XX, Wang F and Xia LP: Autophagy-related proteins Beclin-1 and
LC3 predict cetuximab efficacy in advanced colorectal cancer. World
J Gastroenterol. 17:4779–4786. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Miao Y, Zhang Y, Chen Y, Chen L and Wang
F: GABARAP is over-expressed in colorectal carcinoma and correlates
with shortened patient survival. Hepatogastroenterology.
57:257–261. 2010.PubMed/NCBI
|
|
38
|
White E, Karp C, Strohecker AM, Guo Y and
Mathew R: Role of autophagy in suppression of inflammation and
cancer. Curr Opin Cell Biol. 22:212–217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen N and Karantza V: Autophagy as a
therapeutic target in cancer. Cancer Biol Ther. 11:157–168. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hönscheid P, Datta K and Muders MH:
Autophagy: Detection, regulation and its role in cancer and therapy
response. Int J Radiat Biol. 90:628–635. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
van Lier MG, Leenen CH, Wagner A, Ramsoekh
D, Dubbink HJ, van den Ouweland AM, Westenend PJ, de Graaf EJ,
Wolters LM, Vrijland WW, et al LIMO Study Group: Yield of routine
molecular analyses in colorectal cancer patients ≤70 years to
detect underlying Lynch syndrome. J Pathol. 226:764–774. 2012.
View Article : Google Scholar
|
|
42
|
Jass JR, Do KA, Simms LA, Iino H, Wynter
C, Pillay SP, Searle J, Radford-Smith G, Young J and Leggett B:
Morphology of sporadic colorectal cancer with DNA replication
errors. Gut. 42:673–679. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thibodeau SN, Bren G and Schaid D:
Microsatellite instability in cancer of the proximal colon.
Science. 260:816–819. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lothe RA, Peltomäki P, Meling GI, Aaltonen
LA, Nyström- Lahti M, Pylkkänen L, Heimdal K, Andersen TI, Møller
P, Rognum TO, et al: Genomic instability in colorectal cancer:
Relationship to clinicopathological variables and family history.
Cancer Res. 53:5849–5852. 1993.PubMed/NCBI
|
|
45
|
Choi SW, Lee KJ, Bae YA, Min KO, Kwon MS,
Kim KM and Rhyu MG: Genetic classification of colorectal cancer
based on chromosomal loss and microsatellite instability predicts
survival. Clin Cancer Res. 8:2311–2322. 2002.PubMed/NCBI
|
|
46
|
Halling KC, French AJ, McDonnell SK,
Burgart LJ, Schaid DJ, Peterson BJ, Moon-Tasson L, Mahoney MR,
Sargent DJ, O'Connell MJ, et al: Microsatellite instability and 8p
allelic imbalance in stage B2 and C colorectal cancers. J Natl
Cancer Inst. 91:1295–1303. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hemminki A, Mecklin JP, Järvinen H,
Aaltonen LA and Joensuu H: Microsatellite instability is a
favorable prognostic indicator in patients with colorectal cancer
receiving chemotherapy. Gastroenterology. 119:921–928. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Samowitz WS, Curtin K, Ma KN, Schaffer D,
Coleman LW, Leppert M and Slattery ML: Microsatellite instability
in sporadic colon cancer is associated with an improved prognosis
at the population level. Cancer Epidemiol Biomarkers Prev.
10:917–923. 2001.PubMed/NCBI
|
|
49
|
Watanabe T, Wu TT, Catalano PJ, Ueki T,
Satriano R, Haller DG, Benson AB III and Hamilton SR: Molecular
predictors of survival after adjuvant chemotherapy for colon
cancer. N Engl J Med. 344:1196–1206. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Salazar R, Roepman P, Capella G, Moreno V,
Simon I, Dreezen C, Lopez-Doriga A, Santos C, Marijnen C, Westerga
J, et al: Gene expression signature to improve prognosis prediction
of stage II and III colorectal cancer. J Clin Oncol. 29:17–24.
2011. View Article : Google Scholar
|
|
51
|
Gray RG, Quirke P, Handley K, Lopatin M,
Magill L, Baehner FL, Beaumont C, Clark-Langone KM, Yoshizawa CN,
Lee M, et al: Validation study of a quantitative multigene reverse
transcriptase-polymerase chain reaction assay for assessment of
recurrence risk in patients with stage II colon cancer. J Clin
Oncol. 29:4611–4619. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
O'Connell MJ, Lavery I, Yothers G, Paik S,
Clark-Langone KM, Lopatin M, Watson D, Baehner FL, Shak S, Baker J,
et al: Relationship between tumor gene expression and recurrence in
four independent studies of patients with stage II/III colon cancer
treated with surgery alone or surgery plus adjuvant fluorouracil
plus leucovorin. J Clin Oncol. 28:3937–3944. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Deschoolmeester V, Baay M, Lardon F,
Pauwels P and Peeters M: Immune cells in colorectal cancer:
Prognostic relevance and role of MSI. Cancer Microenviron.
4:377–392. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Eveno C, Nemeth J, Soliman H, Praz F, de
The H, Valleur P, Talbot IC and Pocard M: Association between a
high number of isolated lymph nodes in T1 to T4 N0M0 colorectal
cancer and the microsatellite instability phenotype. Arch Surg.
145:12–17. 2010.PubMed/NCBI
|
|
55
|
Kuperwasser C, Chavarria T, Wu M, Magrane
G, Gray JW, Carey L, Richardson A and Weinberg RA: Reconstruction
of functionally normal and malignant human breast tissues in mice.
Proc Natl Acad Sci USA. 101:4966–4971. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kalluri R: Basement membranes: Structure,
assembly and role in tumour angiogenesis. Nat Rev Cancer.
3:422–433. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Pavlides S, Whitaker-Menezes D,
Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro
MC, Wang C, Fortina P, Addya S, et al: The reverse Warburg effect:
Aerobic glycolysis in cancer associated fibroblasts and the tumor
stroma. Cell Cycle. 8:3984–4001. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bonuccelli G, Tsirigos A, Whitaker-Menezes
D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N,
Howell A, Martinez-Outschoorn UE, et al: Ketones and lactate 'fuel'
tumor growth and metastasis: Evidence that epithelial cancer cells
use oxidative mitochondrial metabolism. Cell Cycle. 9:3506–3514.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Martinez-Outschoorn UE, Balliet RM,
Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes
D, Daumer KM, Lin Z, Witkiewicz AK, et al: Oxidative stress in
cancer associated fibroblasts drives tumor-stroma co-evolution: A
new paradigm for understanding tumor metabolism, the field effect
and genomic instability in cancer cells. Cell Cycle. 9:3256–3276.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Worthley DL, Giraud AS and Wang TC:
Stromal fibroblasts in digestive cancer. Cancer Microenviron.
3:117–125. 2010. View Article : Google Scholar
|