Introduction
Lung adenocarcinoma is the main cause of
cancer-related mortality worldwide (1,2). The
immune response against tumors requires activation of
tumor-specific CD8+ T cells to generate effector
CD8+ cytotoxic T lymphocytes (CTLs) with the ability to
kill tumor cells (3). The antitumor
CD8+ T cells are activated by signals resulting from T
cell receptor (TCR) recognition of specific tumor-derived peptide
antigens presented by major histocompatibility complex class I
(MHC-I) molecules on dendritic cells (DCs) (3,4). The
CTL response to cancer is subordinated to the immunosuppressor
environment that takes place during the progression of tumors
(4). One treatment strategy is
adoptive immunotherapy that involves the transfusion of autologous
CD8+ CTLs to remove the malignant cells (5). Efforts to activate and expand
tumor-specific CTLs in vitro have been focused on in the
search for immunogenic tumor-associated antigens (TAAs) as well as
appropriate tumor antigen-presenting cells (APCs) (5,6). The
most significant antigen expressed in the vast majority of
adenocarcinomas is a hypoglycosylated isoform from human mucin 1
(MUC1) protein, which exhibits immunogenic peptide sequences
(7,8). Among MUC1-derived peptides, the
H-2kb-restricted MUC1-SAPDTRPA (MUC1-8-mer) peptide has
proven to be the most immunogenic epitope for murine T cell
activation (9,10). MHC-binding epitope prediction
analysis showed that the MUC1-8-mer peptide is also restricted to
HLA-A2 molecules (11). The T2 cell
line expresses HLA-A2 molecules; therefore it has been used as an
APC to activate distinctive TAA-specific CD8+ T cells
from healthy volunteers (12).
Additionally, T2 cells have been used to activate cancer-patient
CD8+ T cells specific for TAA-derived peptides, but not
MUC1-derived peptides (13). Our
aim was to evaluate i) whether T2 cells can present the MUC1-8-mer
peptide, and ii) to determine whether MUC1-8-loaded T2 cells
activate and expand CD8+ T cells isolated from lung
adenocarcinoma HLA-A2+ patients.
Materials and methods
Lung adenocarcinoma patients
Nine adult patients with a diagnosis of non-small
cell lung cancer established by clinical history, physical
examination, chest X-rays, and histopathology were included. The
patients were hospitalized at the Oncology Unit at the Instituto
Nacional de Enfermedades Respiratorias 'Ismael Cosío Villegas' in
Mexico City. The patient recruitment criteria included patients
with a diagnosis of lung adenocarcinoma who had not undergone any
previous cancer-associated surgery or medical treatment. Patients
were classified as stage III and IV according to the standard
criteria of the Tumor, Node and Metastasis (TNM) system (14). A peripheral blood sample was
obtained from each patient before the start of anticancer
chemotherapy or radiotherapy. Ten age-matched and clinically
healthy volunteers with no history of cancer were included as
controls. The Science and Bioethics Committee of our Institution in
accordance with the Declaration of Helsinki approved the study, and
patients and healthy volunteers provided informed consent for blood
sampling after written information was provided.
Monoclonal antibodies and reagents
Peridinin chlorophyll protein complex-cyanine 5.5
(PerCP-Cy5.5)-labeled anti-human CD3 (clone SK7) monoclonal
antibody (mAb), phycoerythrin (PE)-labeled anti-human CD4 (clone
OKT4) mAb, fluorescein isothiocyanate (FITC)-labeled anti-human CD8
(clone SK1) and anti-HLA-A2 (clone BB7.2) mAbs, and PerCP-Cy5.5-,
PE-, FITC-labeled isotype control (clone MOPC-21) mAbs, and human
recombinant IL-2 were purchased from BioLegend, Inc. (San Diego,
CA, USA). PE-labeled anti-human CD25 (clone M-A251) mAb and
7-amino-actinomycin-D (7-AAD) were acquired from BD Biosciences
(San Jose, CA, USA). Alexa Fluor 594-labeled goat anti-IgG mouse
antibody was obtained from Molecular Probes-Life Technologies
(Eugene, OR, USA). Human β2 microglobulin
(β2m) and mouse anti-CA 27–29 (clone M4021209, specific
for SAPDTRPA) mAb were obtained from Fitzgerald Industries
International (Acton, MA, USA). Blood DNA isolation and Fastype
HLA-DNA SSP Typing system kits were provided by Bio-Synthesis Inc.
(Lewisville, TX, USA). Lymphoprep™ (Ficoll 1.077 density) was from
Axis-Shield PoC As (Oslo, Norway). CD8+ T cell negative
isolation kit in a magnetic antibody cell sorting (MACS) system
containing biotin-labeled antibodies to human CD4, CD15, CD16,
CD19, CD34, CD36, CD56, CD123, TCR-γδ, CD235a (glycophorin A) and
magnetic microbeads coated with mouse Abs against biotin and human
CD14; as well as mAbs to human CD2, CD3 and CD28 from the T cell
activation/expansion kit were from Miltenyi Biotec (Bergisch
Gladbach, Germany). Fetal bovine serum (FBS; Performance Plus),
penicillin, streptomycin, L-glutamine and recombinant Taq
DNA polymerase were purchased from Gibco-Life Technologies
(Rockville, MD, USA). Carboxyfluorescein succimidyl ester (CFSE)
was from Invitrogen (Camarillo, CA, USA). Vectashield mounting
medium with DAPI was from Vector Laboratories, Inc. (Burlingame,
CA, USA). RPMI-1640 culture medium, bovine serum albumin fraction V
(BSA), ethylenediaminetetraacetic acid (EDTA), dimethyl sulfoxide,
agarose, ethidium bromide, trypan blue dyes, and salt reagents were
obtained from Sigma-Aldrich (St. Louis, MO, USA).
Peptides
The SAPDTRPA-human mucin 1 (MUC1-8-mer peptide),
GILGFVFTL-influenza A virus matrix protein-158–66
(IVMP1-9-mer peptide), and SIINFEKL-chicken
ovalbumin257–264 (OVA-8-mer peptide) (15–17)
were synthesized by the Instituto de Biotecnología at the
Universidad nacional Autónoma de México in Cuernavaca (Morelos,
Mexico) on a 430A multiple peptide synthesizer (Applied Biosystems,
San Diego, CA, USA) according to commercially available
manufacturer's protocols. The affinity of these peptides for the
HLA-A2 molecule was confirmed by NetMHC 3.4 Server software
(18). The purity of the peptides
was >95%, and their molecular weights were assessed by high
performance liquid chromatography and confirmed by mass
spectrometry. The peptides were dissolved in dimethyl sulfoxide at
a concentration of 10 mg/ml and stored at −70°C until required.
Cells
Peripheral blood mononuclear cells (PBMCs) were
isolated from 20 ml heparinized whole blood by Ficoll density
gradient centrifugation for 30 min at 360 g and 10°C (19). After centrifugation, the interface
cells were collected, washed twice in RPMI-1640 medium, and counted
in a neubauer chamber to assess cell viability via the trypan blue
dye exclusion test.
The human T2 cell line was obtained from the
American Type Culture Collection (ATCC, CRL-1992™; Manassas, VA,
USA). T2 cells express an HLA-A2 molecule that lacks TAP function,
so it can easily be loaded with exogenous peptides for
CD8+ T cell recognition (20). The T2 cell line was cultured in
RPMI-1640 medium supplemented with 20% heat-inactivated FBS, 100
µg/ml streptomycin, 100 U/ml penicillin, and 2 mM
L-glutamine (20% FBS supplemented-RPMI medium) at 37°C in a
humidified atmosphere containing 5% CO2.
Purification of CD8+ T
cells
Cytotoxic CD8+ T cells were isolated from
PBMCs by a negative magnetic selection kit (Miltenyi Biotec).
Briefly, PBMCs (1×107 cells) were suspended in 40
µl phosphate-buffered saline (PBS: 0.01 M sodium phosphate,
0.15 M sodium chloride, pH 7.2) supplemented with 0.5% BSA and 2 mM
EDTA and incubated with biotin-antibodies to human leukocyte
phenotype molecules for 10 min at 4°C, followed by a second
incubation with magnetic microbeads coated with mouse Abs against
biotin and human CD14 for an additional 15 min at 4°C. The purity
percentage for magnetically unlabeled CD8+ T cells was
always >95%, as determined by flow cytometry via incubation with
PE-Cy5.5-anti-human CD3, PE-anti-CD4, and FITC-anti-CD8 mAbs for 30
min at 4°C. The magnetically labeled CD8− T cells were
used to identify HLA-A2 alleles.
DNA typing for the HLA-A2 allele
Genomic DNA was extracted from CD8− T
cells by a blood DNA isolation kit (Bio-Synthesis) according to the
manufacturer's instructions. The total DNA concentration was
quantified by spectrophotometry at 260 and 280 nm using an ASP-2680
spectrophotometer (ACTGene Inc., Piscataway, NJ, USA). The DNA was
suspended in 100 µl of the elution buffer and stored at
−20°C until use. Molecular typing was performed with the polymerase
chain reaction (PCR) sequence-specific primer (SSP) technique using
a Fastype HLA-DNA SSP Typing system kit (Bio-Synthesis). For HLA-A
typing, 24 primer pairs were used at a low-resolution modality.
Briefly, PCR amplifications were carried out on 1.8 µg of
genomic DNA in a 24-µl reaction volume containing 50 mM KCl,
10 mM Tris-Cl, pH 8.3 and 1.5 mM MgCl2; 60 µM of
each dNTP (21). Samples were
subjected to 20 cycles at 94°C for 20 sec for denaturing, 20 cycles
at 61°C for 50 sec for annealing, and 20 cycles at 72°C for 30 sec
for extension using an automated thermal cycler (GeneAmp PCR system
9700; Applied Biosystems, Foster City, CA, USA). An additional hold
of 94°C for 20 sec and then 10 cycles at 65°C for 1 min were added
to the denaturing step before the first cycle. After the last
cycle, the extension step received an additional 5 min at 72°C. The
amplifications were achieved using recombinant Taq DNA
polymerase. The integrity of amplified PCR SSP products was
assessed by submarine 2% agarose gel electrophoresis and staining
with 0.01 mg/ml ethidium bromide for 40 min. Each DNA sample was
then visualized in a dual intensity ultraviolet (UV) light
transilluminator (UVP Inc., Upland, CA, USA) and analyzed with the
Kodak EDAS-290 gel documentation system (Kodak, Rochester, NY, USA)
according to the electrophoretic migration of the DNA sample
compared with the internal control primer pair specific for the
human G3PDH gene (Bio-Synthesis). The results were interpreted
following the instructions on the typing sheets from the procedure
guide.
Peptide loading on the T2 cell line
The HLA-A2-binding ability of the MUC1-8-mer peptide
was assessed by HLA-A2 membrane stabilization on the T2 cell line
according to a previously described method (13). In brief, T2 cells (2×105)
were placed in flat-bottomed, 96-well cell culture plates (Thermo
Scientific Nunc, Roskilde, Denmark) in 10% FBS supplemented-RPMI
medium and incubated with both MUC1-8-mer peptide (100
µg/ml) and human β2m (20 µg/ml) for 24 h
at 37°C in 5% CO2 atmosphere. The optimal dose of both
β2m and the MUC1-8 peptide was previously obtained from
different concentrations tested. The T2 cells incubated either with
the HLA-A2 restricted IVMP1-9-mer or the OVA-8-mer peptides were
used as positive controls (16,17),
whereas cells cultured in the absence of the MUC1-8-mer peptide
were considered to be the negative control. After incubation, T2
cells were washed twice with PBS containing 0.2% BSA and sodium
azide (PBS-BSA buffer) and stained with FITC-anti-HLA-A2 mAb for 30
min at 4°C. Finally, the cells were analyzed by flow cytometry
where the HLA-A2 expression on T2 cells was evaluated as the mean
fluorescence intensity (MFI), which was calculated by obtaining the
MFI difference between cells incubated in the presence or absence
of the MUC1-8-mer peptide.
Flow cytometry
Cells were acquired using a FACScan flow cytometer
(Becton Dickinson, San Jose, CA, USA) and analyzed with FlowJo
software version 8.7.7. (Tree Star Inc., Ashland, OR, USA). To
analyze the immunofluorescence staining of cell-surface molecules,
25,000 events were counted in linear mode for both forward scatter
(FSC) and side scatter (SSC). Cells were then gated by their
physical properties (FSC and SSC) and analyzed with log
amplification for immunofluorescence. Data are presented as
histograms or two-dimensional dot-plots. Fluorescent
staining-labeled isotype-matched control mAbs were used to assess
background staining.
Confocal microscopy
T2 cells (2×105) were cultured in 8-well
microchamber glass slides (BD Falcon™, Bedford, MA, USA) in 10% FBS
supplemented-RPMI medium and incubated with MUC1-8 peptide plus
β2m for 24 h under the conditions as described above.
After the culture, the cells were washed twice with PBS containing
1% BSA and 0.2% sodium azide and incubated with Alexa Fluor
594-labeled goat anti-mouse IgG mAb for 30 min after incubation
with the anti-CA 27–29 mAb for 2 h at 4°C. A second staining was
performed with FITC-labeled anti-HLA-A2 mAb for 30 min at 4°C.
Cells incubated with the FITC-isotype control and the Alexa Fluor
594 secondary antibody, were used as controls. For colocalization
analysis, the cells were fixed in 1% p-formaldehyde, and the slides
were mounted in Vectashield with DAPI diluted 1:3 in PBS.
Fluorescence images were acquired with an LSM-510 zeiss confocal
microscope (Carl zeiss, Oberkochen, Germany) using a 63X objective
lens. All images were captured under the same exposure,
magnification and intensification. The digital images were
processed by ImageJ software (Wayne Rasband, NIH, Bethesda, MA,
USA) and analyzed using Mander's coefficient (22). Finally, the images were clarified by
Adobe Photoshop CS2 software (Adobe Systems, Agrate Bianza,
Italy).
In vitro activation and expansion of
CD8+ T cells by MUC1-8-mer peptide-loaded T2 cells
MUC1-8-mer peptide-loaded T2 cells were used as APCs
to activate freshly obtained CD8+ T cells from lung
adenocarcinoma patients or healthy controls, using a modified
method described by Jaramillo et al (13). In brief, T2 cells (2×105)
were cultured with the MUC1-8-mer peptide plus β2m for
24 h, as described above. Cells were then harvested, washed in the
culture medium, and fixed in 1% p-formaldehyde for 30 min at 4°C.
After washing, fixed T2 cells were cultured with CD8+ T
cells (4×105) in 96-well plates in 10% FBS-supplemented
RPMI medium in the presence of 2 µl of bead particle-coupled
anti-CD2 and anti-CD28 mAbs from a T cell activation/expansion
system (Miltenyi Biotec) for 6 days at 37°C in a 5% CO2
atmosphere. Human recombinant IL-2 (20 U/ml) was added after 3 days
of cell culture, when half of the culture medium supernatant was
removed and the well was replenished with fresh 10%
FBS-supplemented RPMI medium. CD8+ T cells cultured with
MUC1-8-mer peptide loaded-T2 cells, and CD8+ T cells
with anti-CD2 and anti-CD28 mAbs were used as the specific-antigen
CD8+ T cell stimulation control. CD8+ T cells
only activated with mAbs to CD3, CD2, and CD28 were used as the
unspecific-antigen CD8+ T cell stimulation positive
control. At the end of the culture, CD8+ T cells were
harvested, washed in a PBS-BSA buffer, and stained with
FITC-anti-CD8 and PE-anti-CD25 mAbs for 30 min at 4°C. To exclude
dying cells before acquisition in a flow cytometer, cells were
additionally incubated in a 7-AAD staining solution for 15 min at
4°C, washed in PBS-BSA buffer and analyzed by flow cytometry.
For clonal expansion detection, the CD8+
T cells suspended in RPMI-1640 medium were stained with 15
µl of 0.5 mM CFSE (prepared from a 5-mM stock solution
dissolved in dimethyl sulfoxide) for 15 min at 37°C in darkness
(23). After incubation, the cells
were washed twice in 10 ml of 10% FBS-supplemented RPMI medium and
the cell viability was evaluated by trypan blue dye exclusion test.
CFSE-labeled CD8+ T cells (4×105) were then
cultured with fixed MUC1-8-mer peptide-loaded T2 cells
(2×105) in the presence of costimulatory antibodies plus
IL-2 (added every third day) for 10 days. An additional stimulation
of CD8+ T cells was carried out through adding fixed
MUC1-8-mer peptide-loaded T2 cells (2×105) on day 7 of
culture after removing half of the culture medium supernatant and
replenishing the well with fresh 10% FBS-supplemented RPMI
medium.
Statistical analysis
The entire analysis was performed by STATA™ 10
software using a Shapiro-Wilk test to identify population
distributions. Because the variables were asymmetrically
distributed, a Wilcoxon rank-sum (Mann-Whitney) test was carried
out for their comparison. Values are shown as median (md) and
interquartile range (iqr). Some data showed symmetry; thus, they
were analyzed using the Student's t-test, and the values are
displayed as mean ± standard deviation. Differences between groups
were considered statistically significant at P<0.05.
Results
Patient characteristics
The mean patient group age was 63.67±10.7 years
(range, 51–74 years); among these, 4 were male (44%) and 5 were
female (56%); 7 out of 9 patients were HLA-A2+. Four of
the patients had metastasis; 6 of the 9 total patients presented
with tumor stage IV, whereas the remaining 3 had tumor stage III
(Table I). The control group mean
age was 59.1±9.3 years (range, 34–64 years); 6 were male and 4
female.
 | Table IGeneral data of the non-small cell
lung adenocarcinoma patients and healthy controls. |
Table I
General data of the non-small cell
lung adenocarcinoma patients and healthy controls.
| Participant | Gender/age
(years) | HLA-A2 |
Stage/classification | Histopathology for
MUC1 |
|---|
| HC1 | M/38 | – | – | – |
| HC2 | M/59 | – | – | – |
| HC3 | F/34 | – | – | – |
| HC4 | F/62 | – | – | – |
| HC5 | M/59 | – | – | – |
| HC6 | F/62 | + | – | – |
| HC7 | F/59 | + | – | – |
| HC8 | M/64 | + | – | – |
| HC9 | M/51 | + | – | – |
| HC10 | M/63 | + | – | – |
| P1 | F/74 | – | T4N3M3 | + |
| P2 | M/53 | – | T4N3M0 | + |
| P3 | F/62 | + | T4N3M0 | + |
| P4 | F/74 | + | T4N3M1 | + |
| P5 | M/53 | + | T4N2M0 | + |
| P6 | M/71 | + | T4N3M2 | + |
| P7 | M/51 | + | T3N2M0 | + |
| P8 | F/69 | + | T3N2M0 | + |
| P9 | F/66 | + | T3N3M1 | + |
MUC1-8-mer peptide plus β2m
increases the expression of HLA-A2 molecules on the T2 cell
surface
The HLA-A2-specific affinity of the MUC1-8-mer
peptide to T2 cells was predicted by NetMHC 3.4 software (18), and compared to the affinity of the
well-recognized HLA-A2-specific IVMP1-9-mer and OVA-8-mer epitopes
(Table II). T2 cells pulsed with
the MUC1-8-mer peptide in the presence of β2m showed an
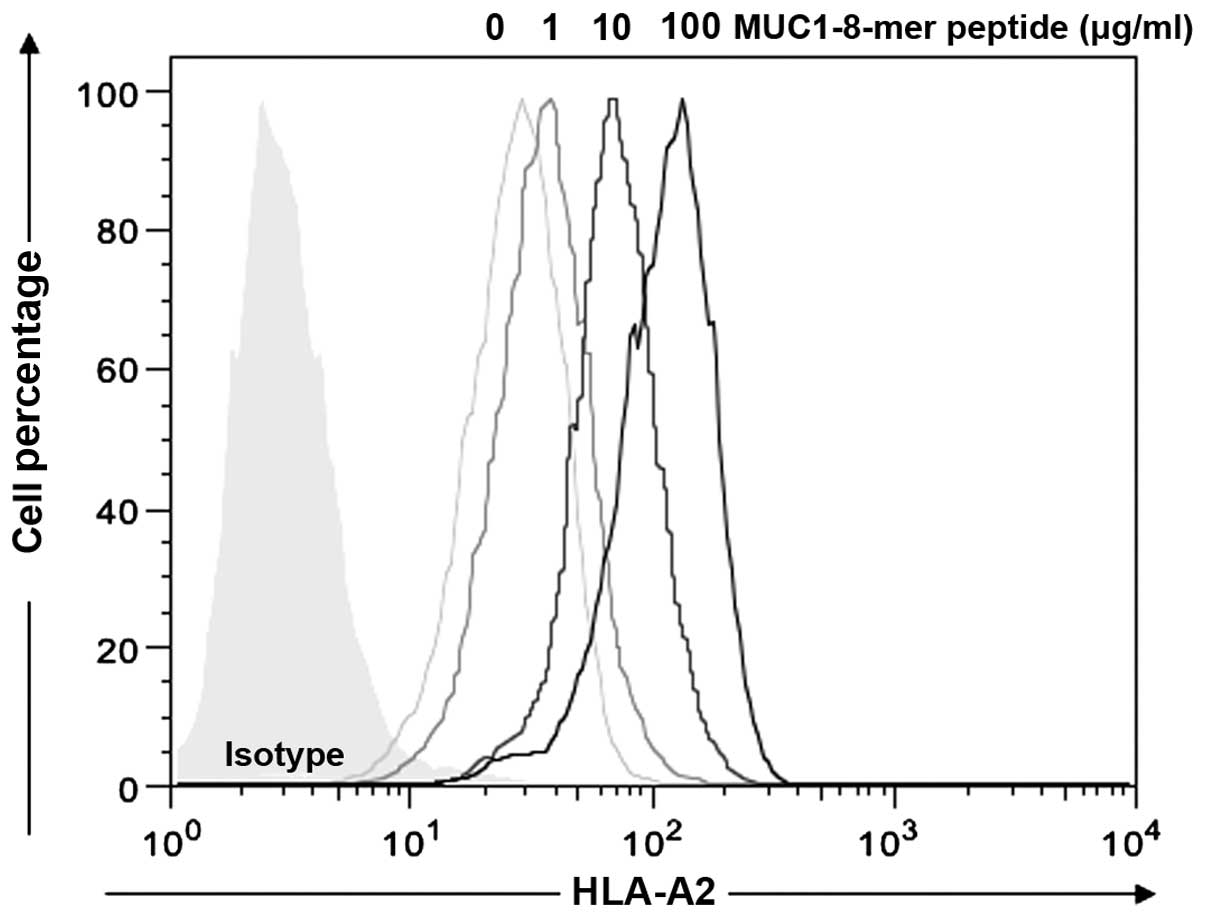
increase in HLA-A2 molecule expression in a dose-dependent manner
(0, 1, 10 and 100 µg); the basal value was 31.6±12.7; for 1
µg/ml peptide the value increased to 40.3±18.9; for 10
µg/ml, 77.1±32.3 and for 100 µg/ml, 123±52,
respectively (Fig. 1).
Concentrations >100 µg/ml of the MUC1-8-mer peptide did
not increase HLA-A2 expression on the T2 cells. Therefore
MUC1-8-mer peptide at 100 µg/ml was considered as the
optimal concentration for expression of HLA-A2 molecules and this
was the concentration used in all the subsequent experiments.
| Table IIHLA-A2 peptide binding predictions of
the NetMHC 3.4 Server software. |
Table II
HLA-A2 peptide binding predictions of
the NetMHC 3.4 Server software.
| Peptide name | Sequence | Logscore | Affinity (nM) | Binding level |
|---|
| MUC1-8-mer | SAPDTRPA | 0.073 | 22,690 | Medium |
| OVA-8 | SIINFEKL | 0.206 | 5,377 | Medium |
| IVMPI-9 | GILGFVFTL | 0.769 | 12 | Strong binder |
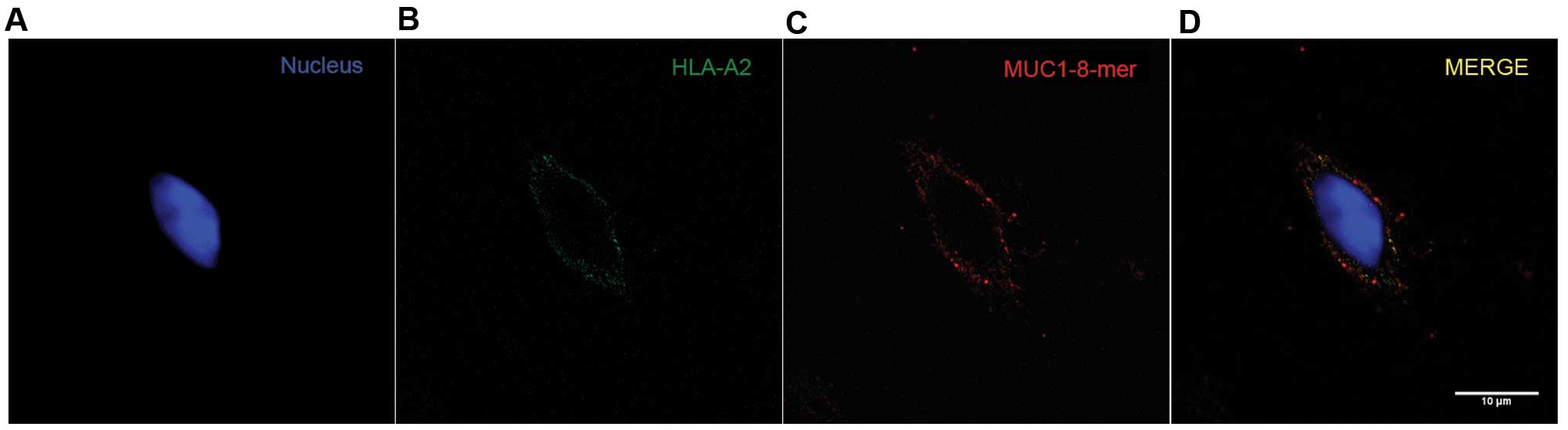
MUC1-8-mer peptide and the HLA-A2
molecule colocalize on the T2 cell surface
To confirm the assembly of the MUC1-8-mer
peptide-HLA-A2 complex onto the T2 cell surface, an anti-CA 27–29
mAb (antibody specific for the SAPDTRPA peptide sequence) was used
together with the anti-HLA-A2 mAb. The analysis of the overlap of
fluorescent emissions from the green channel (HLA-A2) and the red
channel (MUC1-8-mer peptide) on the T2 cell membrane showed
significant values for the Mander's coefficient, which were from
0.04 to 1 (22). As shown in
Fig. 2, one T2 cell is shown
separately in the green and red channels; in the third image, both
fluorescence emissions are merged (yellow color) and distributed
along the cell membrane, indicating that the MUC1-8-mer peptide is
found in the same place as the HLA-A2 molecule (22).
MUC1-8-mer peptide-loaded T2 cells plus
costimulatory antibodies induce activation and expansion of
CD8+ T cells from HLA-A2 patients
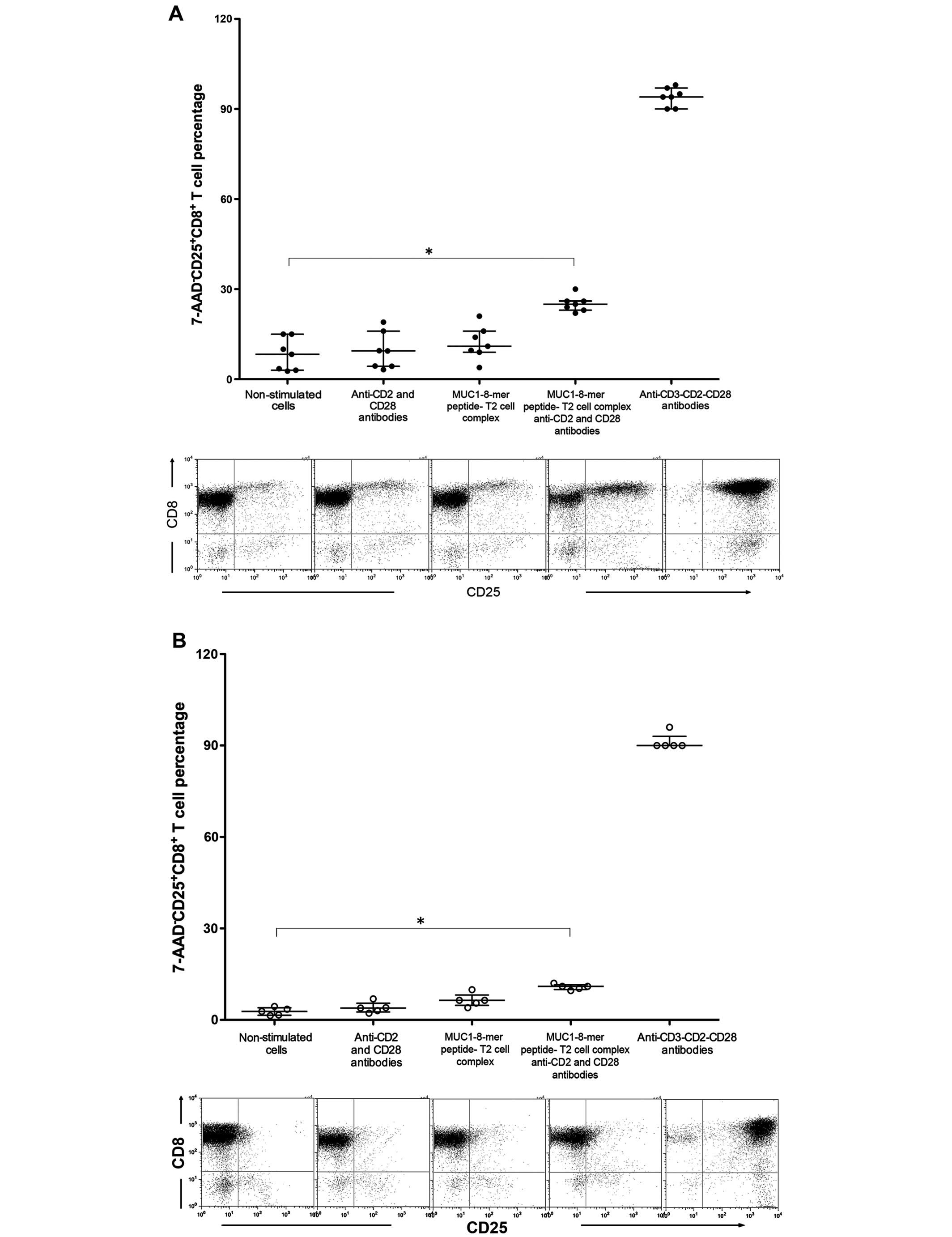
The percentage of CD25+CD8+ T
cells after stimulated with the T2 cell-MUC-1-8-mer complex and
anti-CD2 and CD28 was 4.2-fold higher in the HLA-A2+
patients than those from the HLA-A2− patients (md 25%,
iqr 22–30 vs. md 6%, iqr 5.3–6.7).
In the HLA-A2+ patients, the percentage
of CD25+CD8+ T cells stimulated with anti-CD2
and anti-CD28 or the MUC1-8-mer-T2 cell complex was identical to
that observed in the non-stimulated CD8+ T cells;
notably when the CD8+ T cells were stimulated with the
MUC-1-8-mer T2 cell complex and anti CD2-CD28 mAbs, there was a
3-fold increase compared to the non-stimulated cells (md 25.0%, iqr
22–30 vs. md 8.3%, iqr 3.7–15, P=0.018) (Fig. 3A). Similarly, the proportions of
CD25-expressing CD8+ T cells from HLA-A2+
healthy controls showed an identical behavior but with lower values
(md 11%, iqr 9.6–12 stimulated cells vs. md 2.8%, iqr 1.4–4.4
non-stimulated cells, P=0.018) (Fig.
3B). non-stimulated CD8+ T cells from
HLA-A2+ patients showed that there was dispersion in the
percentages of CD25+CD8+ T cells (Fig. 3A), therefore samples from cancer
patients were divided according to their TnM classification
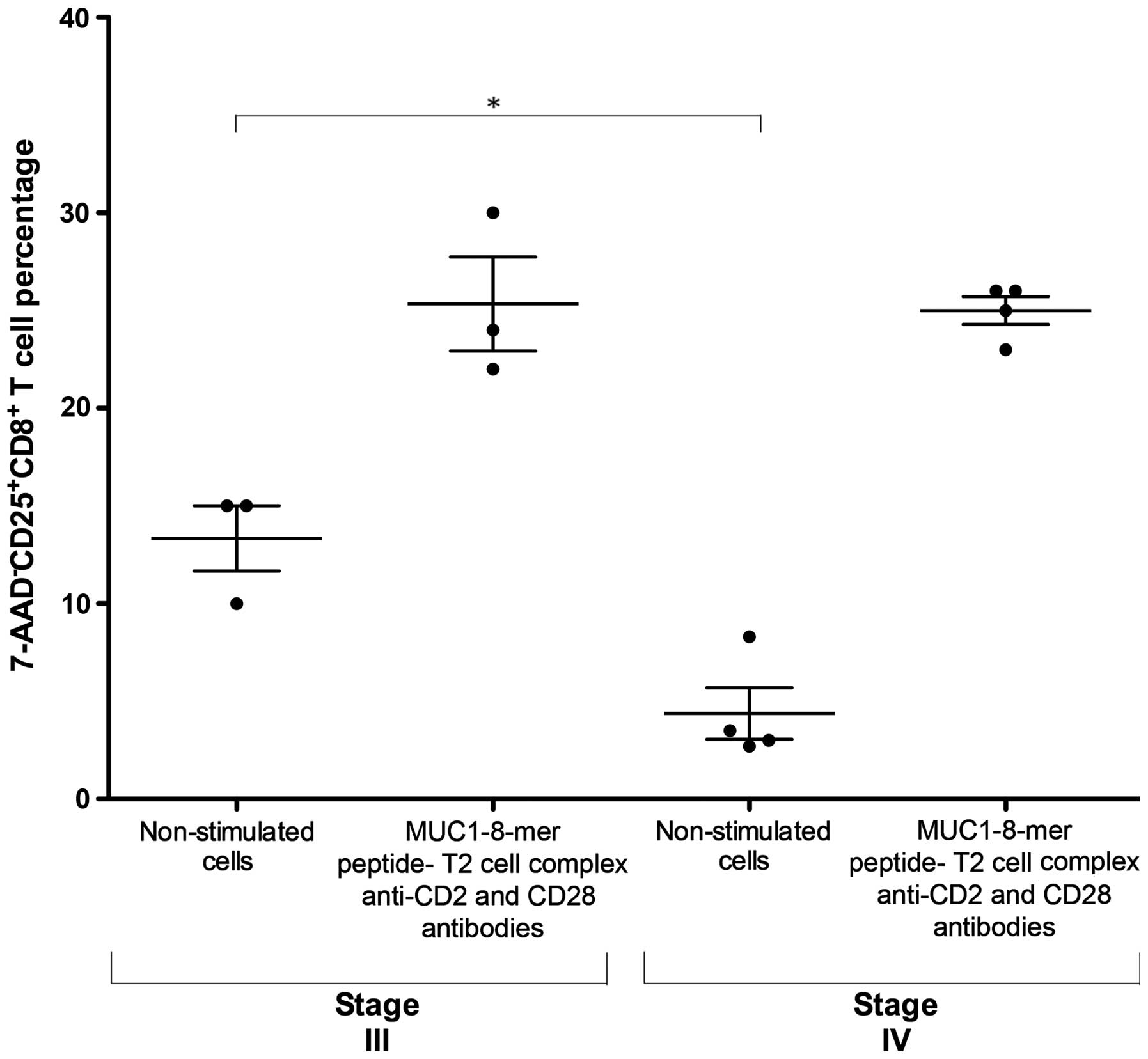
(14). Fig. 4 shows that non-stimulated cells from
patients with stage III cancer exhibited a higher proportion of
CD25 expression than non-stimulated cells from patients with stage
IV (md 15%, iqr 10–15 vs. md 3.2%, iqr 2.7–8.3, P=0.03). notably,
the proportion of CD25+CD8+ T cells was
similar in both patient groups after stimulation with the
MUC1-8-mer peptide-T2 cell complex in the presence of anti-CD2 and
anti-CD28 mAbs plus IL-2 (md 24%, iqr 22–30 vs. md 25.5%, iqr
23–26, respectively). In contrast, the polyclonal activation of
CD8+ T cells from healthy controls and patients with
anti-CD3, anti-CD28 and anti-CD2 showed similar activation values,
but the expression intensity of CD25+ cells was lower in
the cancer patients (Fig. 3).
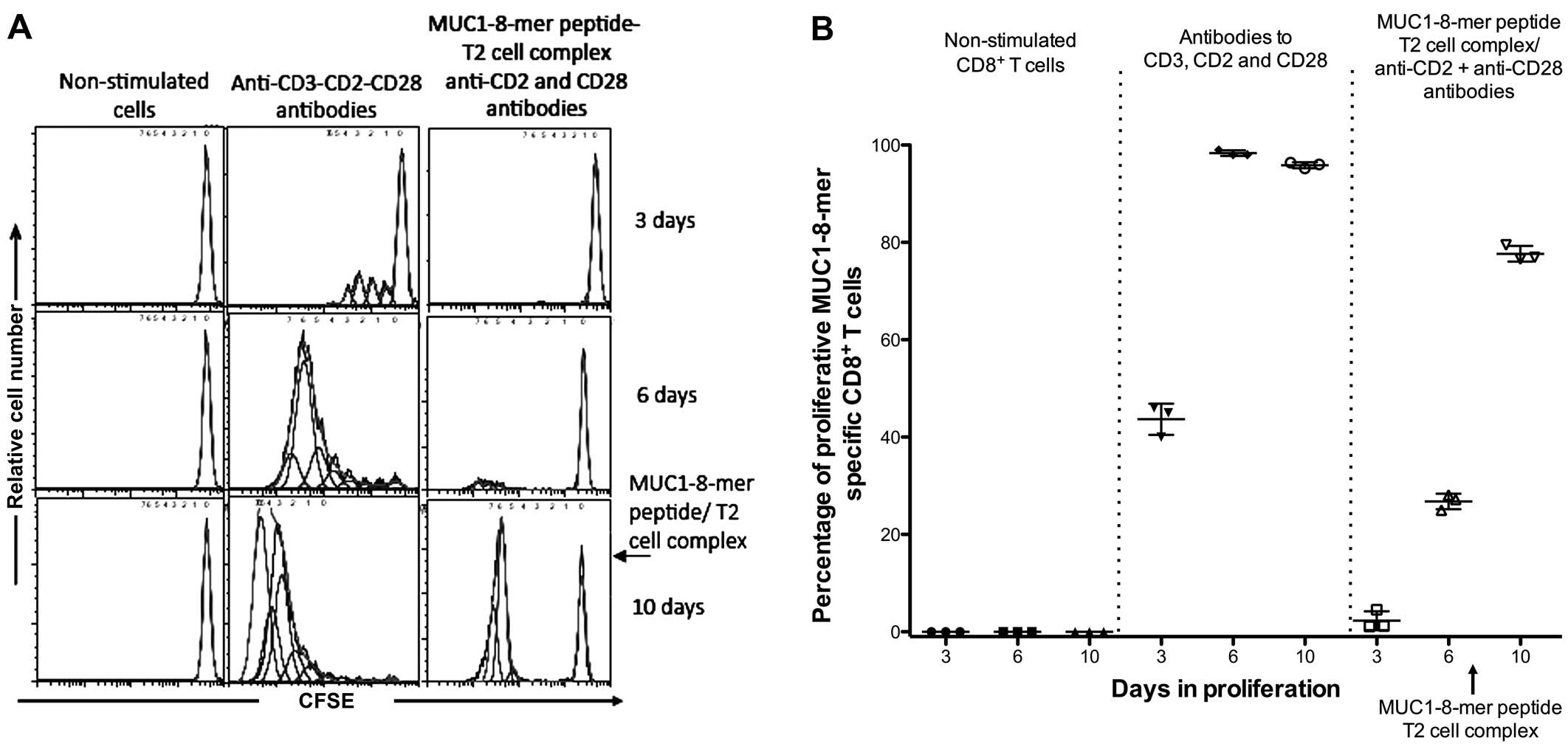
To evaluate the proliferative response of
antigen-specific cells, restimulation of CD8+ T cells
with the MUC1-8-mer peptide-T2 complex was performed after 7 days
of culture. Three days later, the cells were fixed and clonal
expansion was determined by CFSE treatment. Fig. 5A shows the proliferative response of
CFSE-labeled CD8+ T cells specific to the MUC1-8-mer
peptide. This cell population had a 77.6% increase after the
restimulation in comparison to the 26.7% observed in CFSE-labeled
CD8+ T cells activated with the MUC1-8-mer peptide-T2
complex plus anti-CD2 and anti-CD28 antibodies (Fig. 5B).
Discussion
Conventional treatments against lung cancer are
unsatisfactory in most cases, thus a more effective therapy for the
removal of tumor cells is urgent (24,25).
Adoptive T cell therapy has shown promising results as a treatment
strategy for cancer patients (5).
However, the challenge that faces this type of therapy is the ex
vivo activation and expansion of CTLs from a limited number of
peripheral blood mononuclear cells from patients with advanced
cancer (26). The identification of
immunogenic TAA epitopes and selection of appropriated APCs are
crucial to activate tumor-specific CTLs (6,27). Our
results confirmed the binding of the MUC1-8-mer peptide into
β2m established T2 cells according to MHC-binding
epitope predicting analysis (11).
Type 1 mucin (MUC1) is an important TAA in adenocarcinomas; diverse
MUC1-derived peptides have been used to induce a CD8+
CTL response in patients with adenocarcinomas (6). Among MUC1-derived peptides, the
MUC1-8-mer peptide SAPDTRPA has been shown to be an immunogenic
epitope that activates murine T cells (10). The advantage of T2 cells is that
they lack TAP function and consequently they have empty and
unstable HLA-A2 molecules (20).
This characteristic gives them a potential advantage as APC
peptides can be loaded exogenously onto the HLA-A2 molecules to
activate TAA-specific CTLs (12,13).
The use of autologous monocyte-derived DCs as APCs would be ideal
to generate efficient costimulatory and antigen-specific signals
for activation of autologous T cells (11). However, the number of functional
monocyte-derived DCs from cancer patients is low in long lasting
cultures due to their limited replicative potential (5).
Our results indicated that the MUC1-8-mer peptide-T2
complex only induced activation of purified CD8+ T cells
when anti-CD28 and anti-CD2 antibodies plus IL-2 were added to the
cell culture. The additional costimulation generated by beads
coupled with anti-CD2 and anti-CD28 antibodies and recombinant IL-2
allowed clonal expansion of MUC1-8 peptide-specific CD8+
T cells isolated from HLA-A2 adenocarcinoma patients. The
antibodies to costimulatory molecules induce formation of actin
filaments between the beads and CD8+ T cell surface
mimicking the immunological synapsis between APCs and T cells
(28,29). Our results also showed that the
activation of MUC1-8 peptide-specific CD8+ T cells was
similar to those from different activation systems using other
MUC1-derived epitopes (30–33). This finding confirms reports in
which CD8+ T cells from HLA-A2+ healthy
donors recognize distinct MUC1-derived peptides (11,31,33).
Mamaglobulin A-derived peptides loaded into T2 cells stimulated
with soluble anti-CD28 antibodies plus recombinant human IL-2 have
been used to activate HLA-A2+ CD8+ T cells in
breast cancer patients (13).
However, in this system, expansion of CTLs required necessarily,
several weekly restimulations to maintain the activity against
diverse cell lines (13). Our
results were similar but opposed to Jaramillo et al
(13), as we evaluated the
percentage of proliferative cells.
Notably, we observed the preexistence of
CD8+ T cells reactive to the MUC1-8 peptide in
HLA-A2+ patients as well as in HLA-A2+
healthy controls; the latter could be because the MUC1-8 peptide
has been shown to be highly immunogenic in a murine model (10), thus suggesting that there is a basal
T cell population recognizing MUC1 in healthy individuals. The
proportion of CD8+ T cells from HLA-A2+
healthy controls activated by MUC1-8 peptide-loaded T2 cells was
significantly lower than those from HLA-A2+ cancer
patients after 6 days of culture. This suggests that
MUC1-recognizing T cells in healthy individuals are capable of
continuously limiting the development of a tumor whereas in
adenocarcinoma patients this ability has been lost probably as a
results of the immunosuppressive environment that has been well
established (34–36).
Our results also showed that the proportions of
CD25+CD8+ non-stimulated T cells isolated
from stage III and IV patients were significantly different, but
that after stimulation with the MUC1-8-mer peptide T2 complex in
the presence of anti-CD2 and CD28 antibodies the same amount of
CD25+CD8+ T cells was induced. This confirms
that the potential to respond to APCs is maintained in both stages,
but the amount of CD25 expression in the stage IV cells was
diminished, as possibly the cells keep their ability to respond to
an external antigen-presenting system (37). The range of possible explanations
include different cytokine environment (34–36);
rescue of the cell signaling mechanism by the direct and continuous
contact with the anti-CD28 antibody that surpasses the inhibitory
signal mediated by CTLA-4 as observed in CD4+ T cells
(38); lack of contact with Treg
cells as the experiments were performed in vitro, or tumor
progression (35,36,39).
Ongoing experiments are being performed in our laboratory to try to
determine a possible mechanism.
Taken together, we modified an in vitro
system that uses MUC1-8 peptide-pulsed T2 cells and improved it
through the stimulation of CD8+ T cell costimulatory
molecules, such as CD2 and CD28. Under these conditions, we found
that preexistent MUC1-specific CD8+ T cells from
HLA-A2+ lung adenocarcinoma patients and
HLA-A2+ healthy controls were efficiently activated. The
specificity of this system allowed us to distinguish between cancer
patients and healthy individuals. Furthermore, clonal expansion of
MUC1-specific CD8+ T cells from cancer patients occurred
independently of tumor disease progression. This activation system
could be an innovative tool to induce and expand tumor-specific
CTLs from HLA-A2+ patients with adenocarcinoma.
Acknowledgments
We thank Dr Demetrio Bernal-Alcántara and Dr Raúl
Mancilla for technical assistance and helpful discussions. We also
thank Dr Rafael Wong Michell and Ing. Julio César Miranda Amador
for substantial collaboration on the development of the project.
This study was supported by Consejo nacional de Ciencia y
Tecnología (CONACYT) Project SALUD-2012-01-180516 and student
scholarship 245173 from Red Temática Glicociencia en Salud 253596
del CONACYT, Mexico. This article is part of the requirements for
obtaining the degree of PhD for José Agustín Atzin Méndez in the
program of Doctorado en Ciencias Biológicas at the Facultad de
Medicina of the Universidad nacional Autónoma de México,
Mexico.
References
|
1
|
Nakamura H and Saji H: Worldwide trend of
increasing primary adenocarcinoma of the lung. Surg Today.
44:1004–1012. 2014.
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015.
|
|
3
|
Abbas AK, Lichtman AH and Pillai S:
Cellular and Molecular Immunology. 8th edition. Elsevier Saunders;
Philadelphia, PA: pp. 109–220. 2015
|
|
4
|
Vesely MD, Kershaw MH, Schreiber RD and
Smyth MJ: Natural innate and adaptive immunity to cancer. Annu Rev
Immunol. 29:235–271. 2011.
|
|
5
|
June CH: Principles of adoptive T cell
cancer therapy. J Clin Invest. 117:1204–1212. 2007.
|
|
6
|
Turtle CJ and Riddell SR: Artificial
antigen-presenting cells for use in adoptive immunotherapy. Cancer
J. 16:374–381. 2010.
|
|
7
|
Roulois D, Grégoire M and Fonteneau JF:
MUC1-specific cytotoxic T lymphocytes in cancer therapy: Induction
and challenge. Biomed Res Int. 2013:8719362013.
|
|
8
|
Singh R and Bandyopadhyay D: MUC1: A
target molecule for cancer therapy. Cancer Biol Ther. 6:481–486.
2007.
|
|
9
|
Madurga S, Belda I, Llorà X and Giralt E:
Design of enhanced agonists through the use of a new virtual
screening method: Application to peptides that bind class I major
histocompatibility complex (MHC) molecules. Protein Sci.
14:2069–2079. 2005.
|
|
10
|
Koido S, Enomoto Y, Apostolopoulos V and
Gong J: Tumor regression by CD4 T-cells primed with dendritic/tumor
fusion cell vaccines. Anticancer Res. 34:3917–3924. 2014.
|
|
11
|
Ninkovic T, Kinarsky L, Engelmann K,
Pisarev V, Sherman S, Finn OJ and Hanisch FG: Identification of
O-glycosylated decapeptides within the MUC1 repeat domain as
potential MHC class I (A2) binding epitopes. Mol Immunol.
47:131–140. 2009.
|
|
12
|
Bossi G, Gerry AB, Paston SJ, Sutton DH,
Hassan NJ and Jakobsen BK: Examining the presentation of
tumor-associated antigens on peptide-pulsed T2 cells.
Oncoimmunology. 2:e268402013.
|
|
13
|
Jaramillo A, Narayanan K, Campbell LG,
Benshoff ND, Lybarger L, Hansen TH, Fleming TP, Dietz JR and
Mohanakumar T: Recognition of HLA-A2-restricted
mammaglobin-A-derived epitopes by CD8+ cytotoxic T
lymphocytes from breast cancer patients. Breast Cancer Res Treat.
88:29–41. 2004.
|
|
14
|
Sobin LH, Gospodarowicz MK and Witterkind
C: The TNM Classification of Malignant Tumours. 7th edition.
Wiley-Blackwell; Oxford: pp. 211–230. 2009
|
|
15
|
Apostolopoulos V, Yu M, Corper AL, Li W,
McKenzie IF, Teyton L, Wilson IA and Plebanski M: Crystal structure
of a non-canonical high affinity peptide complexed with MHC class
I: A novel use of alternative anchors. J Mol Biol. 318:1307–1316.
2002.
|
|
16
|
Bednarek MA, Sauma SY, Gammon MC, Porter
G, Tamhankar S, Williamson AR and Zweerink HJ: The minimum peptide
epitope from the influenza virus matrix protein. Extra and
intracellular loading of HLA-A2. J Immunol. 147:4047–4053.
1991.
|
|
17
|
Fremont DH, Stura EA, Matsumura M,
Peterson PA and Wilson IA: Crystal structure of an
H-2Kb-ovalbumin peptide complex reveals the interplay of
primary and secondary anchor positions in the major
histocompatibility complex binding groove. Proc Natl Acad Sci USA.
92:2479–2483. 1995.
|
|
18
|
Lundegaard C, Lamberth K, Harndahl M, Buus
S, Lund O and Nielsen M: NetMHC-3.0: Accurate web accessible
predictions of human, mouse and monkey MHC class I affinities for
peptides of length 8–11. Nucleic Acids Res. 36:W509–W512. 2008.
|
|
19
|
Bøyum A: Isolation of lymphocytes,
granulocytes and macrophages. Scand J Immunol. 5(Suppl 5): 9–15.
1976.
|
|
20
|
Luft T, Rizkalla M, Tai TY, Chen Q,
MacFarlan RI, Davis ID, Maraskovsky E and Cebon J: Exogenous
peptides presented by transporter associated with antigen
processing (TAP)-deficient and TAP-competent cells: Intracellular
loading and kinetics of presentation. J Immunol. 167:2529–2537.
2001.
|
|
21
|
Tan J, Tang X and Xie T: Comparison of HLA
class I typing by serology with DNA typing in a Chinese population.
Transplant Proc. 32:1859–1861. 2000.
|
|
22
|
Manders EMM, Verbeek JF and Aten JA:
Measurement of co-localization of objects in dual-colour confocal
images. J Microsc. 169:375–382. 1993.
|
|
23
|
Lyons AB and Parish CR: Determination of
lymphocyte division by flow cytometry. J Immunol Methods.
171:131–137. 1994.
|
|
24
|
Suda K and Mitsudomi T: Successes and
limitations of targeted cancer therapy in lung cancer. Prog Tumor
Res. 41:62–77. 2014.
|
|
25
|
Wangari-Talbot J and Hopper-Borge E: Drug
resistance mechanisms in non-small cell lung carcinoma. J Can Res
Updates. 2:265–282. 2013.
|
|
26
|
Dudley ME and Rosenberg SA:
Adoptive-cell-transfer therapy for the treatment of patients with
cancer. Nat Rev Cancer. 3:666–675. 2003.
|
|
27
|
Disis ML: Immune regulation of cancer. J
Clin Oncol. 28:4531–4538. 2010.
|
|
28
|
Skånland SS, Moltu K, Berge T, Aandahl EM
and Taskén K: T-cell co-stimulation through the CD2 and CD28
co-receptors induces distinct signalling responses. Biochem J.
460:399–410. 2014.
|
|
29
|
Perica K, Kosmides AK and Schneck JP:
Linking form to function: Biophysical aspects of artificial antigen
presenting cell design. Biochim Biophys Acta. 1853:781–790.
2015.
|
|
30
|
Dittmann J, Keller-Matschke K, Weinschenk
T, Kratt T, Heck T, Becker HD, Stevanović S, Rammensee HG and
Gouttefangeas C: CD8+ T-cell response against
MUC1-derived peptides in gastrointestinal cancer survivors. Cancer
Immunol Immunother. 54:750–758. 2005.
|
|
31
|
Gückel B, Rentzsch C, Nastke MD, Marmé A,
Gruber I, Stevanović S, Kayser S and Wallwiener D: Pre-existing
T-cell immunity against mucin-1 in breast cancer patients and
healthy volunteers. J Cancer Res Clin Oncol. 132:265–274. 2006.
|
|
32
|
Kokowski K, Harnack U, Dorn DC and Pecher
G: Quantification of the CD8+ T cell response against a
mucin epitope in patients with breast cancer. Arch Immunol Ther Exp
(Warsz). 56:141–145. 2008.
|
|
33
|
Choi C, Witzens M, Bucur M, Feuerer M,
Sommerfeldt N, Trojan A, Ho A, Schirrmacher V, Goldschmidt H and
Beckhove P: Enrichment of functional CD8 memory T cells specific
for MUC1 in bone marrow of patients with multiple myeloma. Blood.
105:2132–2134. 2005.
|
|
34
|
Hamaï A, Benlalam H, Meslin F, Hasmim M,
Carré T, Akalay I, Janji B, Berchem G, Noman MZ and Chouaib S:
Immune surveillance of human cancer: If the cytotoxic T-lymphocytes
play the music, does the tumoral system call the tune? Tissue
Antigens. 75:1–8. 2010.
|
|
35
|
Finn OJ: Cancer immunology. N Engl J Med.
358:2704–2715. 2008.
|
|
36
|
Hwang I and Nguyen N: Mechanisms of
tumor-induced T cell immune suppression and therapeutics to counter
those effects. Arch Pharm Res. 38:1415–1433. 2015.
|
|
37
|
Quinlin IS, Burnside JS, Dombrowski KE,
Phillips CA, Dolby N and Wright SE: Context of MUC1 epitope:
Immunogenicity. Oncol Rep. 17:453–456. 2007.
|
|
38
|
Hamel ME, Noteboom E and Kruisbeek AM:
Non-responsiveness of antigen-experienced CD4 T cells reflects more
stringent co-stimulatory requirements. Immunology. 93:366–375.
1998.
|
|
39
|
Phillips JD, Knab LM, Blatner NR, Haghi L,
DeCamp MM, Meyerson SL, Heiferman MJ, Heiferman JR, Gounari F,
Bentrem DJ, et al: Preferential expansion of pro-inflammatory Tregs
in human non-small cell lung cancer. Cancer Immunol Immunother.
64:1185–1191. 2015.
|