Introduction
Colorectal cancer (CRC) is the third most common
cancer in males, the second in females and the fourth leading cause
of cancer-related death worldwide. In 2008, more than 1.2 million
new CRC cases and 608,700 deaths were caused by CRC; notably, the
highest incidence rates are found in The Occident, especially in
males (1). However, CRC incidence
rates are rapidly increasing in the Asian countries (2).
Among all the risk factors that may cause CRC
directly, molecular and genic effects are the dominant causes.
Since there are multiple unknown carcinogens and varying genetic
backgrounds, it is difficult to determine which factor is the most
important during the development of CRC (3). Moreover, lack of efficient early
diagnostic biomarkers, such as an actual molecule involved in the
progression of CRC, urges path-breaking studies on these
aspects.
Increasing evidence shows that non-coding genes may
be strictly accountable for the gene expression complexity in
humans (4–6). Non-coding RNAs (ncRNAs) are proven to
be the key regulators in the process of transcription and
expression, which could be divided into small ncRNAs (<200 nt)
and long ncRNAs (lncRNAs) (7,8). An
increasing number of studies have focused on the functional hot
spot of lncRNAs in their roles as regulators of biological
processes, such as genomic imprinting, chromatin modification and
post-transcriptional processing (9–11),
displaying more complex regulatory mechanisms than microRNAs
(12,13). In cis- and
trans-regulatory mechanisms (14,15),
lncRNAs have been shown to play important roles in various human
diseases, especially in malignant tumors (16–19).
Prostate cancer non-coding RNA 1 (PRNCR1), also
known as PCAT8 and CARLo-3, is a ~13 kb intronless lncRNA, which is
transcribed from the 'gene-desert' region of 8q24 (20). It has been reported that PRNCR1 is
associated with prostate cancer susceptibility and PRNCR1 could be
involved in prostate carcinogenesis by modulating androgen receptor
(AR) activity. This mechanism was further described by Yang et
al. Binding of PRNCR1 to the acetylated AR and its association
with DOT1L appear to be required for recruitment of a second
lncRNA, PCGEM1, to the DOT1L-mediated methylated of AR at the
N-terminus. The interactions of these overexpressed lncRNAs may
potentially serve as important regulators in prostate cancer
(21). For CRC, many researchers
have reported that the crucial locus of 8q24 may contribute to
susceptibility to CRC (22,23). It has been gradually recognized that
aberration of PRNCR1 might be a biological signature of CRC, but
its specific expression pattern related to CRC remains unknown.
In this study, we identified the PRNCR1 expression
profile in CRC patients and assessed the association between PRNCR1
expression and clinicopathological features. By using antisense
oligonucleotide (ASO)-mediated inhibition, we evaluated the impact
of PRNCR1 on cancer cell proliferation, apoptosis, migration and
invasion in vitro.
Materials and methods
Patients and tissue samples
This study was approved by the Ethics Committee of
the Cancer Institute of Jiangsu, and written informed consent was
obtained from all patients. A total of 63 pairs of primary CRC
tissues and adjacent normal tissues (5 cm or more from tumor
tissues) were collected from patients who had undergone surgery at
the Colorectal Cancer Center, Cancer Institute of Jiangsu, between
2013 and 2014. Patients who received chemotherapy or radiotherapy
before surgery were excluded. All tumor specimens were collected
immediately after removal from the resected colorectum, frozen and
stored at −80°C. All tumors and paired normal tissues were
ascertained by experienced pathologists. The clinical and
pathological characteristics for each patient were also
collected.
RNA extraction and qRT-PCR analyses
Total RNA was extracted from tissues or cultured
cells with TRIzol reagent (Life Technologies, Gaithersburg,
Scotland, UK) according to the manufacturer's protocol. Total RNA
(1.5 µg) was reverse transcribed in a final volume of 20
µl using random primers under standard conditions using the
PrimeScript R™ Master Mix (cat. no. RR036A; Takara Bio, Inc.,
Dalian, China).
After the RT reaction, qRT-PCR was performed using
the SYBR Select Master Mix (cat. no. 4472908; Applied Biosystems,
Foster City, CA, USA) with 0.5 µl complementary DNA (cDNA)
on an ABI 7300 system (Applied Biosystems) according to the
manufacturer's instructions. By using β-actin as an internal
control, the PRNCR1 expression level was determined by qRT-PCR
using the following primer sequences: forward,
5′-CCAGATTCCAAGGGCTGATA-3′ and reverse, 5′-GATGTTTGGAGGCATCTGGT-3′.
The forward primer sequences for the β-actin primer were
5′-CCAGATTCCAAGGGCTGATA-3′ and reverse for β-actin were
5′-GATGTTTGGAGGCATCTGGT-3′. The qRT-PCR reaction included an
initial denaturation step at 95°C for 10 min, followed by 40 cycles
of 92°C for 15 sec and 60°C for 1 min. The Ct value for each sample
was calculated with the ΔΔCt method, and fold-changes in expression
(tumor vs. normal) were calculated using 2−ΔΔCt methods
(24).
Cell culture and ASO transfection
Three CRC cell lines, SW620, HCT116 and SW480, were
obtained from the Shanghai Institutes for Biological Science,
Shanghai, China. LoVo and HT-29 cells were donated by Dr Zhicheng
Chen (Department of Anorectal Clinic, The Medical School of
Southeast University). A normal human colorectal epithelial cell
line (FHC) was purchased from ScienCell Research Laboratories.
All cell lines were grown in RPMI-1640 medium
(Kaiji, Nanjing, China) at 37°C in a humidified 5% CO2
atmosphere. The CRC cell lines at 50% confluency were transfected
with 100 nM of either the ASO targeting PRNCR1 or scrambled
negative controls (GenePharma, Shanghai, China) using Lipofectamine
RNAimax reagent (Invitrogen Inc., Carlsbad, CA, USA) according to
the instructions provided by the manufacturer. The ASO sequences
were as follows: ASO-1 for PRNCR1, 5′-ACUCUCCTTCTCCACCUCCA-3′;
ASO-2 for PRNCR1, 5′-ACUCCCACACCACCACCACC-3′ and scrambled ASO,
5′-AAGCGCGCACCAGCGCCUCC-3′, which were designed by a professional
website (http://www.idtdna.com/Scitools/Applications/AntiSense/Antisense.aspx?source=menu).
Cell proliferation assay
Cell proliferation was assayed by the Cell Counting
Kit-8 (CCK-8) assay (Promega, Madison, WI, USA) according to the
manufacturer's protocol. The transfected cells were plated in
96-well plates (2,000 cells/well). Following the manufacturer's
protocol, cell proliferation was detected every 24 h. In brief, 10
µl of CCK-8 solution was added to each well and incubation
was carried out for 2 h at 37°C. Then, each solution was measured
spectrophotometrically at 450 nm.
In vitro cell migration and invasion
assays
HT-29 cells transfected with 100 nM ASO-PRNCR1 or
scramble were harvested after 24 h. For the migration assays, the
transfected cells (2.5×105) were plated in the upper
chamber of Transwell assay inserts (Millipore, Billerica, MA, USA)
containing 200 µl of serum-free DMEM with a membrane (8-mm
pores). Then the inserts were placed into the wells of the bottom
chamber of a 24-well plate filled with conditioned medium. After 24
h of incubation, the cells on the filter surface were fixed with
methanol, stained with crystal violet, and photographed with a
digital microscope. Cell numbers were calculated in five random
fields for each chamber.
For the invasion assays, the transfected cells
(4×105) were plated in the top chamber with a
Matrigel-coated membrane (BD Biosciences) in 500 µl
serum-free RPMI-1640 accompanied by 750 µl 10% FBS-RPMI-1640
in the bottom chamber. After a 48-h incubation period, the invasive
ability was assessed as mentioned previously for the migration
assay.
Flow cytometric analysis
Transfected cells were harvested after transfection.
HT-29 cells were stained with Annexin V and propidium iodide (PI)
using Annexin V-FITC/PI apoptosis detection kits (BD Biosciences)
and then examined by flow cytometry (FACScan; BD Biosciences).
Cells were discriminated into viable cells, early apoptotic cells,
apoptotic cells and dead cells. Cells for cell cycle analysis were
stained with PI by the Cycletest™ Plus DNA Reagent kit (BD
Biosciences) and analyzed by FACScan.
Statistical analysis
The Student's t-test, Spearman's test, and one-way
AVONA, the receiver operating characteristic (ROC) curve, binary
correlation analysis and logistic regression were performed to
analyze the data. The ROC curve was calculated to estimate the
diagnostic efficiency of PNRCR1, and the cut-off value of best
diagnostic efficiency was also determined. All statistical analyses
were performed with SPSS software package (version 19.0; SPSS
Inc.). All P-values were two-sided, and P<0.05 was considered to
indicate a statistically significant difference.
Results
Correlation between PRNCR1 expression and
clinical characteristics
The PRNCR1 expression levels were analyzed in 63
paired primary CRC and adjacent non-cancerous tissues. It was found
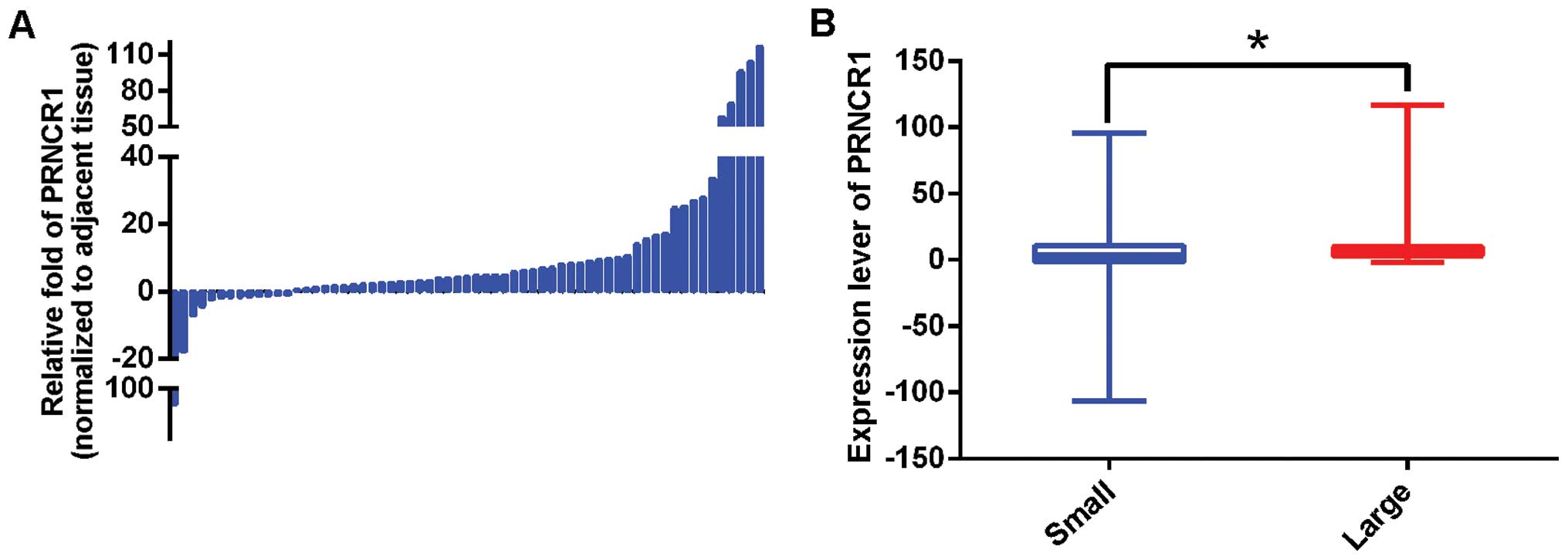
that PRNCR1 was significantly overexpressed in CRC, with an average
fold increase of 10.55 (P=0.006) (Fig.
1A). Then, the association between PRNCR1 expression and
clinicopathological parameters was explored. As shown in Table I, tumor volume was significantly
associated with the expression level of PRNCR1 after univariate and
multivariate analyses. Specifically, the PRNCR1 expression level
was higher in patients with a large volume tumor [Fig. 1B and Table I; large vs. small, 19.17 vs. 10.55;
Punivariate=0.012, Pmultivariate=0.018 (OR=5.227; CI,
1.328–20.581)]. PRNCR1 expression was not correlated with gender,
age, tumor site, differentiation, family history or TNM stage.
 | Table ICorrelation between PRNCR1 expression
and clinicopathological characteristics of the CRC patients. |
Table I
Correlation between PRNCR1 expression
and clinicopathological characteristics of the CRC patients.
|
Characteristics | No. (%) | Fold-change | P-value |
|---|
| Total | 63 (100) | | |
| Age (years) | | | 0.596a |
| ≥60.22d | 34 (53.97) | 10.73 | |
| <60.22d | 29 (46.03) | 15.69 | |
| Gender | | | 0.888b |
| Male | 37 (58.73) | 15.30 | |
| Female | 26 (41.27) | 9.76 | |
| Family history | | | 0.253b |
| Positive | 17 (26.98) | 6.56 | |
| Negative | 46 (73.02) | 15.40 | |
| Tumor site | | | 0.632b |
| Below decending
colon | 49 (77.78) | 14.85 | |
| Above sigmoid
colon | 14 (22.22) | 6.59 | |
| Tumor size
(cm3) | | | 0.012a |
| ≥23.53e | 18 (28.57) | 19.17 | 0.018f |
| <23.53e | 45 (71.43) | 10.55 | |
|
Differentiation | | | 0.491b |
| Poor | 25 (39.68) | 13.87 | |
| Moderate or
high | 38 (60.32) | 12.45 | |
| T stage | | | 0.087b |
| Entire serosal
invasion | 52 (82.54) | 12.60 | |
| Subserous
invasion | 11 (17.46) | 14.94 | |
| N stage | | | 0.775b |
| Positive | 28 (44.44) | 18.81 | |
| Negative | 35 (55.56) | 15.05 | |
| M stage | | | 0.756b |
| Positive | 11 (17.46) | 13.32 | |
| Negative | 52 (82.54) | 12.95 | |
Expression profile of PRNCR1 in CRC cell
lines
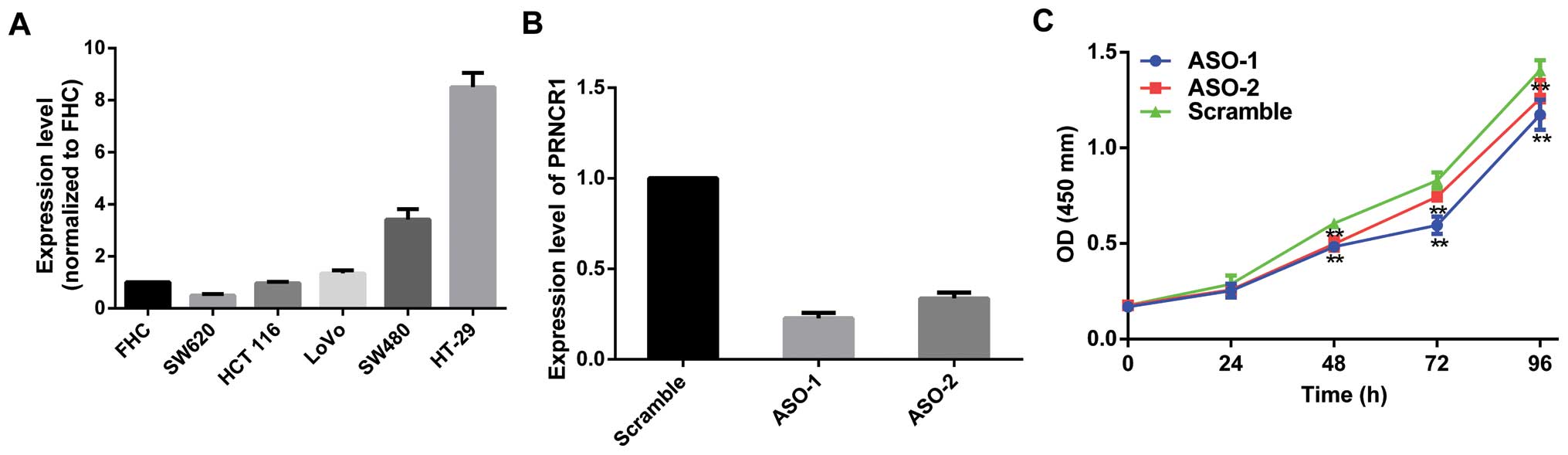
Initially, the expression profile in CRC cell lines
was first assessed by qRT-PCR. When normalized to the FHC cells,
the expression level of PRNCR1 was upregulated in most CRC cell
lines (Fig. 2A). Specifically,
PRNCR1 was upregulated in the HCT116, SW480, LoVo and HT-29 cells
but was downregulated in the SW620 cells.
PRNCR1 promotes the proliferation of CRC
cell lines in vitro
Primarily, two ASOs specifically targeting PRNCR1
were designed to knock down PRNCR1 in vitro. Based on the
relatively higher expression of PRNCR1, HT-29 cells were
transfected with ASO-PRNCR1 or scramble. At 36 h after treatment,
PRNCR1 expression was effectively knocked down (Fig. 2B). CCK8 assay showed that knockdown
of PRNCR1 significantly inhibited cell proliferation in the HT-29
cell line (Fig. 2C). Then, we
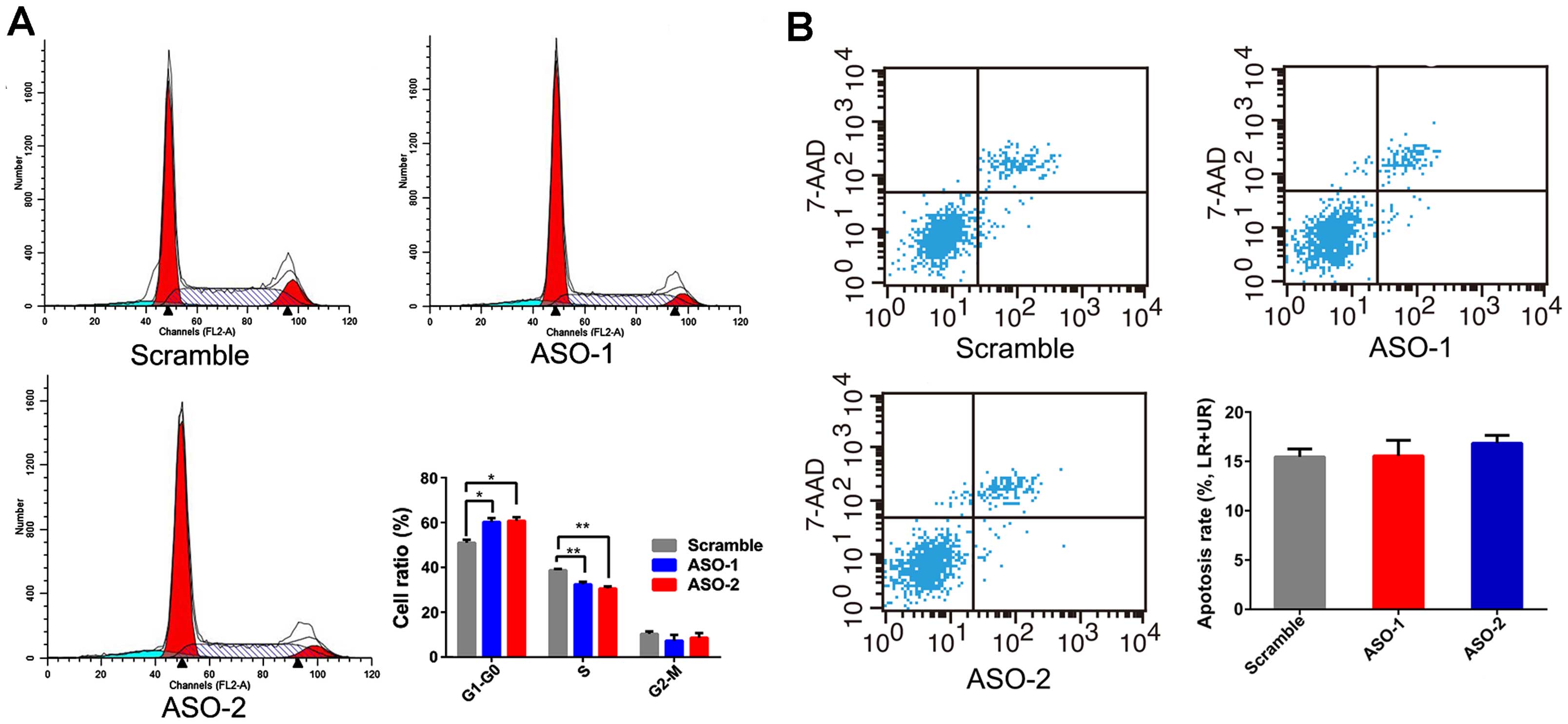
evaluated whether PRNCR1 could impact proliferation of CRC cells by
altering the rate of apoptosis or cell cycle progression. Flow
cytometric analysis was performed, and the results revealed that
ASO treatment blocked HT-29 cells at the G0/G1 phase with a
concomitant decrease of cells in the S phase (Fig. 3A). However, inhibition of PRNCR1 by
ASO did not affect apoptosis (Fig.
3B).
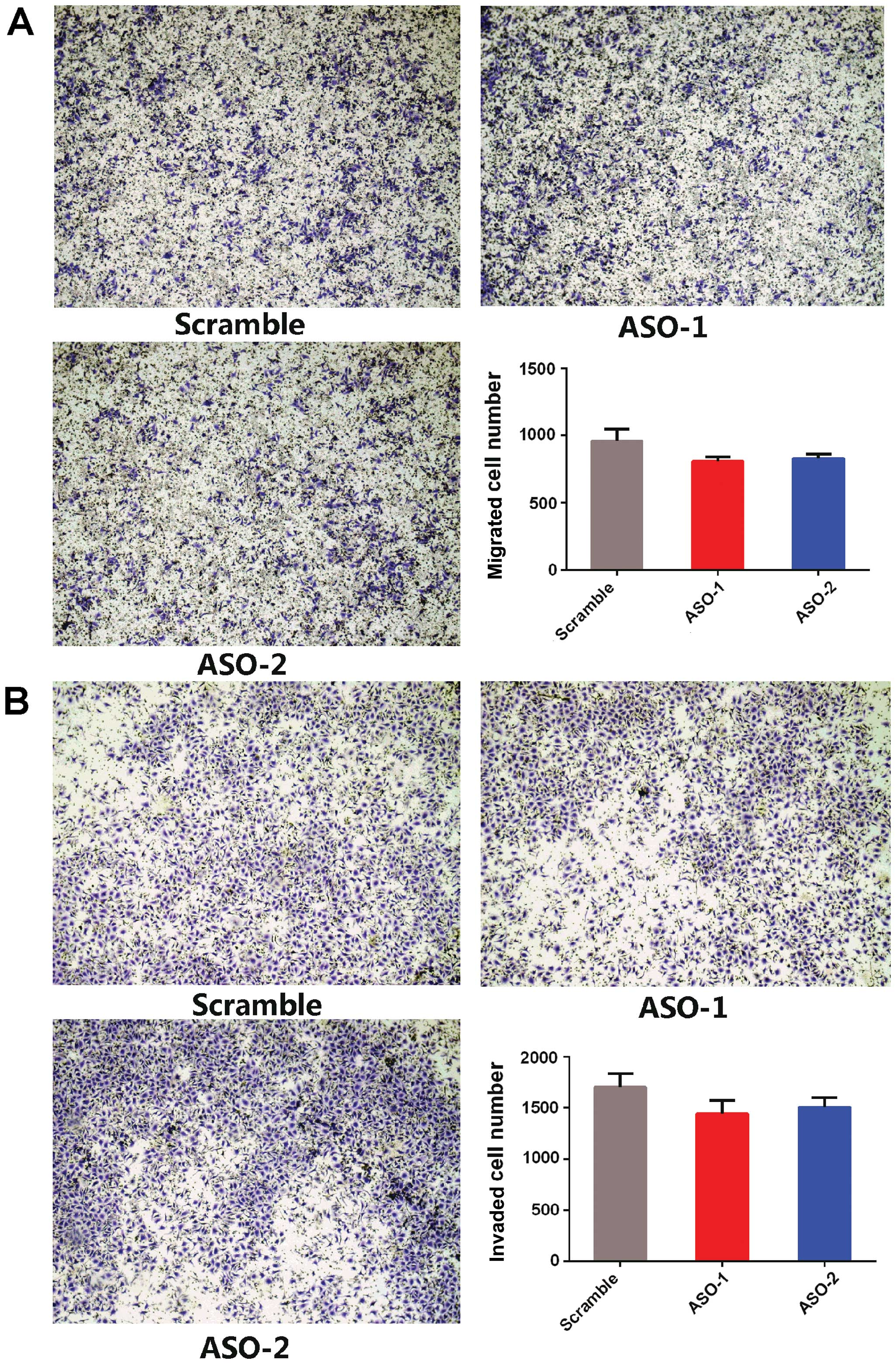
Furthermore, Transwell invasion and Matrigel
invasion assays revealed that ASO-PRNCR1 treatment did not affect
the migration and invasion capacities compared to the scramble
control (Fig. 4). These results
suggest that PRNCR1 promotes the proliferation of CRC cells.
Diagnostic value of PRNCR1
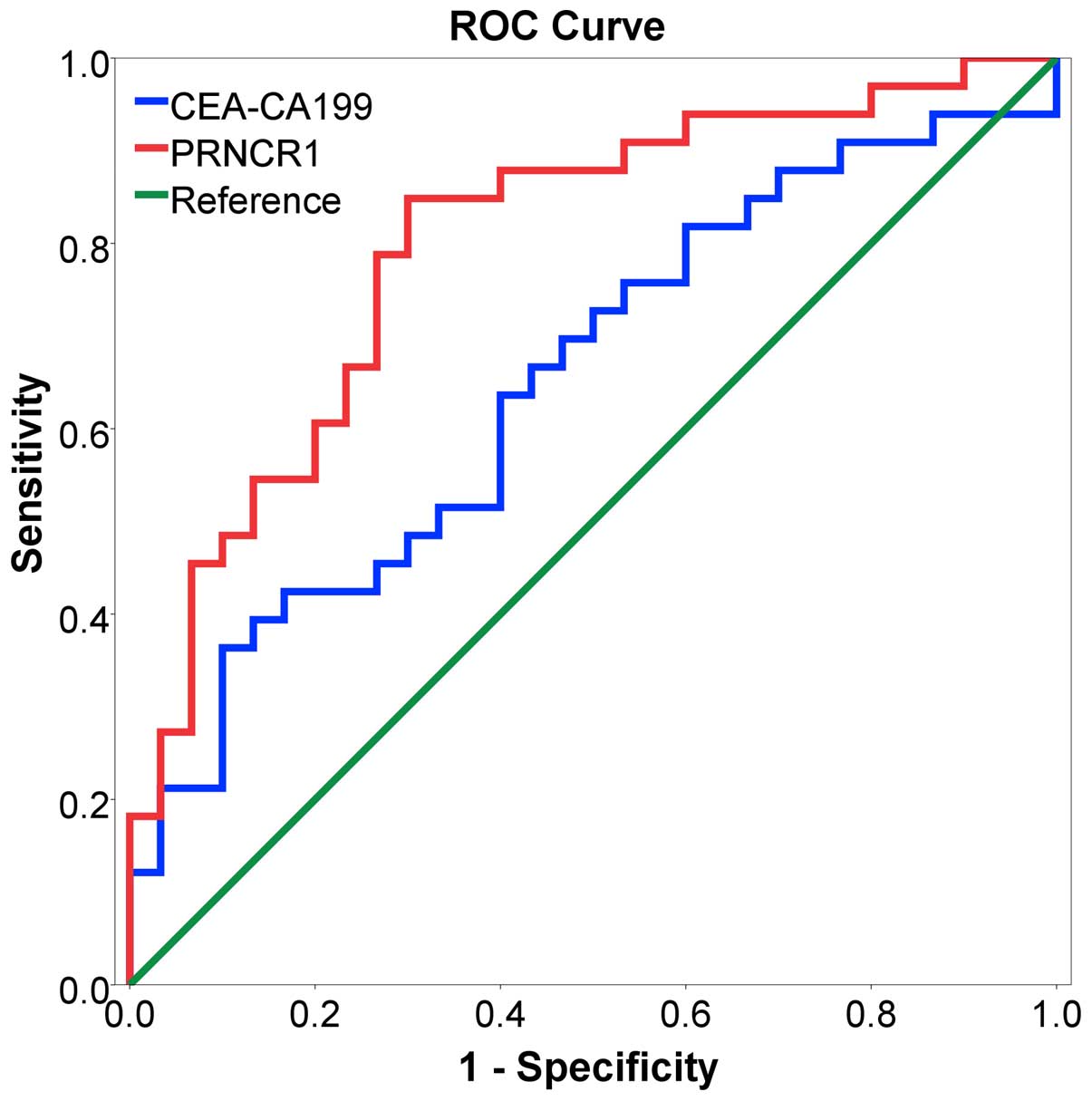
To further evaluate the potential diagnostic value
of PRNCR1, an ROC curve was generated to assess the potential of
PRNCR1 as an early diagnostic biomarker for CRC. As shown in
Fig. 5, the area under the curve
(AUC) was 0.799>0.5, which was higher than CEA-CA199
(AUC=0.651), indicating that PRNCR1 might have adequate potential
as a biomarker. According to -the evaluation of the ROC curve, a
cut-off value of 2.934 of a fold-change of PRNCR1 maximized the
sensitivity (84.8%) and specificity (70.0%) in predicting the risk
of CRC, and showed better efficiency than CEA-CA199.
Discussion
lncRNAs are involved in every aspect of cancer
progression, such as initiation, cancer metastasis, and could
function as oncogenes and tumor suppressors (22). PRNCR1 can promote the progression of
prostate cancer, yet the function and molecular mechanism in CRC
remain unknown (20,21,25,26).
In the present study, we assessed the relationship
between PRNCR1 expression and clinical characteristics of CRC
patients and the possible function of PRNCR1 was probed by
ASO-mediated inhibition in CRC cells. PRNCR1 expression was found
to be increased in CRC tissues compared to the level in paired
adjacent normal tissues. ASO-mediated silencing of PRNCR1 in HT-29
cells showed that silencing of PRNCR1 inhibited the proliferation
and blocked cell cycle progression in the HT-29 cells. These data
suggest that PRNCR1 may play an important role in CRC.
In addition, our results also indicated that a high
level of PRNCR1 expression was markedly correlated with large tumor
volume. In addition, it has been found that patients carrying the
rs1456315G polymorphism have a tumor of much larger size (22). This could be explained by the
following reasons. Primarily, the predicted secondary structure of
PRNCR1 mRNA might be influenced by SNPs in PRNCR1, shifting the
stability of the lncRNA PRNCR1 (20). Secondly, in terms of our findings,
the silencing of PRNCR1 blocked the cell cycle at the G0/G1 phase
indicating that PRNCR1 promotes the proliferation of CRC cells, and
eventually may bring about larger tumor volume eventually.
Based on a literature review, five SNPs, rs13252298,
rs1456315, rs1456315G, rs7007694C and rs16901946G, located in the
lncRNA PRNCR1 exhibit a strong relationship with CRC tumorigenesis
and development (22). Moreover,
emerging genome-wide association studies (GWAS) show that a CpG
site at Chr8: 128167809 in PRNCR1 and CRC susceptibility SNP
rs1456315G have been found to be highly correlated with each other
(27–29). In addition, correlations of these
DNA methylation levels at CpG sites with each other within or
nearby the GG genotype of rs6983267 in colon cancer associated
transcript 2 (CCAT2) could be linked with c-MYC by enhancing Wnt
signaling to upregulate transcription of CCAT2 (30–33),
suggesting the homologous impact on PRNCR1. In agreement with our
hypothesis, a previous study identified correlations for DNA
methylation levels at CpG sites located near one another,
especially those within distances equivalent to 1–2 kb apart
(34). Moreover, compared with the
functional mechanism of PRNCR1 in prostate cancer cells (20,21), a
similarity may exist in CRC; however, much more research is still
required to scrutinize all feasible targets of the characteristic
site.
Disappointedly, no correlation between PRNCR1 and
invasion, migration, apoptosis, differentiation and TNM stages of
CRC was noted. The likely cause of this could be explained as
follows. Poor differentiation of CRC cells furthers high metastatic
and malignant potentials (35).
Patients with SNPs in PRNCR1 have decreased risks to develop poorly
differentiated CRC, while others with SNPs in PRNCR1 have increased
risks (22). This may be why
increased PRNCR1 expression may not relate to worse clinical
characteristic of CRC.
Likewise, we detected the diagnostic value of PRNCR1
by ROC analysis, and PRNCR1 showed better predictive efficiency
than serum biomarker CEA-CA199, indicating that PRNCR1 may be a
potential biomarker for CRC diagnosis.
To sum up, we found that PRNCR1 is upregulated in
CRC tissues and the upregulation of PRNCR1 is associated with large
tumor volume. In addition, knockdown of PRNCR1 significantly
inhibited proliferation and induced cell cycle arrest at the G0/G1
phase in the CRC cells, Moreover, PRNCR1 potentially could be an
efficient predictive biomarker.
Acknowledgments
We thank Dr Zhicheng Chen (Department of Anorectal
Clinic, Medical School of Southeast University) for gifting the CRC
cells. This study was funded by the Natural Science Foundation of
China (81372321 to L.X.; 81201830 and 81472200 to R.Y.), the
Natural Science Foundation for High Education of Jiangsu Province
(13KJB320010 to R.Y.), and Jiangsu Provincial Special Program of
Medical Science (BL2012030 to L.X.).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Center MM, Jemal A and Ward E:
International trends in colorectal cancer incidence rates. Cancer
Epidemiol Biomarkers Prev. 18:1688–1694. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Han Y, Yang YN, Yuan HH, Zhang TT, Sui H,
Wei XL, Liu L, Huang P, Zhang WJ and Bai YX: UCA1, a long
non-coding RNA up-regulated in colorectal cancer influences cell
proliferation, apoptosis and cell cycle distribution. Pathology.
46:396–401. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kapranov P, Willingham AT and Gingeras TR:
Genome-wide transcription and the implications for genomic
organization. Nat Rev Genet. 8:413–423. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kapranov P, Cheng J, Dike S, Nix DA,
Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermüller J,
Hofacker IL, et al: RNA maps reveal new RNA classes and a possible
function for pervasive transcription. Science. 316:1484–1488. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Esteller M: Non-coding RNAs in human
disease. Nat Rev Genet. 12:861–874. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mattick JS: RNA regulation: A new
genetics? Nat Rev Genet. 5:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee JT and Bartolomei MS: X-inactivation,
imprinting and long noncoding RNAs in health and disease. Cell.
152:1308–1323. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yoon JH, Abdelmohsen K and Gorospe M:
Posttranscriptional gene regulation by long noncoding RNA. J Mol
Biol. 425:3723–3730. 2013. View Article : Google Scholar :
|
|
11
|
Marchese FP and Huarte M: Long non-coding
RNAs and chromatin modifiers: Their place in the epigenetic code.
Epigenetics. 9:21–26. 2014. View Article : Google Scholar :
|
|
12
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: Functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wapinski O and Chang HY: Long noncoding
RNAs and human disease. Trends Cell Biol. 21:354–361. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huarte M, Guttman M, Feldser D, Garber M,
Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M,
et al: A large intergenic noncoding RNA induced by p53 mediates
global gene repression in the p53 response. Cell. 142:409–419.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nagano T and Fraser P: No-nonsense
functions for long noncoding RNAs. Cell. 145:178–181. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li J, Xuan Z and Liu C: Long non-coding
RNAs and complex human diseases. Int J Mol Sci. 14:18790–18808.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deng K, Guo X, Wang H and Xia J: The
lncRNA-MYC regulatory network in cancer. Tumour Biol. 35:9497–9503.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He X, Tan X, Wang X, Jin H, Liu L, Ma L,
Yu H and Fan Z: C-Myc-activated long noncoding RNA CCAT1 promotes
colon cancer cell proliferation and invasion. Tumour Biol.
35:12181–12188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xue Y, Ma G, Gu D, Zhu L, Hua Q, Du M, Chu
H, Tong N, Chen J, Zhang Z, et al: Genome-wide analysis of long
noncoding RNA signature in human colorectal cancer. Gene.
556:227–234. 2015. View Article : Google Scholar
|
|
20
|
Chung S, Nakagawa H, Uemura M, Piao L,
Ashikawa K, Hosono N, Takata R, Akamatsu S, Kawaguchi T, Morizono
T, et al: Association of a novel long non-coding RNA in 8q24 with
prostate cancer susceptibility. Cancer Sci. 102:245–252. 2011.
View Article : Google Scholar
|
|
21
|
Yang L, Lin C, Jin C, Yang JC, Tanasa B,
Li W, Merkurjev D, Ohgi KA, Meng D, Zhang J, et al:
lncRNA-dependent mechanisms of androgen-receptor-regulated gene
activation programs. Nature. 500:598–602. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li L, Sun R, Liang Y, Pan X, Li Z, Bai P,
Zeng X, Zhang D, Zhang L and Gao L: Association between
polymorphisms in long non-coding RNA PRNCR1 in 8q24 and risk of
colorectal cancer. J Exp Clin Cancer Res. 32:1042013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barry KH, Moore LE, Sampson J, Yan L,
Meyer A, Oler AJ, Chung CC, Wang Z, Yeager M, Amundadottir L, et
al: DNA methylation levels at chromosome 8q24 in peripheral blood
are associated with 8q24 cancer susceptibility loci. Cancer Prev
Res (Phila). 7:1282–1292. 2014. View Article : Google Scholar
|
|
24
|
VanGuilder HD, Vrana KE and Freeman WM:
Twenty-five years of quantitative PCR for gene expression analysis.
Biotechniques. 44(Suppl 5): 619–626. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pestell RG and Yu Z: Long and noncoding
RNAs (lnc-RNAs) determine androgen receptor dependent gene
expression in prostate cancer growth in vivo. Asian J Androl.
16:268–269. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pomerantz MM, Beckwith CA, Regan MM, Wyman
SK, Petrovics G, Chen Y, Hawksworth DJ, Schumacher FR, Mucci L,
Penney KL, et al: Evaluation of the 8q24 prostate cancer risk locus
and MYC expression. Cancer Res. 69:5568–5574. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Haiman CA, Patterson N, Freedman ML, Myers
SR, Pike MC, Waliszewska A, Neubauer J, Tandon A, Schirmer C,
McDonald GJ, et al: Multiple regions within 8q24 independently
affect risk for prostate cancer. Nat Genet. 39:638–644. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu J, Mo Z, Ye D, Wang M, Liu F, Jin G, Xu
C, Wang X, Shao Q, Chen Z, et al: Genome-wide association study in
Chinese men identifies two new prostate cancer risk loci at 9q31.2
and 19q13.4. Nat Genet. 44:1231–1235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bensen JT, Xu Z, Smith GJ, Mohler JL,
Fontham ET and Taylor JA: Genetic polymorphism and prostate cancer
aggressiveness: A case-only study of 1,536 GWAS and candidate SNPs
in African-Americans and European-Americans. Prostate. 73:11–22.
2013. View Article : Google Scholar
|
|
30
|
Tuupanen S, Turunen M, Lehtonen R,
Hallikas O, Vanharanta S, Kivioja T, Björklund M, Wei G, Yan J,
Niittymäki I, et al: The common colorectal cancer predisposition
SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt
signaling. Nat Genet. 41:885–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ling H, Spizzo R, Atlasi Y, Nicoloso M,
Shimizu M, Redis RS, Nishida N, Gafà R, Song J, Guo Z, et al:
CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic
progression and chromosomal instability in colon cancer. Genome
Res. 23:1446–1461. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sugimachi K, Niida A, Yamamoto K,
Shimamura T, Imoto S, Iinuma H, Shinden Y, Eguchi H, Sudo T,
Watanabe M, et al: Allelic imbalance at an 8q24 oncogenic SNP is
involved in activating MYC in human colorectal cancer. Ann Surg
Oncol. 21(Suppl 4): S515–S521. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wright JB, Brown SJ and Cole MD:
Upregulation of c-MYC in cis through a large chromatin loop linked
to a cancer risk-associated single-nucleotide polymorphism in
colorectal cancer cells. Mol Cell Biol. 30:1411–1420. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen J, Zhang JG, Li J, Pei YF and Deng
HW: On combining reference data to improve imputation accuracy.
PLoS One. 8:e556002013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Paraf F and Jothy S: Colorectal cancer
before the age of 40: A case-control study. Dis Colon Rectum.
43:1222–1226. 2000. View Article : Google Scholar : PubMed/NCBI
|