Introduction
Cancer is a major public health issue worldwide
because of its substantially high rates of morbidity and mortality.
Lung cancer is currently the second most common cause of
cancer-related deaths in Taiwan (1). More than 75% of lung cancers are
diagnosed as non-small cell lung cancers (NSCLC). Chemotherapy is
always the first choice for NSCLC patients who have a positive
performance status. An appropriate chemotherapy regimen can
significantly remit clinical symptoms and improve the quality of
life of patients with advanced NSCLC. However, with currently
available chemotherapeutics, only a modest increase in the
five-year survival rate can be achieved in patients with advanced
NSCLC (2,3). Thus, enhanced chemotherapeutics are
required to improve the five-year survival rate.
Numerous cytotoxic chemicals, including anticancer
drugs, cause cell death in sensitive cells by inducing apoptosis,
at least partially (4). The two
major cellular routes involved in cytotoxic chemical-induced
apoptosis have been identified: namely, the death receptor pathway
and the mitochondrial pathway (5).
In the death receptor pathway, the binding of death receptors with
ligands, such as Fas or cross-linking antibodies, results in
receptor trimerization. This is followed by the binding of the
adaptor molecule, the Fas-associated death domain, to the
cytoplasmic domain of the receptor. Subsequent activation of the
caspase-3 family includes several effectors and pro-caspases in the
caspase cascade (6). In addition,
the mitochondrial pathway relies on the release of cytochrome
c from the mitochondria into the cytosol. This process is
initiated by the interaction of the mitochondria with one or more
of the Bcl-2 family proteins. Thus, numerous Bcl-2 family proteins
have been considered to be the major regulators of the apoptotic
process, and represent a critical checkpoint within apoptotic
pathways, acting upstream of such irreversible damage to cellular
constituents (7). The Bcl-2 family
of homologous proteins comprises pro-apoptotic and anti-apoptotic
molecules (8). For instance, Bcl-2
and Bcl-XL have been demonstrated to inhibit apoptosis,
whereas Bad, Bak and Bax have been reported to enhance apoptosis
(9,10). Their anti-apoptotic or pro-apoptotic
functions rely on constituting the heterodimers (7). Furthermore, the ratio of
anti-apoptotic to pro-apoptotic proteins also partially determines
how a cell responds to apoptotic or survival signals (11). In certain human cancer cell lines,
the expression levels of Bcl-2 and Bcl-XL result in a
positive correlation with the prevention of apoptosis induced by
various cytotoxic drugs (12).
However, overexpression of the Bax protein sensitizes cancer cells
to several chemotherapeutic agents resulting in enhanced apoptosis
(12). These observations suggest
that modulation of intrinsic targets, such as Bcl-family proteins,
can be a potential strategy for developing anticancer agents.
Cyclooxygenase (COX)-2 is a crucial rate-limiting
enzyme involved in inflammatory processes because of its role in
producing prostaglandins from arachidonic acid. COX-2 is a
downstream target of PPARγ, and PPARγ ligands have been shown to
induce COX-2 expression in mammary epithelial cells (13), monocytes (14) and human synovial fibroblasts
(15). Unlike COX-1, which is the
constitutive housekeeping isoform, COX-2 is an inducible isoform
that can be upregulated by growth factors, cytokines and
lipopolysaccharides in a wide variety of cells (16). Tumor necrosis factor α (TNFα) is a
pleiotropic cytokine that plays a critical role in numerous
biological effects, including inflammation, by inducing the
production of cytokines and pro-inflammatory mediators such as
COX-2. Previous studies have shown that TNFα can induce
NF-κB-dependent COX-2 expression in A549 cells (16,17).
ROS have been demonstrated to activate several mitogen-activated
protein serine/threonine kinases (MAPKs) that transduce diverse
extracellular stimuli (mitogenic growth factors, environmental
stresses and pro-apoptotic agents) to the nucleus through kinase
cascades to regulate a wide array of cellular processes, including
proliferation, differentiation and apoptosis (18–20).
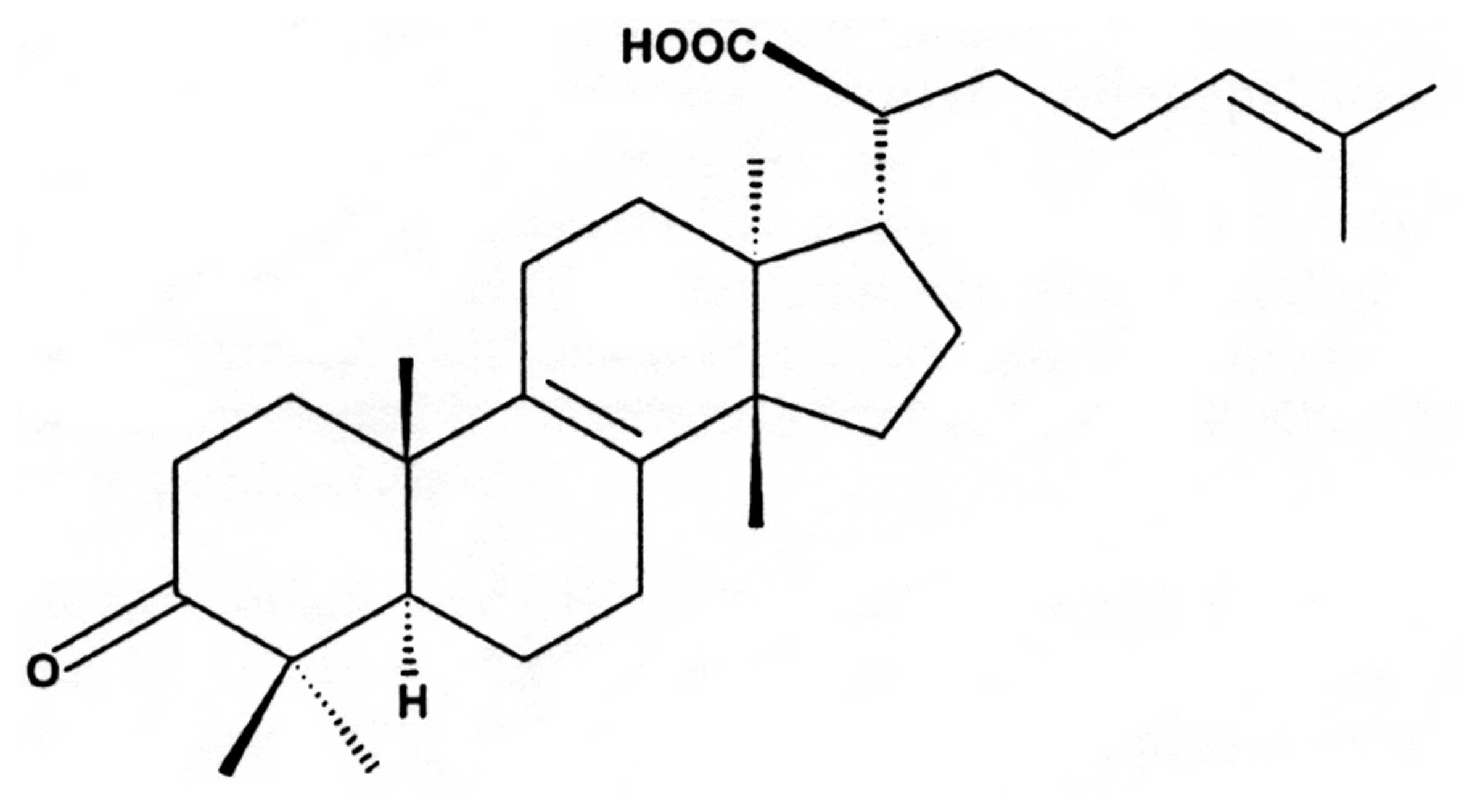
β-elemonic acid (13α,14β,17α,20S,-3-Oxolanosta-8,
24-dien-21-oic acid) is a known triterpene that has not yet been
used in therapy or investigated for its pharmaceutical effects
(21). Few studies have
demonstrated the anti-inflammatory activities of β-elemonic acid
(22). Despite extensive analysis
of the antitumor activities of β-elemonic acid, its ability to
modulate lung cancer growth has not been effectively characterized.
In the present investigation, we studied the quantitative and
qualitative changes of several known effectors in the β-elemonic
acid-induced apoptotic process in human NSCLC A549 cells.
Materials and methods
Materials and cell culture
β-elemonic acid (Fig.
1) was obtained from Sigma-Aldrich (St. Louis, MO, USA), and it
was dissolved in pure grade dimethyl sulfoxide (DMSO). Human NSCLC
A549 cells were obtained from the American Type Culture Collection
(ATTC; Manassas, VA, USA). Annexin V/propidium iodide (PI)
apoptosis detection kits were purchased from R&D systems
(Minneapolis, MN, USA). Monoclonal antibodies for p-Bcl-2,
Bcl-XL, Bax, MAPK family, COX-2 and anti-rabbit IgGs
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA). All chemicals were of the highest pure grade available.
Human NSCLC A549 cells were maintained in Dulbecco's
modified Eagle's medium (DMEM), supplemented with heat inactivated
10% fetal bovine serum (FBS), 100 μg/ml of penicillin, 100
μg/ml of streptomycin and 100 μg/ml of ampho-tericin
B (all from Hyclone, Logan, UT, USA). The cells were grown in a
humidified incubator at 37°C in a 5% CO2/95% air
atmosphere. For each experiment, 3×105 cells were seeded
in 1 ml of fresh medium in a 24-well plate and incubated with or
without chemicals for the indicated time. For the cytotoxicity
study, the cells were treated with β-elemonic acid during the
exponential cell growth phase.
Cytotoxicity assay
The general viability of human NSCLC A549 cells
treated with or without β-elemonic acid was determined using the
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide]
method (23). The MTT was dissolved
in phosphate-buffered saline (PBS) at a concentration of 5 mg/ml
and filtered. From the stock solution, 100 μl per 1 ml of
medium was added to each well. The plates were incubated at 37°C
for 1 h with gentle agitation. After the reaction with MTT, the
medium was replaced with 1 ml of pure DMSO for color development,
and the 24-well plate was read by an enzyme-linked immunosorbent
assay (ELISA) reader at 570 nm to obtain absorbance density values
to determine the cell viability. The viable cells after MTT
treatment produced a dark blue formazan product, whereas no such
staining formed in the dead cells.
Apoptotic assay by Annexin V
staining
The apoptotic status of the cells was evaluated by
measuring the exposure of phosphatidylserine on the cell membranes,
using apoptosis detection kits (R&D systems). Cells were
harvested and resus-pended in a staining solution containing PI (50
mg/ml) and Annexin V-FITC (25 mg/ml) for 15 min at room temperature
in the dark. The cell apoptosis was examined under a fluorescence
microscopic system (Olympus TX 4).
DNA content and cell cycle analysis
Human A549 cells were collected and rinsed with PBS,
after being cultured with 0, 1, 3, or 10 μM β-elemonic acid
for 24 h and suspended in 75% ethanol at −20°C overnight. The fixed
cells were centrifuged at 1,200 × g and washed twice with PBS. To
detect DNA content, the cells were contained in the dark with PI 50
mg/l and 0.1% RNase A in 400 μl PBS at 25°C for 30 min.
Stained cells were analyzed on FACSort (Becton Dickinson, San
Diego, CA, USA). The percentage of apoptotic cells was determined
using CellQuest software.
Measurement of reactive oxygen species
(ROS)
The cells were incubated in the absence or presence
(1, 3, or 10 μM) of β-elemonic acid for 24 h. Thirty minutes
before terminating the incubation period, DCF-DA (final
concentration of 10 mM) was added to the cells and incubated for
the last 30 min at 37°C. The cells were subsequently harvested to
detect ROS accumulation by using the ELISA immunoassay method.
Western blot analysis
After exposure to the indicated concentrations of
β-elemonic acid, human NSCLC A549 cells were washed with cold PBS.
Whole cell extracts were prepared by incubating the cells with a
cold lysis buffer (20 mM Tris-HCl; pH 7.5, 150 mM NaCl, 1 mM EDTA,
1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM
β-glycerophosphate, 1 mM Na3VO4, 1
μg/ml of leupeptin and 1 mM PMSF). The protein content of
the lysates was determined using the detergent-compatible
colorimetric (DC) protein assay kit (Bio-Rad, Berkeley, CA, USA).
The cell lysates (25 μg of protein/lane) were
electrophoresed on 12% SDS-polyacrylamide gels. The cellular
proteins were subsequently transferred to polyvinylidene difluoride
(PVDF) membranes by electroblotting for 2 h and western blot
analysis was conducted as previously described (24). The protein levels were visualized
using an enhanced chemiluminescence detection kit (Amersham
Pharmacia Biotech, Piscataway, NJ, USA).
Statistical analysis
All experimental data are presented as the mean and
standard error (mean ± SEM) from four to five experiments. The
statistical analysis of data was performed using a one-way analysis
of variance (ANOVA), followed by the Schefft test and P<0.05 was
considered to indicate a statistically significant difference.
Results
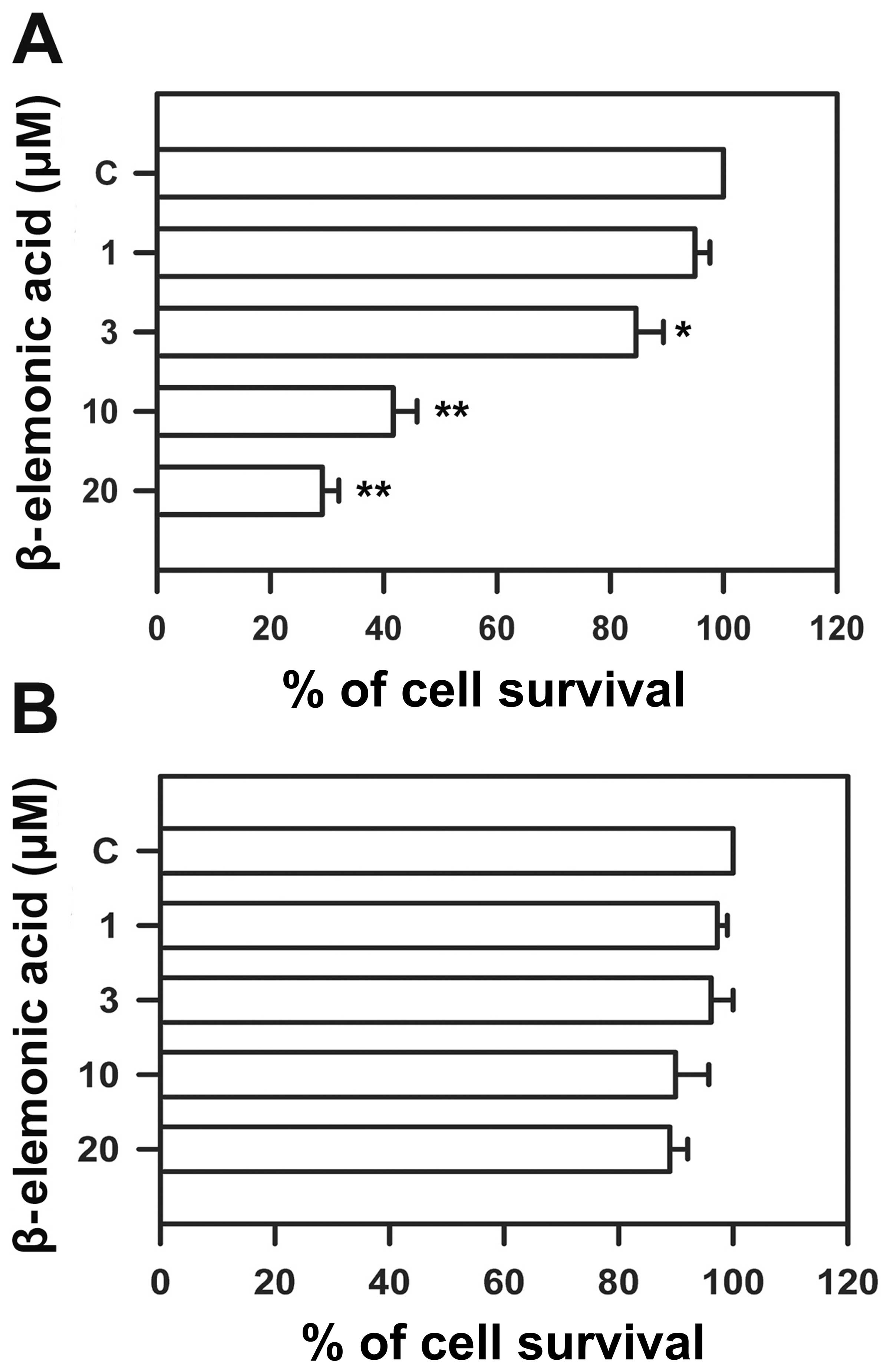
β-elemonic acid induces cell cytotoxicity
in A549 cells but not in normal lung WI-38 cells
Human NSCLC A549 cells and normal epithelial WI-38
cells were treated with various doses of β-elemonic acid, and cell
viability was measured using the MTT assay to assess the
cytotoxicity of β-elemonic acid. β-elemonic acid exerted potent
cytotoxic effects on human NSCLC A549 cells in a dose-dependent
manner (Fig. 2A). The
IC50 value following a 24-h exposure to β-elemonic acid
was 6.92 ± 0.83 μM. The same dose as that for treating the
NSCLC A549 cells did not appear to exert inhibitory effects on
human WI-38 cells (Fig. 2B). In
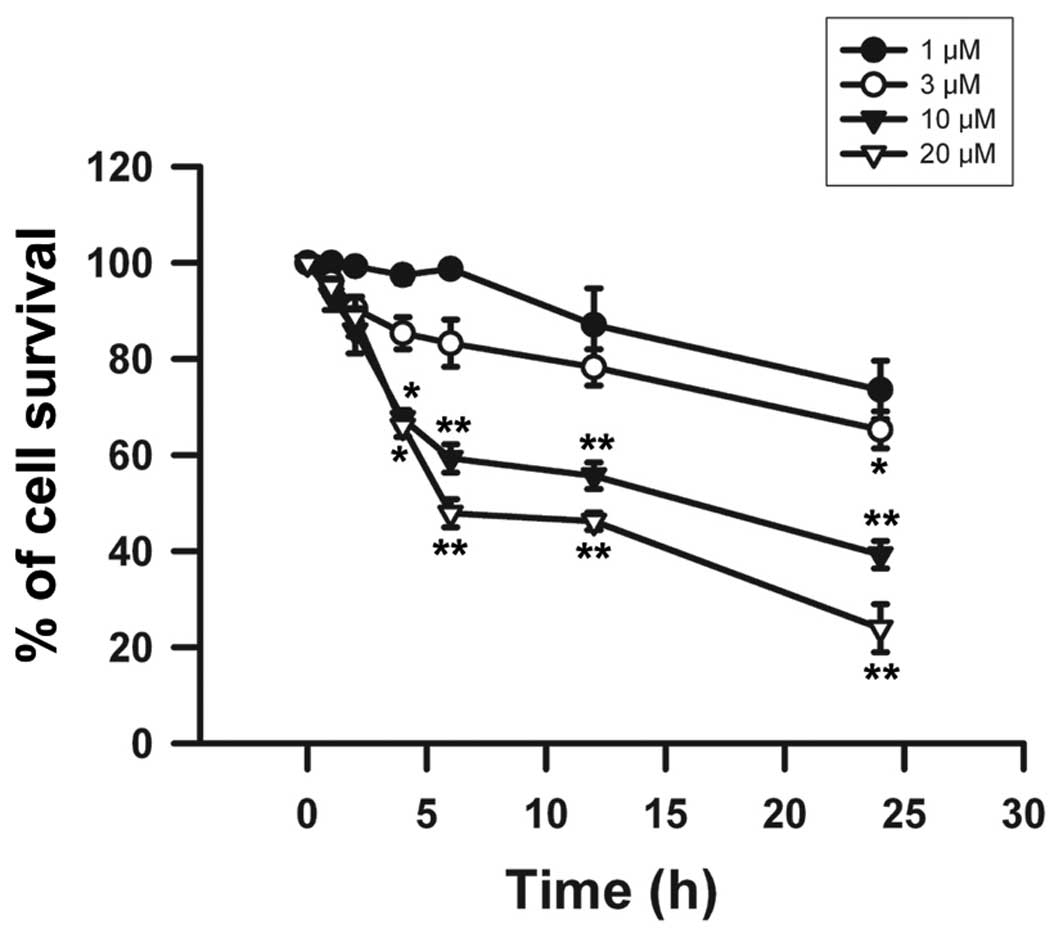
addition, β-elemonic acid also exhibited a decreased cell survival
rate in the A549 cells in a time-dependent manner at each of the
treatment doses tested (Fig. 3).
Significant cell death was observed at 10 and 20 μM
β-elemonic acid after a 2-h treatment and sharply declined after a
6-h incubation. After a 24-h treatment with 10 and 20 μM
β-elemonic acid, the cell survival rate dropped to <40% and 30%,
respectively. The exposure of β-elemonic acid for 24 h induced a
significant cytotoxic effect on A549 cells in a dose-dependent
manner. Therefore, we adopted these conditions in the following
experiments to investigate the mechanisms related to β-elemonic
acid-induced cytotoxicity.
Characterization of β-elemonic
acid-induced apoptotic cell death in human NSCLC A549 cells
To characterize whether β-elemonic acid-induced cell
death was caused by apoptosis, we examined several hallmarks of
apoptosis; namely, early apoptosis and DNA fragmentation, using
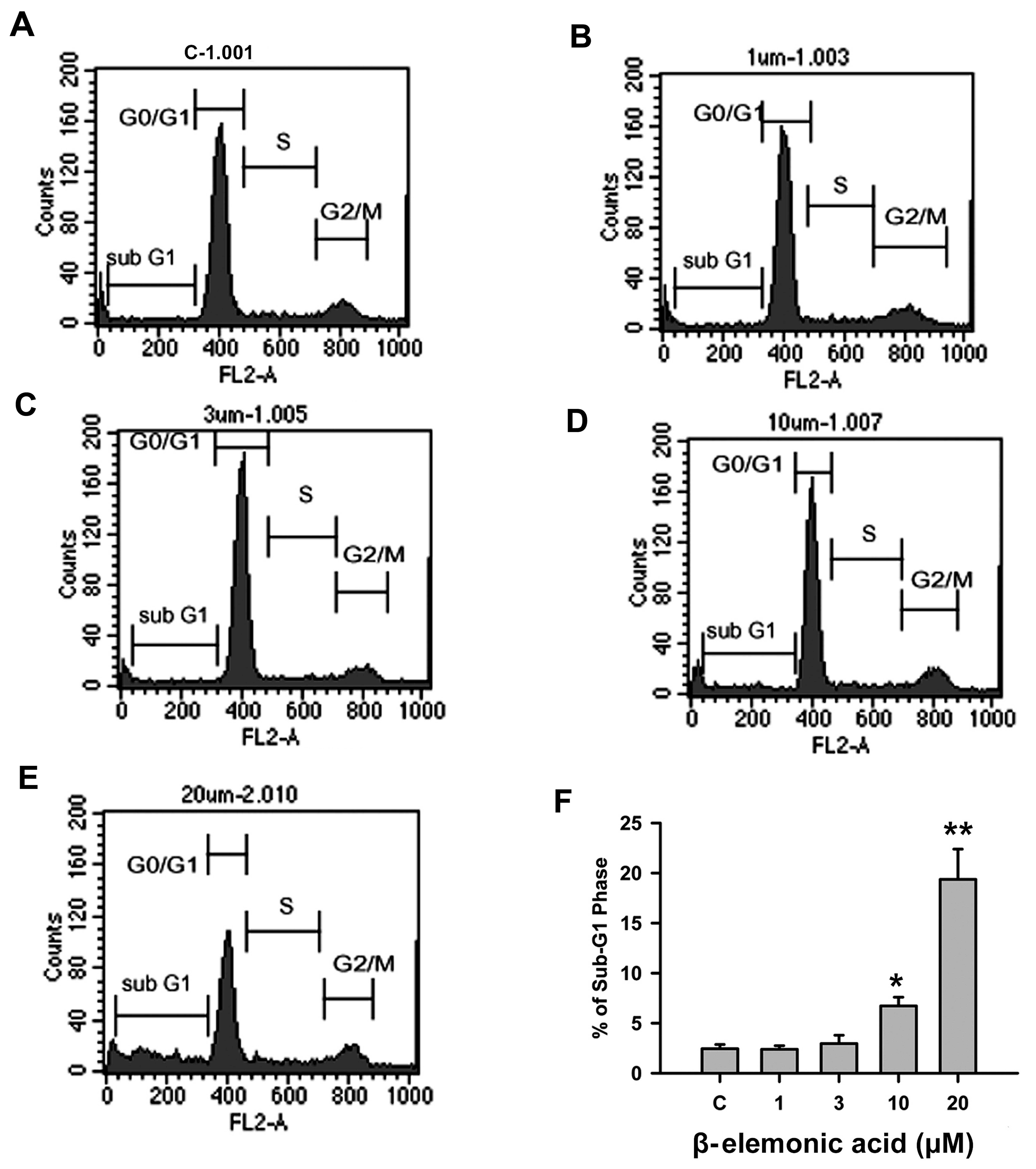
Annexin V staining or flow cytometric analysis. Fig. 4 illustrates the change in DNA
content distribution in cells treated with β-elemonic acid for 24
h. We examined these cells for degraded DNA characteristic of
apoptosis, indicated by hypoploid DNA (sub-G1 phase) content, using
PI staining. Exposure of human NSCLC A549 cells to 0 or 3 μM
β-elemonic acid promoted approximately the same percentage of
hypoploid cells observed in the DMSO-treated control (Fig. 4A–C and Table I). As the treatment dose increased,
the percentage of cells in the hypoploid (sub-G1) phase increased
accordingly. Following treatment with 10 and 20 μM
β-elemonic acid for 24 h, the percentage of sub-G1 phase cells
increased by 6.72±0.87 and 19.37±3.02%, respectively (Fig. 4D and E; Table I). The number of apoptotic cells,
which carry fragmented nuclear particles, was significantly
increased in the β-elemonic acid-treated cells in a dose-dependent
manner (Fig. 4F).
 | Table IEvaluation of β-elemonic acid-induced
apoptosis in human NSCLC A549 cells. |
Table I
Evaluation of β-elemonic acid-induced
apoptosis in human NSCLC A549 cells.
| β-elemonic acid
(μM) | Cell cycle phase
(%)
|
|---|
| Sub-G1 | G0/G1 | S | G2/M |
|---|
| C | 2.45±0.42 | 76.99±0.60 | 6.92±0.43 | 13.48±0.17 |
| 1 | 2.39±0.36 | 76.04±0.76 | 7.78±0.20 | 13.53±0.39 |
| 3 | 2.96±0.84 | 79.21±0.29 | 6.40±0.16 | 12.05±0.45 |
| 10 | 6.72±0.87 | 69.64±1.40 | 8.81±0.33 | 14.84±0.35 |
| 20 | 19.37±3.02a | 58.10±3.51 | 10.33±0.36 | 15.33±0.70 |
In addition, treatment with 20 μM β-elemonic
acid resulted in a cell percentage of 58.01±3.51% in the G0/G1
phase, compared with 76.99±0.60% in the vehicle-treated cells
(Table I). The percentage of cells
in the S phase increased from 6.92±0.43% in the vehicle-treated
cells to 10.33±0.36% in the cells treated with 20 μM
β-elemonic acid after 24 h of exposure. We also observed a slight
increase in the proportion of cells in the G2/M phase from
13.48±0.17% in the vehicle-treated cells to 15.33±0.70% in the
cells treated with 20 μM β-elemonic acid after 24 h of
exposure, but with no significant difference. These results
demonstrated that β-elemonic acid induced human NSCLC A549 cell
cytotox-icity by inducing apoptosis.
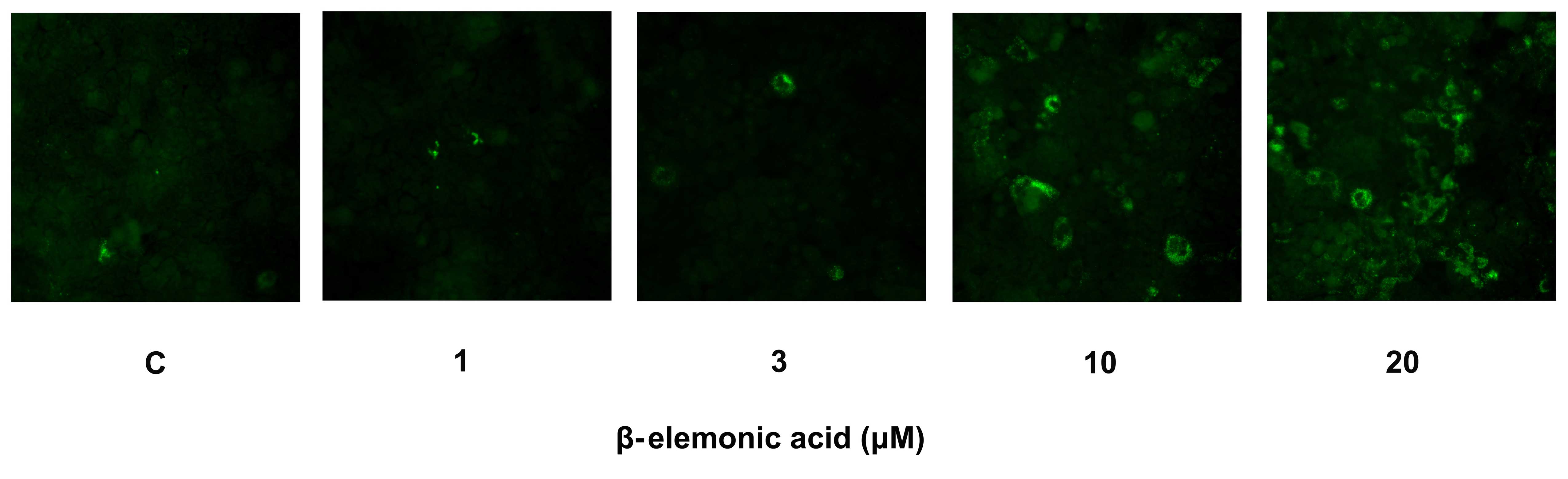
Exposure of phosphatidylserine on the cell surface
is an early event in the onset of apoptosis, which has strong
binding affinity for Annexin V in the presence of calcium. Human
NSCLC A549 cells were incubated with various concentrations of
β-elemonic acid, and the cells were stained with Annexin V-FITC to
assess the apoptotic population. The fluorescence examination
revealed that the β-elemonic acid-treated cells showed early
apoptotic morphological change and increased fluorescence intensity
(Fig. 5). The induction of early
apoptosis was significantly increased in the β-elemonic
acid-treated cells in a dose-dependent manner. Thus, β-elemonic
acid induced human NSCLC A549 cell death via an apoptotic
pathway.
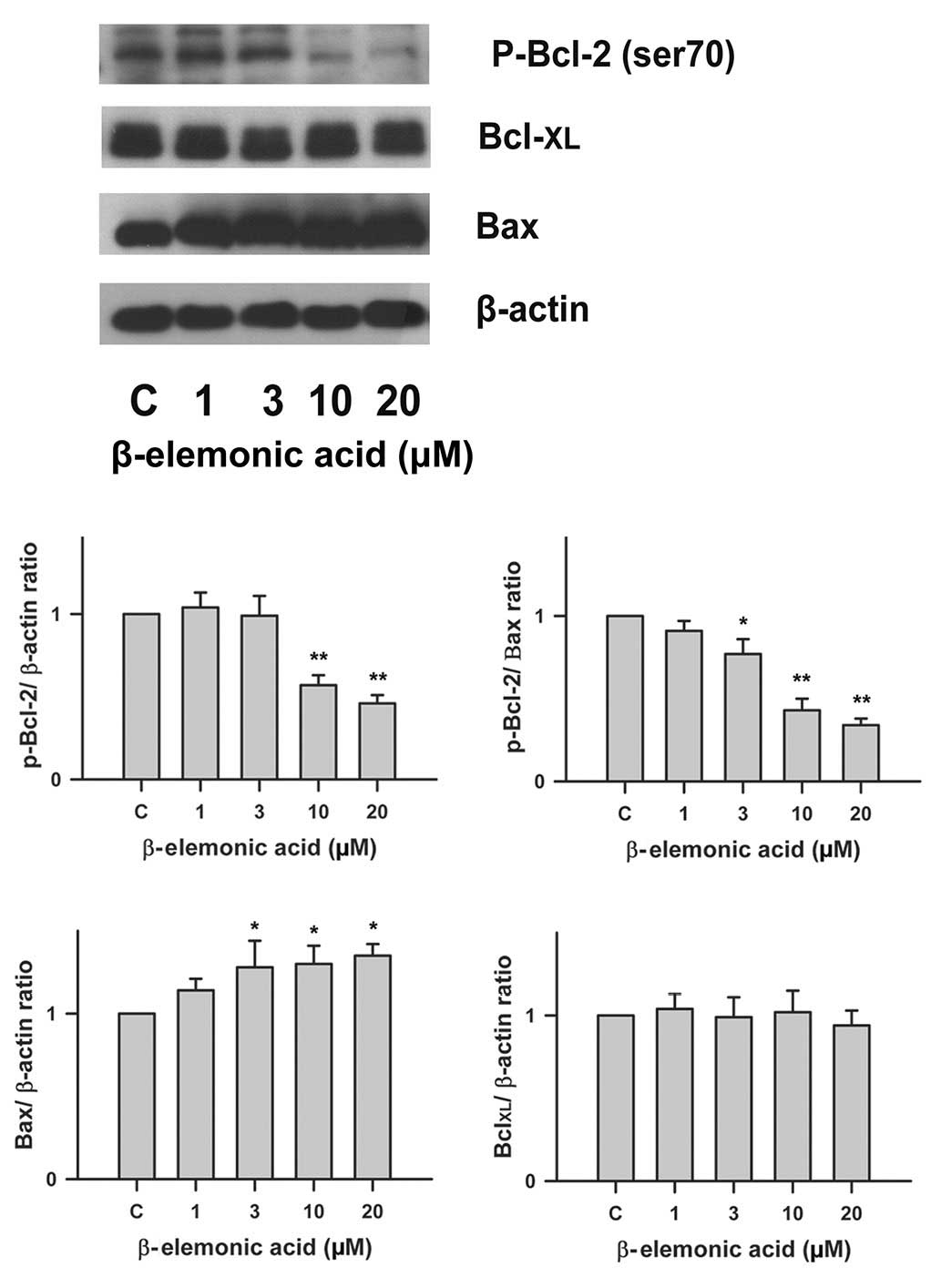
Regulation of Bcl-2 family proteins in
β-elemonic acid-treated non-small cell lung cancer cells
To determine whether Bcl-2 family proteins are
modulated in the β-elemonic acid-induced apoptosis in human A549
cells, we examined the expression of several members of the Bcl-2
family proteins using western blot analysis. Fig. 6 indicates that the exposure of human
A549 cells to 1-20 μM β-elemonic acid resulted in a
significant decrease in Bcl-2 protein expression, and a marked
increase in Bax protein expression. However, the level of
Bcl-XL protein was not affected by β-elemonic acid
treatment.
Regulation of the MAPK signaling pathway
in β-elemonic acid-treated A549 lung cancer cells
Using western blot analysis, we investigated the
effect of β-elemonic acid on the phosphorylation of p42/44,
MAPK/JNK and p38 MAP kinase. β-elemonic acid inhibited
phosphorylation of p42/44, MAPK/JNK and p38 in the A549 cells
(Fig. 7) in vitro. The
phosphorylation of p42/44 MAP, MAPK/JNK and p38kinase were
inhibited by 10 μM β-elemonic acid.
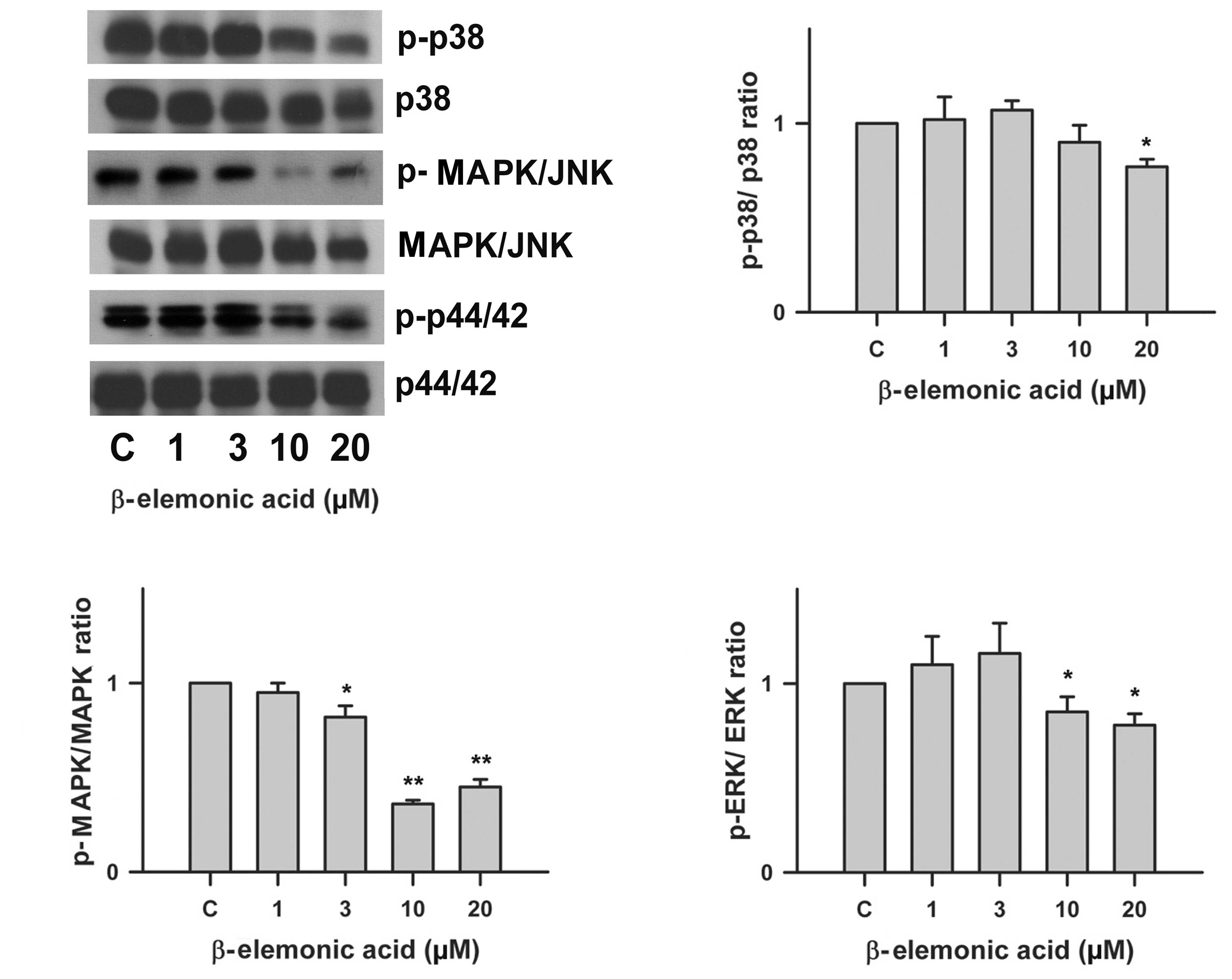 | Figure 7Effects of β-elemonic acid on p42/44,
MAPK/JNK and p38 MAPK signaling pathway in A549 cells. Human A549
cells were treated with 1-20 μM β-elemonic acid for 24 h,
and p42/44, MAPK/JNK, p38 MAPK phosphorylation was assessed by
immunoblotting with an antibody specific for phosphorylated p42/44,
MAPK/JNK, p38 MAPK (p-p42/44, p-MAPK/JNK, and p-p38, respectively).
Quantification of p-p38/p38 ratio (right upper panel), p-MAPK/MAPK
ratio (left lower panel), and p-ERK/ERK ratio (right lower panel)
are also represent. Equal loading in each lane is shown by the
similar intensities of p42/44, MAPK/JNK, p38 MAPK (p42/44, MAPK/JNK
and p38, respectively). The data are representative of three
independent experiments. |
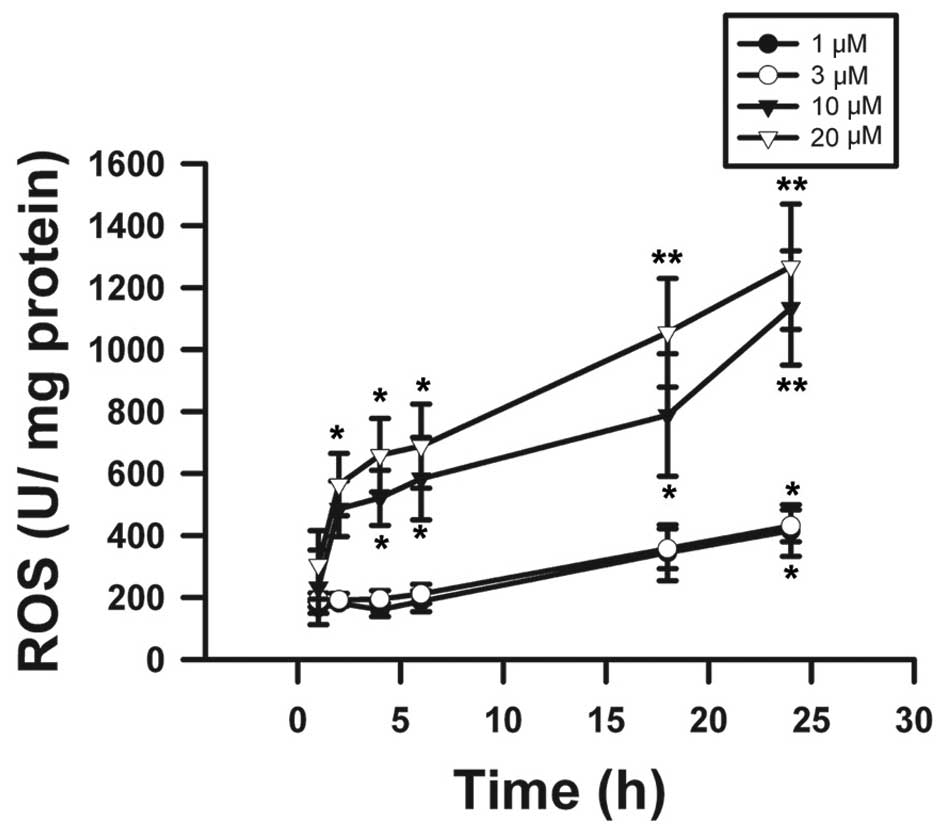
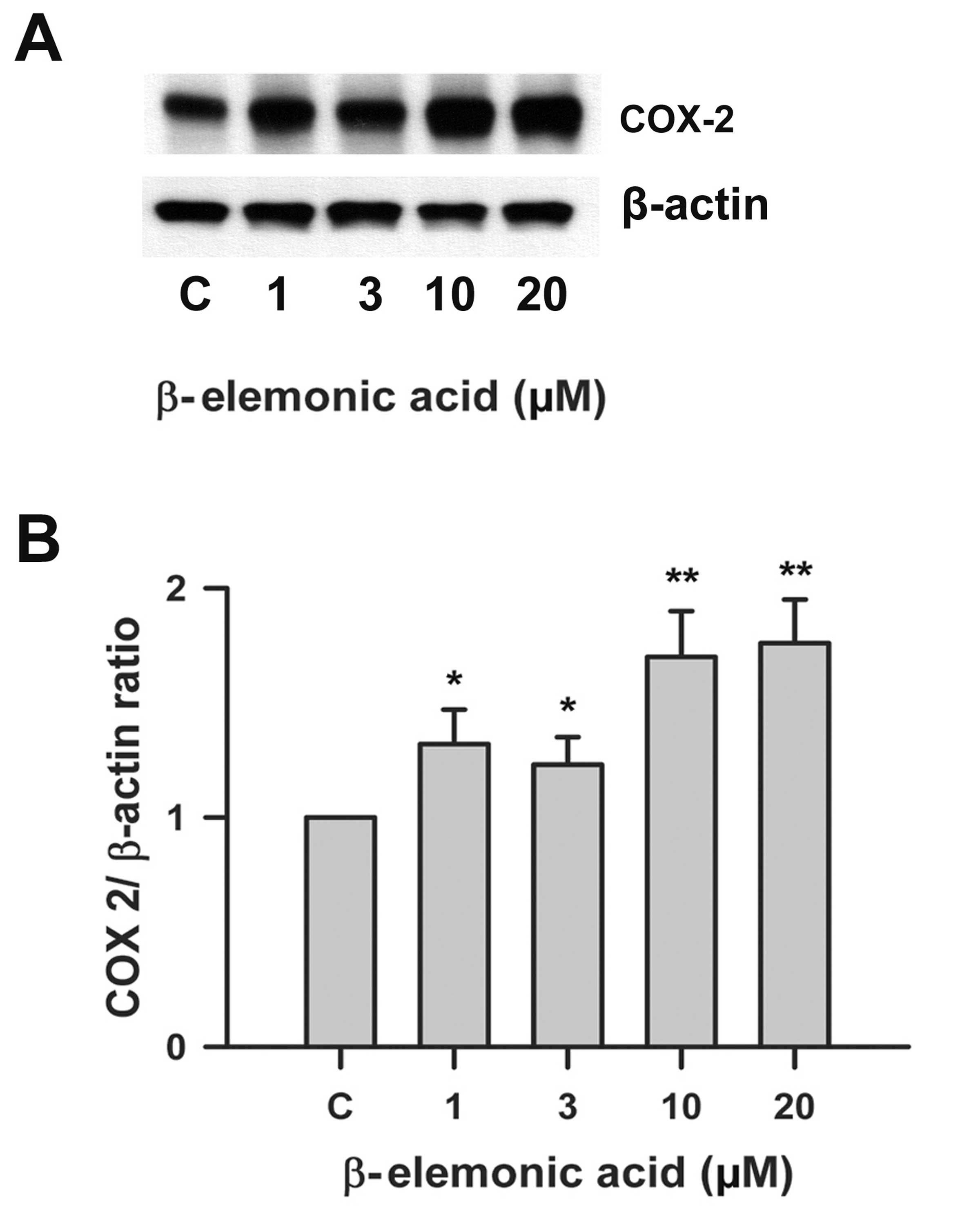
Effects on ROS activity and COX-2 protein
expression by β-elemonic acid in human A549 lung cancer cells
The results obtained in the present study provided
profound evidence that β-elemonic acid exhibited oxidative injury
in A549 cells. Using the DCFH-DA assay, the results revealed that
β-elemonic acid induced ROS levels (Fig. 8). We examined the effects of
β-elemonic acid on COX-2 expression in A549 cells, and the
expression of COX-2 proteins was examined using western blot
analysis. Fig. 9 indicates that the
exposure of human lung A549 cells to 1-20 μM β-elemonic acid
resulted in a marked increase in COX-2 expression. COX-2 protein
was modulated in the β-elemonic acid-induced apoptosis in human
lung A549 cells.
Discussion
We demonstrated that β-elemonic acid strongly
inhibited the growth of human NSCLC A549 cells. This is a
pioneering study of β-elemonic acid-induced cell cytotoxicity in
human lung cancer cells. We observed that β-elemonic acid exerted
no effect on the growth of human normal epithelial WI-38 cells,
consistent with data showing that β-elemonic acid exerts a growth
inhibitory effect on cancer cells, but not on normal cells.
Therefore, β-elemonic acid is an excellent candidate for human
NSCLC therapy without toxicity for normal cells.
Our results revealed that human A549 cells treated
with β-elemonic acid exhibited characteristic morphological
features of apoptosis, such as membrane shrinkage and chromosomal
condensation. The hypothesis that β-elemonic acid-treated cells
undergo apoptosis rather than necrosis was further supported by the
results from the cell cycle analysis. The proportion of hypoploid
cells (sub-G1) was markedly increased after β-elemonic acid
treatment. These results support the finding that β-elemonic acid
induces cell death via an apoptotic pathway.
Cytotoxicity disrupts the cell cycle progression
followed by an accumulation of cells in one or more cell cycle
phases. An accumulation of human A549 cells at the S phase occurred
in a concentration-dependent manner after treatment of the cells
with 20 μM β-elemonic acid after 24 h of exposure. These
results demonstrated that β-elemonic acid induced S phase arrest in
the cell cycle progression in human NSCLC A549 cells, and strongly
implies impaired mitosis due to arrest in the S phase. This cell
cycle behavior after exposure to β-elemonic acid was relatively
similar to the dictamnine cytotoxicity effect on A549 cells
(25). The cell cycle disruption
might partially be explained by the checkpoint response from DNA
damage or other mechanisms that require further investigation.
Human NSCLC A549 cells treated with β-elemonic acid
exhibited characteristic morphological features of apoptosis, such
as membrane shrinkage and chromosomal condensation. Apoptosis is
also accompanied by a loss of membrane phospholipid asymmetry,
resulting in phosphatidylserine exposure at the cell surface.
Phosphatidylserine expression at the cell surface plays a crucial
role in the recognition and removal of apoptotic cells by
macrophages (26). The hypothesis
that β-elemonic acid-treated cells undergo apoptosis rather than
necrosis was further supported by the result from the binding of
fluorescein isothiocyanate-labeled Annexin V to
phosphati-dylserine. Exposure of phosphatidylserine on the cell
surface is an early event in the onset of apoptosis, which has
strong binding affinity with Annexin V in the presence of calcium.
In our results, the number of Annexin V-positive human NSCLC A549
cells was markedly increased after β-elemonic acid treatment in a
dose-dependent manner. Therefore, we inferred that β-elemonic acid
induced cell death via an apoptotic pathway.
Several mechanisms for activating apoptosis under
various physiological or pathological conditions in cells have been
proposed and studied intensively (27). Numerous factors, such as the
expression of Bcl-2 family proteins and the MAPK signaling pathway,
have been suggested to play an essential role in the apoptotic
process in cancer cells. The Bcl-2 family of proteins controls a
critical step in commitment to apoptosis by regulating the
permeability of the mitochondrial outer membrane. The family is
divided into three classes: i) the BH3 proteins that sense cellular
stress and activate, ii) the executioner proteins Bax or Bak that
oligomerize and break down the mitochondrial outer membrane
potential and iii) the anti-apoptotic members such as Bcl-2 that
impede the overall process by inhibiting both BH3 and executioner
proteins (28). As evidenced by
western blot analysis, our results demonstrated that β-elemonic
acid-mediated apoptosis was involved in Bcl-2 downregulation rather
than Bcl-XL to execute Bax oligomerization. Activated
Bax enters the mitochondrial outer membrane, forming oligomers that
lead to membrane poration, release of cytochrome c and
apoptosis.
The MAPKs are a family of kinases that transduce
signals from the cell membrane to the nucleus in response to
various insults. MAPKs are serine/threonine kinases that
phosphorylate substrates that regulate gene expression, mitosis,
proliferation, motility, metabolism and programmed apoptotic death
(19). ERK1 and ERK2 are
well-characterized MAPKs activated in response to growth stimuli.
Both JNKs and p38-MAPK are simultaneously activated in response to
a wide range of cellular and environmental stresses, such as
changes in either osmolarity or metabolism, DNA damage, heat shock,
ischemia, inflammatory cytokines, or oxidative stress (29). In our experiment, presented in
Fig. 7, β-elemonic acid inhibited
phosphorylation of these MAPK pathways in a dose-dependent manner
and the difference was significant at a concentration of 10
μM.
Numerous in vitro studies have revealed that
cancer cells exhibit increased intrinsic ROS stress, increased
metabolic activity and mitochondrial malfunction. Because the
mitochondrial respiratory chain (electron transport complexes) is a
primary source of cellular ROS generation, the vulnerability of the
mitochondrial DNA to ROS-mediated damage appears to be a mechanism
to amplify ROS stress in cancer cells (30). To assess the cytotoxicity produced
by β-elemonic acid on NSCLC A549 cells, we investigated the role of
oxida-tive stress in ROS generation. The effect of β-elemonic acid
on oxidative stress was analyzed using various concentrations at
distinct incubation periods. The time-dependent increase in ROS
formation presented in Fig. 8
suggests that ROS generation is a crucial mechanism of action,
which might contribute to the cytotoxicity of β-elemonic acid in
NSCLC A549 cells. Generation of intracellular ROS depletes
glutathione (GSH), reduces superoxide dismutase (SOD) enzyme
activity and increases lipid peroxidation in cells, which
inevitably results in cell death.
The NSCLC A549 cells possess the capacity of
expressing COX-2 activity during tumorigenesis by enhancing the
expression of angiogenic chemokines CXCL8 and CXCL5 (31). COX-2 is a downstream target of PPARγ
and its correlation between NSCLC A549 cells has emerged in
numerous studies (32).
Transcriptional upregulation of COX-2 activity was also observed in
an interleukin-1β-treated A549 cell line (33). Consequently, strong evidence
supports the positive correlation between enhanced COX-2 activity
and tumor growth. The western blot analysis presented in Fig. 9 revealed that β-elemonic acid
increased COX-2 activity in the NSCLC A549 cells. Despite the
unexpected result from our study, we deduced diverse COX-2 activity
responses after various stimuli were applied to NSCLC A549
cells.
In summary, our findings suggest that treatment with
the anticancer drug, β-elemonic acid, caused cell death through
apoptosis rather than necrosis, inducing cell cycle arrest at the S
phase and apoptosis via the MAPK signaling pathway in NSCLC cells.
Our findings provide insight into the molecular action of
β-elemonic acid involving COX-2 activity and ROS generation in A549
cells and provides clues for understanding the biological
activities of β-elemonic acid for the treatment of NSCLC.
Acknowledgments
The present study was supported by grant TCPH97-002
from the Taipei County Hospital and grant CTH97-1-2A34 from the
Catholic Cardinal Tien Hospital. We also thank C.F. Yang for her
excellent technical assistance.
References
|
1
|
Anonymous. Analysis in cancer incidence
and mortality - Taiwan. Epidermal Bull. 381–391. 1999.In
Chinese.
|
|
2
|
Sandler AB, Nemunaitis J, Denham C, von
Pawel J, Cormier Y, Gatzemeier U, Mattson K, Manegold C, Palmer MC,
Gregor A, et al: Phase III trial of gemcitabine plus cisplatin
versus cisplatin alone in patients with locally advanced or
metastatic non-small-cell lung cancer. J Clin Oncol. 18:122–130.
2000.PubMed/NCBI
|
|
3
|
Bonomi P, Kim K, Fairclough D, Cella D,
Kugler J, Rowinsky E, Jiroutek M and Johnson D: Comparison of
survival and quality of life in advanced non-small-cell lung cancer
patients treated with two dose levels of paclitaxel combined with
cisplatin versus etoposide with cisplatin: Results of an Eastern
Cooperative Oncology Group trial. J Clin Oncol. 18:623–631.
2000.PubMed/NCBI
|
|
4
|
Fisher DE: Apoptosis in cancer therapy:
Crossing the threshold. Cell. 78:539–542. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaufmann SH and Earnshaw WC: Induction of
apoptosis by cancer chemotherapy. Exp Cell Res. 256:42–49. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Medema JP, Scaffidi C, Kischkel FC,
Shevchenko A, Mann M, Krammer PH and Peter ME: FLICE is activated
by association with the CD95 death-inducing signaling complex
(DISC). EMBO J. 16:2794–2804. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Antonsson B and Martinou JC: The Bcl-2
protein family. Exp Cell Res. 256:50–57. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kausch I and Böhle A: Antisense
oligonucleotide therapy in urology. J Urol. 168:239–247. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McDonnell TJ, Beham A, Sarkiss M, Andersen
MM and Lo P: Importance of the Bcl-2 family in cell death
regulation. Experientia. 52:1008–1017. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Farrow SN and Brown R: New members of the
Bcl-2 family and their protein partners. Curr Opin Genet Dev.
6:45–49. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schmitt E, Sané AT, Steyaert A, Cimoli G
and Bertrand R: The Bcl-xL and Bax-alpha control points: Modulation
of apoptosis induced by cancer chemotherapy and relation to
TPCK-sensitive protease and caspase activation. Biochem Cell Biol.
75:301–314. 1997.PubMed/NCBI
|
|
13
|
Meade EA, McIntyre TM, Zimmerman GA and
Prescott SM: Peroxisome proliferators enhance cyclooxygenase-2
expression in epithelial cells. J Biol Chem. 274:8328–8334. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pontsler AV, St Hilaire A, Marathe GK,
Zimmerman GA and McIntyre TM: Cyclooxygenase-2 is induced in
monocytes by peroxisome proliferator activated receptor gamma and
oxidized alkyl phospholipids from oxidized low density lipoprotein.
J Biol Chem. 277:13029–13036. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kalajdzic T, Faour WH, He QW, Fahmi H,
Martel-Pelletier J, Pelletier JP and Di Battista JA: Nimesulide, a
preferential cyclooxygenase 2 inhibitor, suppresses peroxisome
prolif-erator-activated receptor induction of cyclooxygenase 2 gene
expression in human synovial fibroblasts: Evidence for receptor
antagonism. Arthritis Rheum. 46:494–506. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mitchell JA, Belvisi MG, Akarasereenont P,
Robbins RA, Kwon OJ, Croxtall J, Barnes PJ and Vane JR: Induction
of cyclo-oxygenase-2 by cytokines in human pulmonary epithelial
cells: Regulation by dexamethasone. Br J Pharmacol. 113:1008–1014.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen CC, Sun YT, Chen JJ and Chiu KT:
TNF-alpha-induced cyclooxygenase-2 expression in human lung
epithelial cells: Involvement of the phospholipase C-gamma 2,
protein kinase C-alpha, tyrosine kinase, NF-kappa B-inducing
kinase, and I-kappa B kinase 1/2 pathway. J Immunol. 165:2719–2728.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kyriakis JM and Avruch J: Mammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammation. Physiol Rev. 81:807–869.
2001.PubMed/NCBI
|
|
21
|
Atta-ur-Rahman, Naz H, Fadimatou, Makhmoor
T, Yasin A, Fatima N, Ngounou FN, Kimbu SF, Sondengam BL and
Choudhary MI: Bioactive constituents from Boswellia papyrifera. J
Nat Prod. 68:189–193. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee HJ and Soliman MR: Anti-inflammatory
steroids without pituitary-adrenal suppression. Science.
215:989–991. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jow GM, Wu YC, Guh JH and Teng CM:
Armepavine oxalate induces cell death on CCRF-CEM leukemia cell
line through an apoptotic pathway. Life Sci. 75:549–557. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
An FF, Liu YC, Zhang WW and Liang L:
Dihydroartemisinine enhances dictamnine-induced apoptosis via a
caspase dependent pathway in human lung adenocarcinoma A549 cells.
Asian Pac J Cancer Prev. 14:5895–5900. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Koopman G, Reutelingsperger CP, Kuijten
GA, Keehnen RM, Pals ST and van Oers MH: Annexin V for flow
cytometric detection of phosphatidylserine expression on B cells
undergoing apoptosis. Blood. 84:1415–1420. 1994.PubMed/NCBI
|
|
27
|
Vaux DL and Korsmeyer SJ: Cell death in
development. Cell. 96:245–254. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shamas-Din A, Kale J, Leber B and Andrews
DW: Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb
Perspect Biol. 5:a0087142013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pelicano H, Carney D and Huang P: ROS
stress in cancer cells and therapeutic implications. Drug Resist
Updat. 7:97–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Põld M, Zhu LX, Sharma S, Burdick MD, Lin
Y, Lee PP, Põld A, Luo J, Krysan K, Dohadwala M, et al:
Cyclooxygenase-2-dependent expression of angiogenic CXC chemokines
ENA-78/CXC Ligand (CXCL) 5 and interleukin-8/CXCL8 in human
non-small cell lung cancer. Cancer Res. 64:1853–1860. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bakhle YS: COX-2 and cancer: A new
approach to an old problem. Br J Pharmacol. 134:1137–1150. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Newton R, Kuitert LM, Bergmann M, Adcock
IM and Barnes PJ: Evidence for involvement of NF-kappaB in the
transcriptional control of COX-2 gene expression by IL-1beta.
Biochem Biophys Res Commun. 237:28–32. 1997. View Article : Google Scholar : PubMed/NCBI
|