Introduction
Salivary gland adenoid cystic carcinoma (ACC) is the
most common epithelial malignant neoplasm of salivary glands
(1,2), which is characterized by slow but
aggressive growth, intensive local invasion, distant metastasis to
the lungs at early or late stages, multiple recurrence, and poor
long-term survival rates (3,4). The
primary treatment of ACC is radical surgery, which can be followed
by post-operative radiotherapy. Unfortunately, surgery,
chemotherapy, and radiation therapy provide little improvement in
survival (5). Therefore, there is a
need for better understanding of the biology of ACC and the
development of therapeutic approaches based on relevant
targets.
Recently, many efforts have been made to examine the
antitumor effect of TGF-β superfamily co-receptor, the type III
TGF-β receptor (TβRIII, also known as betaglycan) in both in
vitro and in vivo cancer models, including breast
(6,7), lung (8,9),
prostate (10,11), pancreas (12), ovary (13) and oral squamous cell carcinomas
(14). Conversely, a report by
Gatza et al (15) showed
that TβRIII enhances colon cancer cell migration and growth. A
recent study by Jovanović et al (16) also demonstrated that TβRIII is a
tumor promoter in mesenchymal-stem like triple-negative breast
cancer. These findings suggest that TβRIII exerts dual action on
cancer progression depending upon the cell type. To date, no
studies have investigated the expression and role of TβRIII in
progression of ACC disease.
Here, we demonstrated that the expression of TβRIII
is reduced in ACC. Transient transfection of TβRIII significantly
decreased viability, migration and induced apoptosis in human high
metastasis cell lines of ACCs (ACC-M), with significant inhibition
of nuclear factor κB (NF-κB) signaling through its interaction with
β-arrestin2.
Materials and methods
Tissue samples
Human adenoid cystic carcinoma and their adjacent
non-cancer tissues were collected immediately after surgery from
the Department of Oral and Maxillofacial Surgery at the Second
Affiliated Hospital of Harbin Medical University in China, and
informed consent was obtained from all participating patients or
their guardians. The study was approved by the Institutional Review
Board of Harbin Medical University.
Cell culture
Human high metastasis cell lines of ACCs (ACC-M)
were cultured as previously described (17). ACC-M cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
(v/v) fetal bovine serum and 100 U/ml penicillin and
streptomycin.
Plasmids and transfections
Cells were transfected with pc-DNA3.1-mTβRΙΙΙ
plasmid (GeneChem Co., Ltd., Shanghai, China) with DNA
concentrations of 0.5 and 1 µg/ml. The pc-DNA3.1-plasmid was
used as an empty vector. Transient transfections were carried out
by Fugene 6 (Roche Molecular Biochemicals, Mannheim, Germany)
according to the manufacturer's instructions.
Cell viability assay
Cells were seeded in a 96-well microplate at a
density of 1×104 cells/well and treated as designated.
Cells were incubated with 20 µl of MTT (5 mg/ml;
Sigma-Aldrich, St. Louis, MO, USA) for 4 h at 37°C in the dark.
After medium was removed, 150 µl of DMSO was added to the
wells. The absorbance was measured using a microplate
spectrophotometer (Tecan, Austria) at 490 nm. The cell viability
was calculated as a ratio of the mean OD value in treated versus
untreated cells.
Assessment of electron microscopy
ACC-M cells were cultured in 60-mm plates, collected
in phosphate-buffered saline (PBS) solution and fixed with 2% (v/v)
paraformaldehyde (PFA) containing 2.5% (w/v) glutaraldehyde
(Paesel-Lorei) buffered in HMSS (Hank's modified salt solution) at
4°C for 4 h. The cells were further fixed in 1% (w/v)
OsO4 solution buffered by 0.1 M cacodylate (pH 7.2) at
4°C for 2 h. Then, the cells were scraped off from the plastic and
dehydrate dinethanol. Dehydration was completed in propylene oxide.
The specimens were embedded in Epon medium and dissected into
60–70-nm sections. Specimens were analyzed and documented with a
JEOL 1200 electron microscope (JEOL Ltd., Tokyo, Japan).
Quantification of apoptosis and cell
cycle by flow cytometry
Apoptosis in ACC-M cells, transfected with pcDNA3.1
expressing empty vector or TβRIII for 24 h, was quantified using
the Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection
kit (Invitrogen, Carlsbad, CA, USA). Briefly, the cells were
harvested, washed in cold PBS, and resuspended in Annexin
V-labeling solution. After a 15-min incubation at room temperature,
cells were then stained with 5 µg/ml propidium iodide (PI)
and immediately subjected to Coulter Epics XL flow cytometer
(Beckman Coulter, Miami, FL, USA). The effect of TβRIII
overexpression on cell cycle distribution was determined by flow
cytometry of DNA content from the nuclei of the cells. After
treatment, ACC-M cells were harvested, washed with PBS and fixed in
70% ethanol and treated with 80 mg/ml RNase A at 37°C for 30 min.
DNA was stained with 50 mg/ml PI and analyzed using a Coulter Epics
XL flow cytometer (Beckman Coulter).
RNA interference
RNA interference was performed with siRNA duplexes
(GenePharma, Shanghai) with sequences specifically targeting
β-arrestin2 (5′-AAGGACCGCAAAGU GUUUGUG-3′), or control non-specific
siRNA using Lipofactamine 2000 (Invitrogen) according to the
manufacturer's protocol. Knockdown of expression of the target was
determined by western blot analysis.
Co-immunoprecipition (Co-IP) and western
blot analysis
Total protein samples were extracted from the
cultured cells. Approximately 5 µg of antibody specific to
TβRIII or β-arrestin2 protein was added to cell lysates and then
incubated for 12 h at 4°C. The antibody-protein immune complexes
were precipitated together with protein A/G PLUS-Agarose (rabbit
polyclonal; Santa Cruz Biotechnology), which binds most antibodies,
then incubated at 4°C overnight. When TβRIII or β-arrestin2 protein
binds to one of them, TβRIII or β-arrestin2 protein can be
identified by western blot analysis. Briefly, proteins were
resolved by SDS-polyacrylamide gel electrophoresis, and transferred
to PVDF membranes (Amersham Biosciences). All the blots were
blocked with 5% non-fat dry milk powder in Tris-buffered saline
with Tween-20 for 2 h and then incubated with either primary rabbit
anti-TβRIII, anti-p-p65, anti-β-arrestin2, or anti-IκBα (1:1,000;
Cell Signaling Technology, Beverly, MA, USA). Western blot bands
were quantified using Odyssey v1.2 software by measuring the band
intensity (area × OD) for each group and normalizing it to GAPDH or
β-actin (Zhongshan, Beijing, China) as an internal control.
Scratch wound-healing assay
For the scratch assay, ACC-M cells were seeded at
1×105 cells/6-well plate and allowed to grow overnight.
Cells were transiently transfected with pcDNA3.1 expressing empty
vector or TβRIII. After a 24-h transfection, a scratch wound was
applied using a pipette tip, and a baseline image was obtained.
Scratch wound closure was monitored over a 24 h period. The healing
of the wounds through cell migration was quantified by measuring
the wound distance.
Transwell migration assay
Transwell migration and invasion assay were
performed using the 24-well cell culture inserts without Matrigel
and Matrigel invasion chambers (8-µm pore; BD Biosciences),
respectively. Briefly, 5×104 cells were resuspended in
250 µl serum-free RPMI-1640 and added into the inserts. A
total of 500 µl DMEM with 10% FBS was added to the lower
chamber. After allowing cells to migrate for 4 h or invasion for 22
h, cells on the upper surface of the membrane were removed using a
cotton swab, and the membranes were fixed with methanol and stained
with crystal violet. The number of migrating or invading cells was
determined by averaging cell counts from nine randomly selected
×100 fields.
Immunohistochemistry (IHC)
Serial sections (5–6-µm thick) were made from
paraffin-embedded tissue blocks and mounted on silane-coated glass
slides (Matsunami Glass, Osaka, Japan). One section from each
tissue block was stained with hematoxylin and eosin (H&E), and
the others were used for IHC. IHC staining was performed using the
standard streptavidin-biotin-peroxidase complex method. Briefly,
paraffin sections of ACC tissues were deparaffinized, blocked with
10% normal goat serum for 10 min, and incubated with anti-TβRIII
overnight at 4°C. The tissue section was then incubated with
biotinylated goat anti-rabbit immunoglobulin at a concentration of
1:75 at 37°C for 30 min. The status of TβRIII expression was
assessed by two independent investigators without prior knowledge
of the clinicopathological data.
Statistical analysis
The results are expressed as the mean ± standard
deviation (SD). Statistical significance was evaluated using the
unpaired Student's t-test. A p-value <0.05 was considered
statistically significant.
Results
Tumor characteristics
Table I summarizes
the clinical attributes of the patients, who provided the 12 ACC
tumor samples. All the tumors had arisen sporadically. Of these, 8
occurred in men, and the median age at the time of presentation was
61 years (range, 48–77 years). Tumors arose at the following sites:
submandibular gland (7 tumors), parotid gland (3 tumors), and
tongue (2 tumors). The tumors were classified as follows based on
morphologic subtype: combined tubular and cribriform (8 tumors),
cribriform (2 tumors), combined tubular and solid (2 tumors).
 | Table IClinicopathological features of ACC
samples. |
Table I
Clinicopathological features of ACC
samples.
| Case | Gender | Age (years) | Tumor location | Histological
type |
|---|
| 1 | M | 74 | Submandibular
gland | Tubular +
cribriform |
| 2 | M | 69 | Submandibular
gland | Tubular +
cribriform |
| 3 | F | 57 | Parotid gland | Tubular +
cribriform |
| 4 | F | 77 | Submandibular
gland | Tubular +
cribriform |
| 5 | M | 63 | Tongue | Cribriform |
| 6 | M | 51 | Parotid gland | Tubular +
solid |
| 7 | M | 48 | Parotid gland | Tubular +
solid |
| 8 | F | 66 | Tongue | Cribriform |
| 9 | M | 54 | Submandibular
gland | Tubular +
cribriform |
| 10 | F | 66 | Submandibular
gland | Tubular +
cribriform |
| 11 | M | 59 | Submandibular
gland | Tubular +
cribriform |
| 12 | M | 49 | Submandibular
gland | Tubular +
cribriform |
Decreased TβRIII expression in human
adenoid cystic carcinoma
Loss or reduced expression of TβRIII has been
demonstrated in multiple human cancers. To confirm the expression
of TβRIII in human adenoid cystic carcinoma, we performed IHC
analysis for TβRIII expression in matched normal human salivary
glands and adenoid cystic carcinoma specimens. Representative
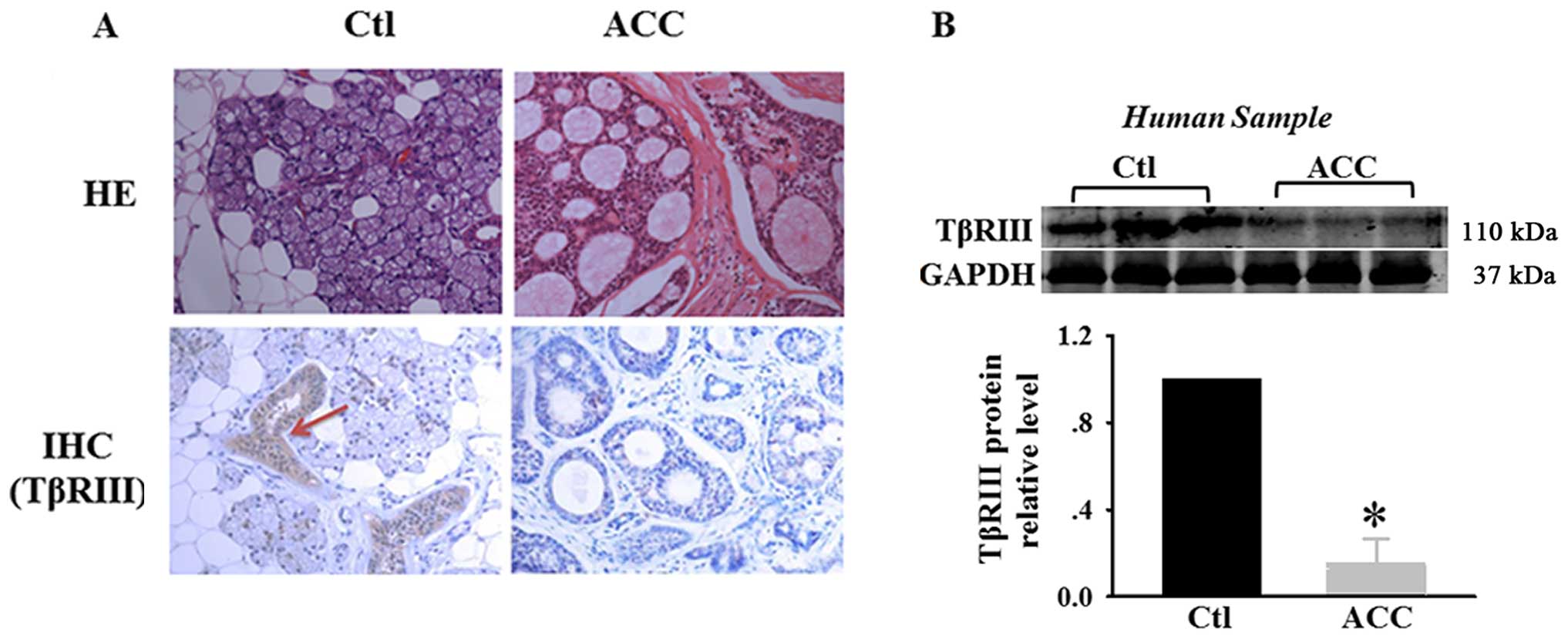
samples are shown in Fig. 1A.
H&E staining showed that adenoid cystic carcinoma exhibited
cribriform architecture with pseudocystic spaces filled with
eosiniphilic coagulum. IHC showed moderate to strong TβRIII
expression in normal salivary glands and negative TβRIII expression
in ACC specimens. We also examined the expression of TβRIII in
matched normal human salivary glands and adenoid cystic carcinoma
specimens. Lower levels of TβRIII protein were observed in human
adenoid cystic carcinoma specimens (Fig. 1B). These results suggest that TβRIII
expression is significantly decreased in adenoid cystic carcinoma,
with loss of TβRIII expression correlating with adenoid cystic
carcinoma progression.
TβRIII overexpression suppresses the
viability of ACC-M cells
To validate the positive functional involvement of
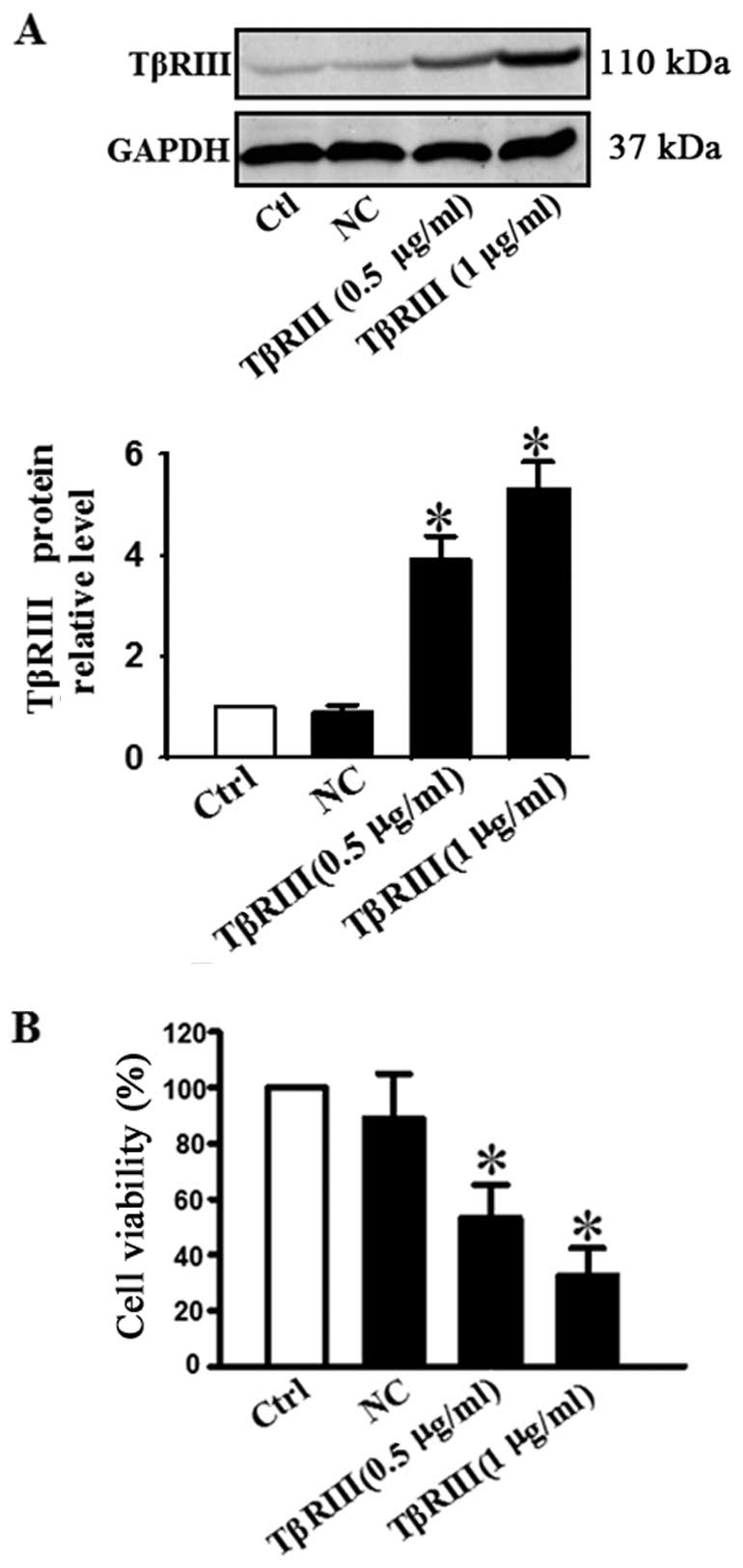
TβRIII in adenoid cystic carcinoma, we transfected ACC-M cells with
a plasmid encoding the constitutively active TβRIII. Successful
transfection of TβRIII was verified by our data shown in Fig. 2A. Western blot analysis showed that
the TβRIII protein level was increased in a concentration-dependent
manner in cells treated with 0.5 and 1 µg/ml of TβRIII
plasmid DNA (Fig. 2A). Cell
viability was determined by MTT assay. Fig. 2B shows that the viability of ACC-M
cells transfected with 0.5 or 1 µg/ml TβRIII plasmid was
reduced dose-dependently.
TβRIII overexpression induces apoptosis
in ACC-M cells
TGF-β signaling pathway induces programmed cell
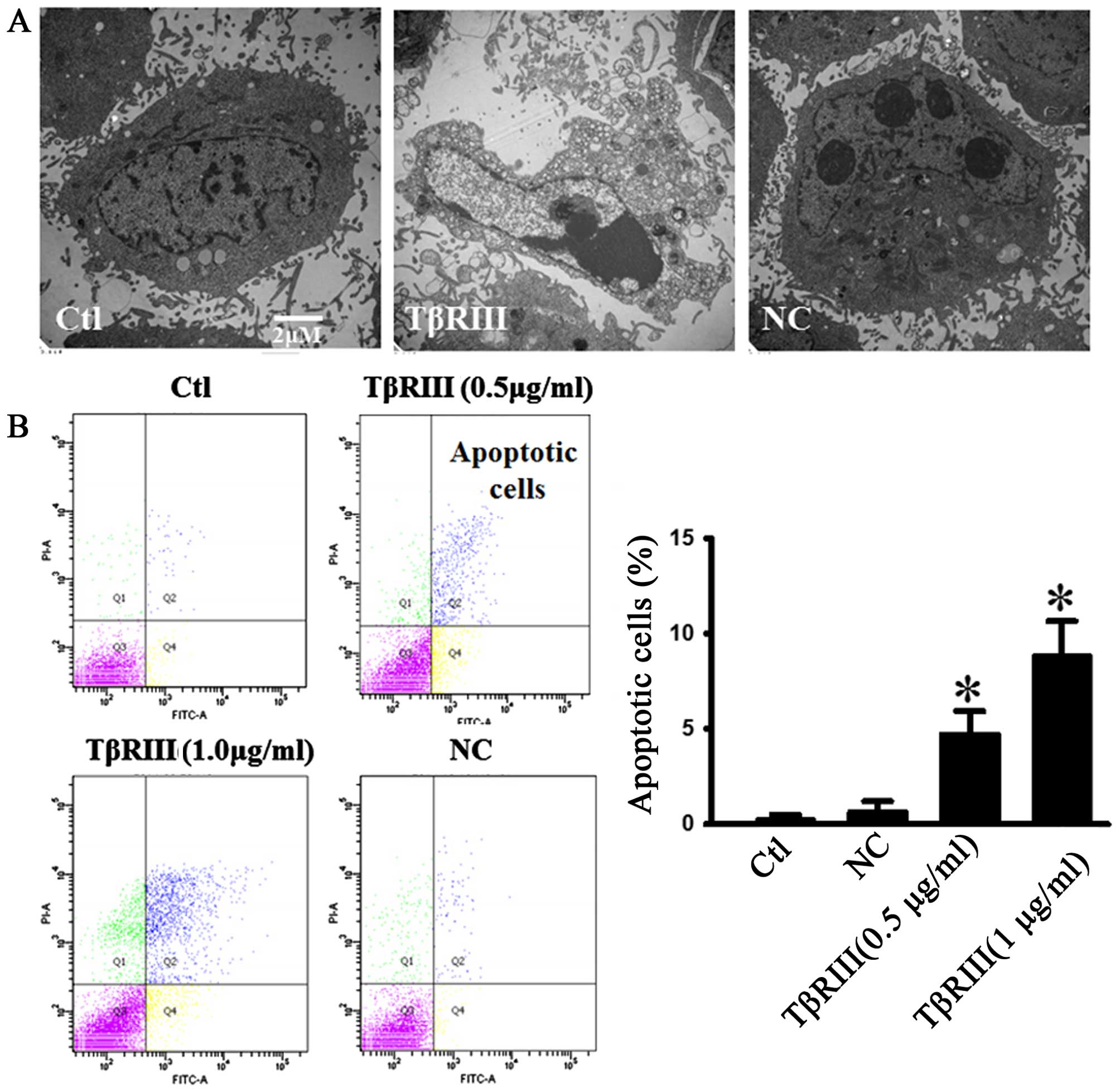
death in a variety of cell types. To determine the effects of
TβRIII overexpression on ACC-M cell apoptosis, we used electron
microscopy and flow cytometry to confirm the apoptotic changes.
Under an electron microscope the cells with TβRIII overexpression
exhibited robust changes in microstructure, including cell surface
microvilli reduction, nuclear chromatin condensation, margination,
and membrane blistering (Fig. 3A).
We transfected ACC-M cells with a plasmid encoding TβRIII or NC
plasmid, and apoptosis was assessed by flow cytometry in live cells
stained with Annexin V-FITC/PI. As shown in Fig. 3B, TβRIII overexpression induced
apoptosis in ACC-M cells in a dose-dependent manner.
TβRIII affects cell cycle progression in
ACC-M cells
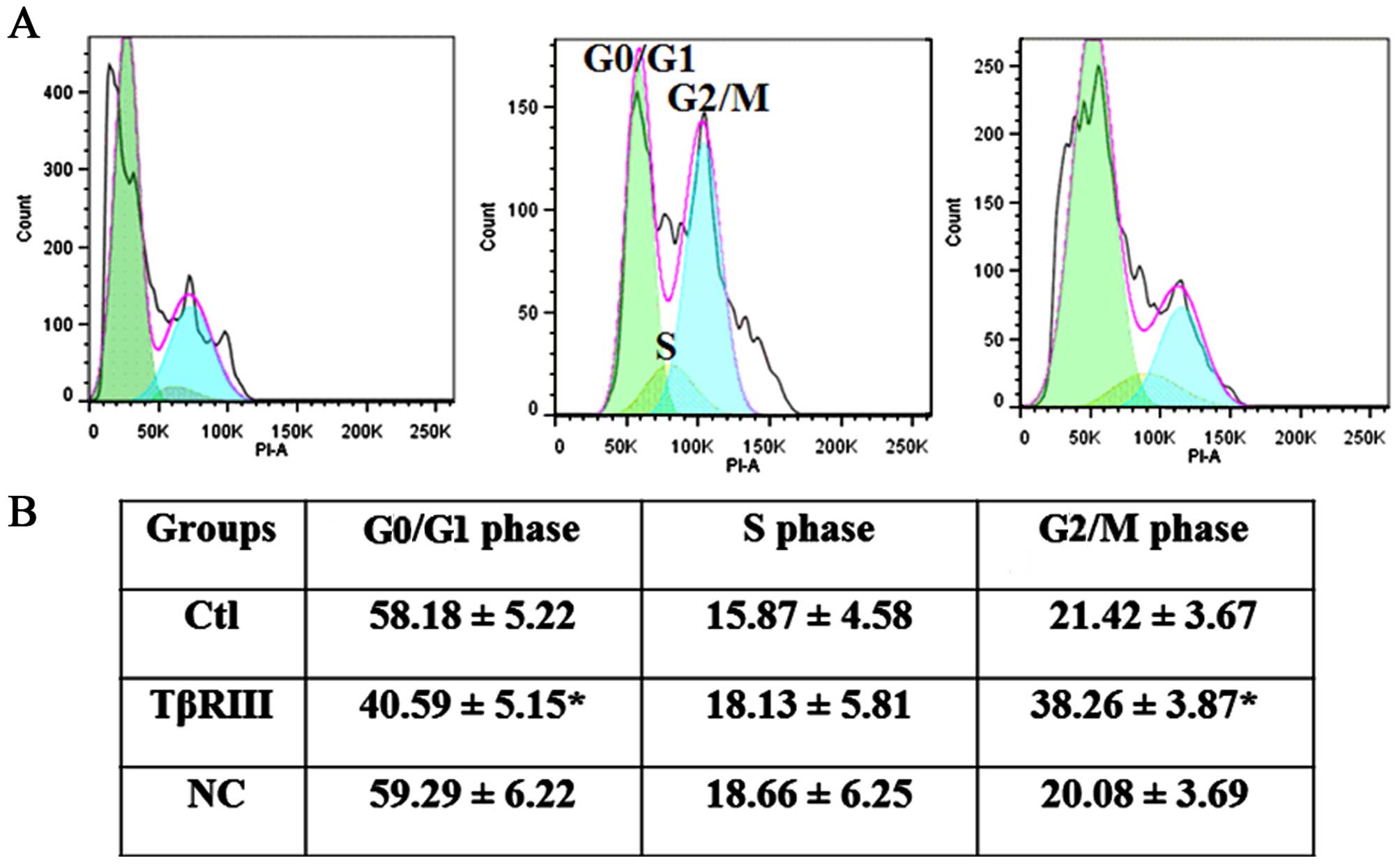
To define the mechanisms by which TβRIII regulated
viability of ACC-M cells further, we examined the effect of
increasing TβRIII expression on the cell cycle progression in ACC-M
cells in vitro. Cell cycle analysis revealed that TβRIII
over-expression resulted in a remarkable G2/M arrest in ACC-M cells
(Fig. 4).
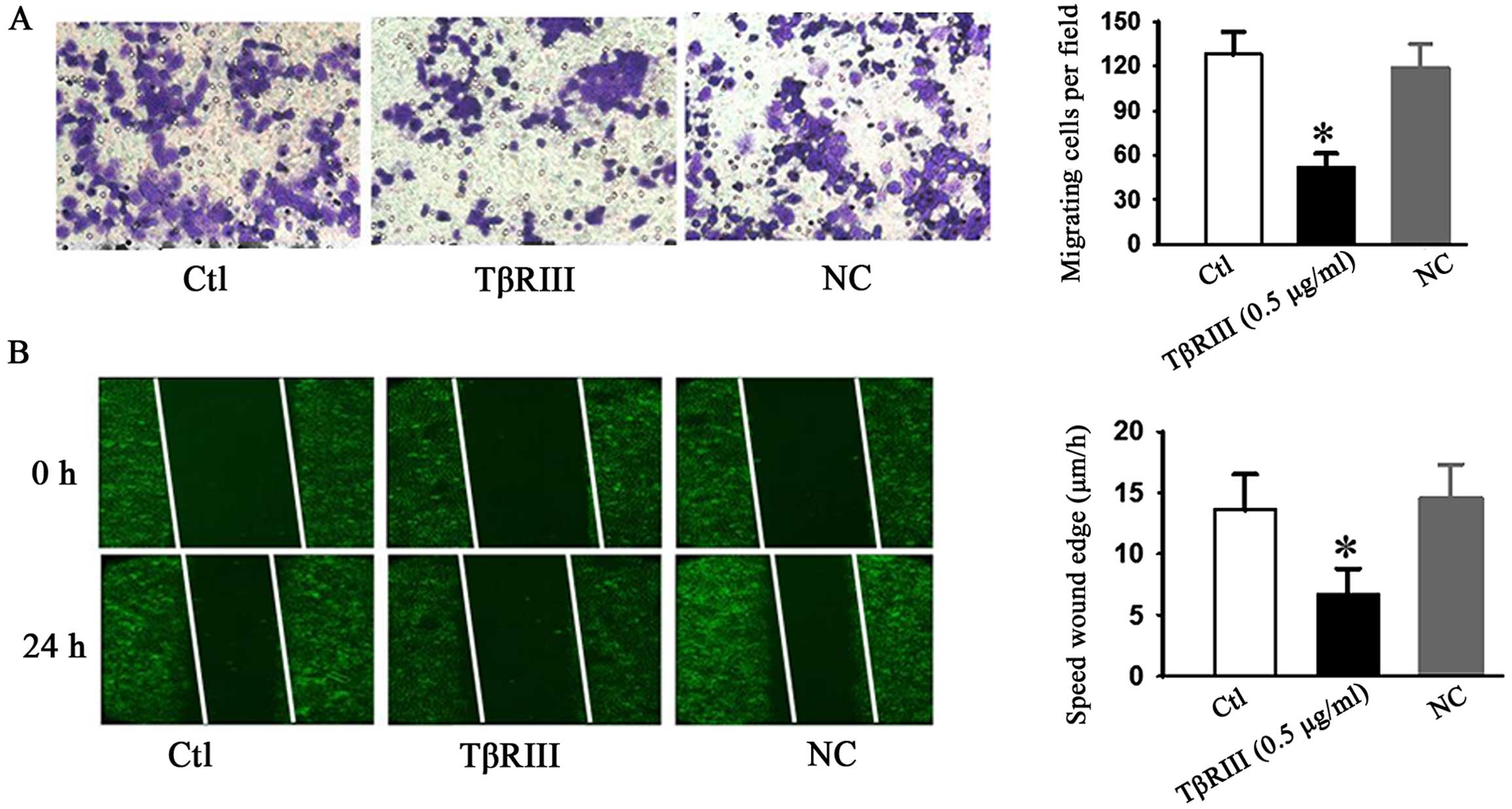
TβRIII delays and decreases ACC-M cells
migration in vitro
We further examined whether TβRIII overexpression
affected cell migration ability in ACC-M cells. Wound healing and
Transwell assays were evaluated. As shown in Fig. 5, both wound healing and Transwell
assays showed that TβRIII over-expression significantly inhibited
the cellular transmigration ability compared with controls. These
results strongly indicated that TβRIII may also regulate the cell
migration ability of adenoid cystic carcinoma.
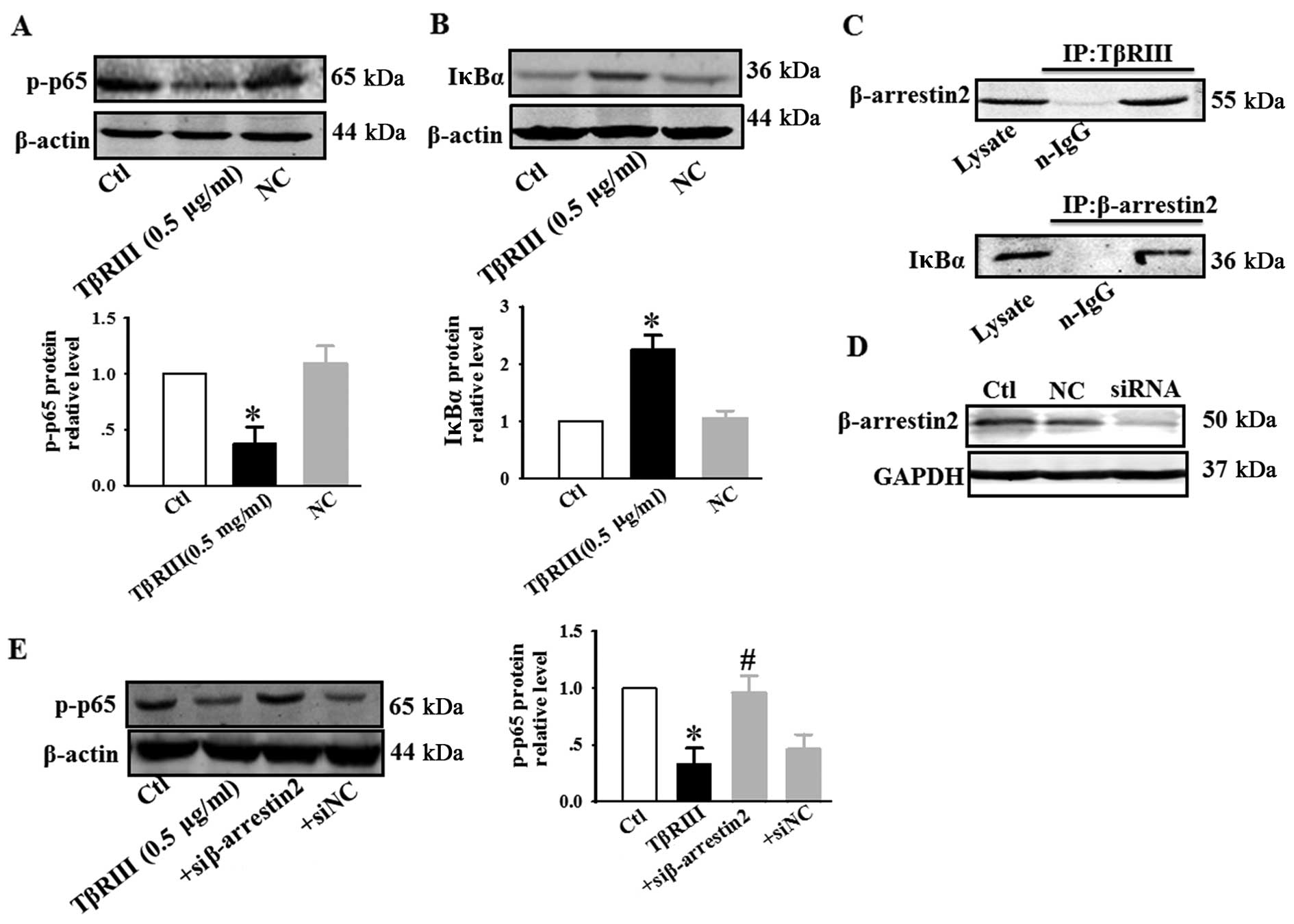
TβRIII negatively regulates NF-κB
signaling through interacting with β-arrestin2 in ACC-M cells
NF-κB is a dimeric transcription factor that
regulates genes involved in cell survival and proliferation
(18). Increased NF-κB activity has
been demonstrated in diverse human malignancies, including ACC-M,
which is believed to enhance tumor cell survival by accelerating
mitosis and inhibiting apoptosis (19). TGF-β has been reported to activate
(20) or inhibit (21) NF-κB signaling through mechanisms yet
to be fully defined. You et al (22) reported that TβRIII was involved in
NF-κB regulation via its interaction with β-arrestin2 in MCF10A
breast epithelial and MDA-MB-231 breast cancer cells. We next
investigated the effect of TβRIII overexpression on NF-κB activity
in ACC-M. Our results indicated that transiently increasing TβRIII
expression decreased p-p65 expression (Fig. 6A) and increased IκBα expression
(Fig. 6B). These results suggest
that inhibition of NF-κB signaling represents a potential mechanism
for TβRIII-mediated inhibition of cell apoptosis, migration, and
mitotic arrest in ACC-M. Our results also confirmed that TβRIII
interacted with β-arrestin2, and β-arrestin2 interacted with IκBα
(Fig. 6C). TβRIII overexpression
decreased p-p65 expression, and co-transfection of TβRIII and siRNA
of β-arrestin2 resulted in an increase in phosphorylation of p65
(Fig. 6E). These results suggest
that TβRIII through its interaction with β-arrestin2, negatively
regulates NF-κB signaling in ACC-M.
Discussion
In the present study, we report the expression
status of TβRIII in salivary gland adenoid cystic carcinoma for the
first time. We define an important role for TβRIII expression in
salivary gland adenoid cystic carcinoma as an inhibitor of cell
growth and migration through its inhibitory effects on NF-κB
signaling via its interaction with β-arrestin2.
Here, we demonstrated that the TβRIII expression was
markedly repressed in adenoid cystic carcinoma specimens compared
with matched normal human salivary glands, suggesting that a low
TβRIII expression level is associated with salivary gland adenoid
cystic carcinoma progression. Loss of TβRIII expression has also
been described in breast, lung, prostate, pancreas, ovary, and oral
squamous cell carcinomas as mentioned above. Turley et al
(10) reported that the restoration
of TβRIII expression in prostate cancer cells inhibits migration
and invasion. Zheng et al (23) reported that transient overexpression
of TβRIII induces apoptosis in human nasopharyngeal carcinoma
CNE-2Z cells. Forced overexpression of TβRIII upregulated
pro-apoptotic Bad and Bax protein, and downregulated anti-apoptotic
p-Bad, Bcl-2, and XIAP protein. These results support that the
TβRIII regulated multiple targets involved in cell proliferation by
mediating the TGF-β superfamily ligand independent signaling.
Furthermore, some studies reported that TβRIII had direct effects
on regulating tumor migration, invasion, and proliferation without
any cytokines and ligands (23,24).
These data indicated that gene therapy of TβRIII may be a powerful
new approach for cancer.
How does decreased TβRIII expression promote
salivary gland adenoid cystic carcinoma progression? The great
potential for hematogenous metastasis at an early stage is one of
the unique characteristics of ACCs. Zhang and Peng (19) reported that the high expression
levels of NF-κB were significantly correlated with ACC metastasis.
Our results showed that the reason of high levels of NF-κB in ACC
may be correlated with reduced expression of TβRIII. Regulation of
TβRIII expression occurs at multiple levels. At the transcriptional
level, TβRIII expression is negatively regulated by TGF-β1 through
inhibition of the promoter in multiple cell types. In some tumors,
especially late-stage tumors, a large amount of TGF-β1 was
secreted, and inhibited the expression of TβRIII (24).
Zanotto-Filho et al (25) demonstrated that the pharmacological
NF-κB inhibitors BAY117082 and MG132 induce cell arrest and
apoptosis in leukemia cells through ROS-mitochondria pathway
activation. Other groups also confirmed the result that inhibition
of NF-κB pathways induces G2/M arrest and apoptosis in different
cancer cells, including prostate cancer (26), glioblastoma (27) and melanoma (28). We studied the effects of TβRIII
overexpression on cell viability, apoptosis, cell cycle, and in
vitro cell migration in highly metastatic cell lines of human
ACC-M. We demonstrated that TβRIII inhibited ACC-M cell growth and
migration through its inhibitory effects on NF-κB signaling. As
NF-κB activation should be accompanied by proteasome-mediated
degradation of IκBα, we also investigated IκBα protein expression
in response to TβRIII stimulation. Our results indicated that
transiently increasing TβRIII expression decreased p-p65 expression
and increased IκBα expression in ACC-M.
We also showed a novel interaction of TβRIII with
the scaffolding protein, β-arrestin2, which results in TβRIII
internalization and downregulation of TGF-β signaling. β-arrestin2
also scaffolds interacting receptors with the IκBα (Fig. 6C). The results are consistent with
other groups. TβRIII has been shown to interact with β-arrestin2
through its cytoplasmic domain (29). Gao et al (30) have shown that β-arrestin2 directly
interacts with IκBα and prevents phosphorylation and degradation of
IκBα. Similar results have also been obtained from Witherow et
al (31). You et al
(22) demonstrated that TβRIII,
through its interaction with β-arrestin2, negatively regulates
NF-κB signaling in breast cancer.
In summary, we demonstrated that the TβRIII
expression was markedly repressed in adenoid cystic carcinoma.
Transient TβRIII overexpression induced apoptosis and G2/M arrest,
inhibited cell growth and migration in ACC-M cells. We established
the TβRIII/β-arrestin2 as an important negative regulator of NF-κB
signaling in ACC-M and a potential mechanism for TβRIII-mediated
inhibition of ACC-M migration and ACC progression. The present
study defines TβRIII as a biomarker exerting antitumor action on
ACC progression, thus, gene therapy of TβRIII may be a new approach
for ACC disease.
Acknowledgments
This study was supported by the Natural Science
Foundation for Youth of HeilongJiang Province (QC2011C006).
Abbreviations:
|
ACC
|
adenoid cystic carcinoma
|
|
TGF-β
|
transforming growth factor β
|
|
NF-κB
|
nuclear factor κB
|
|
BMPs
|
bone morphogenetic proteins
|
|
GDFs
|
growth and differentiation factors
|
|
TβRIII
|
the type III TGF-β receptor
|
|
ACC-M
|
high metastasis cell lines of ACCs
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium
bromide
|
|
FITC
|
fluorescein isothiocyanate
|
|
PI
|
propidium iodide
|
|
H&E
|
hematoxylin and eosin
|
References
|
1
|
Tian Z, Li L, Wang L, Hu Y and Li J:
Salivary gland neoplasms in oral and maxillofacial regions: A
23-year retrospective study of 6982 cases in an eastern Chinese
population. Int J Oral Maxillofac Surg. 39:235–242. 2010.
View Article : Google Scholar
|
|
2
|
Li J, Wang BY, Nelson M, Li L, Hu Y, Urken
ML and Brandwein-Gensler M: Salivary adenocarcinoma, not otherwise
specified: A collection of orphans. Arch Pathol Lab Med.
128:1385–1394. 2004.PubMed/NCBI
|
|
3
|
Spiro RH, Huvos AG and Strong EW: Adenoid
cystic carcinoma: Factors influencing survival. Am J Surg.
138:579–583. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang CY, Zhao YX, Xia RH, Han J, Wang BS,
Tian Z, Wang LZ, Hu YH and Li J: RASSF1A promoter hypermethylation
is a strong biomarker of poor survival in patients with salivary
adenoid cystic carcinoma in a Chinese population. PLoS One.
9:e1101592014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Laurie SA and Licitra L: Systemic therapy
in the palliative management of advanced salivary gland cancers. J
Clin Oncol. 24:2673–2678. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dong M, How T, Kirkbride KC, Gordon KJ,
Lee JD, Hempel N, Kelly P, Moeller BJ, Marks JR and Blobe GC: The
type III TGF-beta receptor suppresses breast cancer progression. J
Clin Invest. 117:206–217. 2007. View
Article : Google Scholar
|
|
7
|
Bandyopadhyay A, López-Casillas F, Malik
SN, Montiel JL, Mendoza V, Yang J and Sun LZ: Antitumor activity of
a recombinant soluble betaglycan in human breast cancer xenograft.
Cancer Res. 62:4690–4695. 2002.PubMed/NCBI
|
|
8
|
Finger EC, Turley RS, Dong M, How T,
Fields TA and Blobe GC: TbetaRIII suppresses non-small cell lung
cancer invasiveness and tumorigenicity. Carcinogenesis. 29:528–535.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hempel N, How T, Cooper SJ, Green TR, Dong
M, Copland JA, Wood CG and Blobe GC: Expression of the type III
TGF-beta receptor is negatively regulated by TGF-beta.
Carcinogenesis. 29:905–912. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Turley RS, Finger EC, Hempel N, How T,
Fields TA and Blobe GC: The type III transforming growth
factor-beta receptor as a novel tumor suppressor gene in prostate
cancer. Cancer Res. 67:1090–1098. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sharifi N, Hurt EM, Kawasaki BT and Farrar
WL: TGFBR3 loss and consequences in prostate cancer. Prostate.
67:301–311. 2007. View Article : Google Scholar
|
|
12
|
Gordon KJ, Kirkbride KC, How T and Blobe
GC: Bone morphogenetic proteins induce pancreatic cancer cell
invasiveness through a Smad1-dependent mechanism that involves
matrix metalloproteinase-2. Carcinogenesis. 30:238–248. 2009.
View Article : Google Scholar :
|
|
13
|
Mythreye K and Blobe GC: The type III
TGF-beta receptor regulates epithelial and cancer cell migration
through beta-arrestin2-mediated activation of Cdc42. Proc Natl Acad
Sci USA. 106:8221–8226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Meng W, Xia Q, Wu L, Chen S, He X, Zhang
L, Gao Q and Zhou H: Downregulation of TGF-beta receptor types II
and III in oral squamous cell carcinoma and oral
carcinoma-associated fibroblasts. BMC Cancer. 11:882011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gatza CE, Holtzhausen A, Kirkbride KC,
Morton A, Gatza ML, Datto MB and Blobe GC: Type III TGF-β receptor
enhances colon cancer cell migration and anchorage-independent
growth. Neoplasia. 13:758–770. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jovanović B, Beeler JS, Pickup MW, Chytil
A, Gorska AE, Ashby WJ, Lehmann BD, Zijlstra A, Pietenpol JA and
Moses HL: Transforming growth factor beta receptor type III is a
tumor promoter in mesenchymal-stem like triple negative breast
cancer. Breast Cancer Res. 16:R692014. View
Article : Google Scholar
|
|
17
|
Chu WF, Wu DM, Liu W, Wu LJ, Li DZ, Xu DY
and Wang XF: Sulforaphane induces G2-M arrest and apoptosis in high
metastasis cell line of salivary gland adenoid cystic carcinoma.
Oral Oncol. 45:998–1004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vlantis K, Wullaert A, Sasaki Y,
Schmidt-Supprian M, Rajewsky K, Roskams T and Pasparakis M:
Constitutive IKK2 activation in intestinal epithelial cells induces
intestinal tumors in mice. J Clin Invest. 121:2781–2793. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J and Peng B: In vitro angiogenesis
and expression of nuclear factor kappaB and VEGF in high and low
metastasis cell lines of salivary gland adenoid cystic carcinoma.
BMC Cancer. 7:952007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu T, Tian L, Han Y, Vogelbaum M and Stark
GR: Dose-dependent cross-talk between the transforming growth
factor-beta and interleukin-1 signaling pathways. Proc Natl Acad
Sci USA. 104:4365–4370. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Monteleone G, Mann J, Monteleone I,
Vavassori P, Bremner R, Fantini M, Del Vecchio Blanco G, Tersigni
R, Alessandroni L, Mann D, et al: A failure of transforming growth
factor-beta1 negative regulation maintains sustained NF-kappaB
activation in gut inflammation. J Biol Chem. 279:3925–3932. 2004.
View Article : Google Scholar
|
|
22
|
You HJ, How T and Blobe GC: The type III
transforming growth factor-beta receptor negatively regulates
nuclear factor kappa B signaling through its interaction with
beta-arrestin2. Carcinogenesis. 30:1281–1287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zheng F, He K, Li X, Zhao D, Sun F, Zhang
Y, Nie D, Li X, Chu W, Sun Y, et al: Transient overexpression of
TGFBR3 induces apoptosis in human nasopharyngeal carcinoma CNE-2Z
cells. Biosci Rep. 33:e000292013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gatza CE, Oh SY and Blobe GC: Roles for
the type III TGF-beta receptor in human cancer. Cell Signal.
22:1163–1174. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zanotto-Filho A, Delgado-Cañedo A,
Schröder R, Becker M, Klamt F and Moreira JC: The pharmacological
NFkappaB inhibitors BAY117082 and MG132 induce cell arrest and
apoptosis in leukemia cells through ROS-mitochondria pathway
activation. Cancer Lett. 288:192–203. 2010. View Article : Google Scholar
|
|
26
|
Yan X, Shen H, Jiang H, Zhang C, Hu D,
Wang J and Wu X: External Qi of Yan Xin Qigong induces G2/M arrest
and apop tosis of androgen-independent prostate cancer cells by
inhibiting Akt and NF-kappa B pathways. Mol Cell Biochem.
310:227–234. 2008. View Article : Google Scholar
|
|
27
|
Zanotto-Filho A, Braganhol E, Battastini
AM and Moreira JC: Proteasome inhibitor MG132 induces selective
apoptosis in glioblastoma cells through inhibition of PI3K/Akt and
NFkappaB pathways, mitochondrial dysfunction, and activation of
p38-JNK1/2 signaling. Invest New Drugs. 30:2252–2262. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zheng M, Ekmekcioglu S, Walch ET, Tang CH
and Grimm EA: Inhibition of nuclear factor-kappaB and nitric oxide
by curcumin induces G2/M cell cycle arrest and apoptosis in human
melanoma cells. Melanoma Res. 14:165–171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen W, Kirkbride KC, How T, Nelson CD, Mo
J, Frederick JP, Wang XF, Lefkowitz RJ and Blobe GC: Beta-arrestin
2 mediates endocytosis of type III TGF-beta receptor and
down-regulation of its signaling. Science. 301:1394–1397. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B
and Pei G: Identification of beta-arrestin2 as a G protein-coupled
receptor-stimulated regulator of NF-kappaB pathways. Mol Cell.
14:303–317. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Witherow DS, Garrison TR, Miller WE and
Lefkowitz RJ: Beta-arrestin inhibits NF-kappaB activity by means of
its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc
Natl Acad Sci USA. 101:8603–8607. 2004. View Article : Google Scholar : PubMed/NCBI
|