Introduction
Betaglycan (β-glycan) [transforming growth factor β
receptor type III (TGFβR3)] belongs to the membrane-bound accessory
receptors involved in signal initiation and propagation in cellular
pathway, activated by transforming growth factor-β types (TGFβs)
(1). Biochemically, this receptor
is a proteoglycan encoded by 225,660 bp TGFBR3, which is
located on chromosome 1p33-p32 (NCBI reference sequence:
NG_027757.1). It has two promoters but in the majority of tissues,
the proximal type dominates over a distal one. TGFBR3 is
divided into 18 exons and encodes a protein of 851 amino acids
(2). The molecular weight of
β-glycan protein is approximately 300 kDa, due to heparin and
chondroitin sulphate modifications (3–5). It is
ubiquitously expressed in almost all cell types, in approximately
200,000 particles/cell, and forms the non-covalently linked
homodimers (6).
Although, structural analysis of β-glycan revealed
lack of any well-known signaling motif in its amino-acid sequence,
it plays a vital role in signal mediation in the TGFβ pathway.
Studies on TGFβ binding caused by β-glycan demonstrated its
affinity to TGFβ isoforms and inhibin A, BMP-4 and -7, as well as
to GDF-5. β-glycan shows the highest effect on TGFβ2-induced signal
initiation, which itself binds poorly to the TGFβ type II receptor
(TGFβRII). This suggests the particular role of β-glycan in TGFβ2
signaling (7–10). Signal mediation via β-glycan is
based on TGFβ factor binding by its extracellular domain, with
simultaneous ligand concentration on the cell surface and complex
formation with TGFβRII receptor. As a consequence, the β-glycan
cytoplasmic region promotes interaction of intracellular domains of
heterodimeric TGFβRII and I receptors, as well as
trans-phosphorylation of TGFβRI by TGFβRII. After activation of
TGFβRII and TGFβRI receptors, β-glycan dissociates from this
complex and phosphorylated TGFβRI receptor propagate signal
downstream to the cellular TGFβ effector, Smad proteins (11). In addition to the above-mentioned
mechanism of action, in physiological conditions, the β-glycan
extracellular domain undergoes proteolytic cleavage (ectodomain
shedding) by metalloproteases (MT1-MMP and MT3-MMP) and plasmin,
resulting in the sequestration of a soluble form of β-glycan
(sol-β-glycan) in the extracellular matrix (ECM). Sol-β-glycan is
thought to modulate the signal induced by TGFβ factors, competing
with TGFβ receptors for their ligands. Sol-β-glycan functions as an
antagonist of TGFβ signaling, suggesting a potential role as an
antitumor agent in future therapies (12–17).
Another proposed mechanism of signaling orchestration in the TGFβ
pathway is the steric effect, which is a result of β-glycan
modification by residues of glycosaminoglycans (GAG). Oncogenic
Ki-ras seems to be involved in post-translational GAG
attachment, resulting in increased responsiveness to TGFβ
proliferative stimuli and downregulation of p21 in colon cancer
(18). Mythreye and Blobe (19) reported that GAG modifications are
necessary for inhibition of cell migration. Compared to controls,
TGFBR3ΔGAG mutants displayed increased migratory properties,
as shown either in ovarian cancer (Ovca429) or in normal ovarian
surface epithelial (NOSE007) cell lines. The GAG chains inhibit
TGFβ induced signaling by preventing formation of TGFβRII-TGFβRI
complexes, as confirmed in an LLC-PK1 model (20).
Some studies suggest an unquestioned role of
alterations in the TGFβ signaling pathway in many human diseases,
in particular cancer (10,21–23). A
large number of factors activating the TGFβ cascade result in the
regulation of opposed processes, what is known as the pleiotropic
effect on cells. TGFβs inhibit cancer development and progression
early in neoplastic transformation, whereas they contribute to
acquisition of a metastatic phenotype in more advanced clinical
stages (11). Our recent data
clearly demonstrated downregulation of β-glycan mRNA and its
relationship with clinical and pathological parameters (24,25).
It suggests that in the case of ECs, TGFβ deregulation may be a
result of impaired TGFβ2 signaling, which is caused by β-glycan.
However, a literature search (Medline® database) has
revealed that there are no studies evaluating the role of allelic
loss of β-glycan (TGFβR3) in primary human ECs.
The results obtained are the basis for the search of
potential molecular mechanisms responsible for the β-glycan decline
in primary human ECs. The aim of our present study was to evaluate
loss of heterozygosity (LOH) as a potential mechanism responsible
for downregulation of β-glycan in primary human ECs. We also
correlated the prevalence of allelic loss with clinical and
pathological variables of uterine malignancies.
Materials and methods
Patient material
Tissue samples from women having undergone surgery
for primary ECs were collected in the Second Department of
Gynecology, Lublin Medical University, Lublin, Poland, between 2010
and 2014. The study group consisted of 48 EC specimens and matched
48 normal tissue samples. None of the patients had received
hormonal therapy, radiation therapy or chemotherapy before surgery.
The mean age of the patients was 62 years, ranging from 46 to 81
years. At surgery, tissue specimens were immediately subdivided
into two portions; one was fixed in buffered formalin (pH 7.4) for
routine pathological examination, and the other was stored at −70°C
until further analysis. The clinical stage was assigned according
to a recently established FIGO classification system (26). World Health Organization
classification was applied to determine the pathological grading.
Myometrial and lymph node invasion and VSI (vascular space
invasion) were evaluated as well. Clinicopathological variables of
the EC samples are depicted in Table
I. The study cohort was subdivided into two age groups: the
first group consisted of women <60 years of age (n=21; 44%), the
second group of women ≥60 years (n=27; 56%). The Independent Ethics
Committee of the Lublin Medical University, Lublin, Poland approved
the tissue collection and subsequent experiments, and all the women
enrolled provided their informed consent.
 | Table IClinicopathological parameters of the
patients with endometrial cancer. |
Table I
Clinicopathological parameters of the
patients with endometrial cancer.
| Clinicopathological
parameters | No. of patients
(%) |
|---|
| Patients' age
(years) |
| <60 | 21 (44) |
| ≥60 | 27 (56) |
| Clinical
stagea |
| I | 22 (46) |
| II | 15 (31) |
| III | 7 (15) |
| IV | 4 (8) |
| Histological
gradeb |
| G1 | 9 (19) |
| G2 | 34 (71) |
| G3 | 5 (10) |
| Depth of myometrial
invasion |
| <1/2 | 21 (44) |
| >1/2 | 27 (56) |
| Vascular space
invasion |
| Not present | 37 (77) |
| Present | 11 (23) |
| Lymph node
invasion |
| Not present | 36 (75) |
| Present | 1 (2) |
| Not assessed | 11 (23) |
RNA isolation and real-time polymerase
chain reaction (PCR)
Total RNA was extracted according to a modified
Chomczynski and Sacchi protocol. Afterwards, RNA (1 µg) was
retro-transcribed using RevertAid™ H Minus First Strand cDNA
Synthesis kit (Fermentas, Canada) according to manufacturer's
recommendation. Real-time PCR was performed using
TaqMan® probes (Life Technologies, Carlsbad, CA, USA),
and in line with the protocol on Mastercycler®
Epgradient S Realplex (Eppendorf, Hamburg, Germany). GAPDH
(glyceraldehyde 3-phosphate dehydrogenase) served as an
internal control. The catalogue numbers of probes were: Hs00234259_
m1 for TGFBR3 (β-glycan) and Hs99999905_m1 for GAPDH. The
relative expression level was normalized to GAPDH, and calculated
using the following equation: 2−ΔCt x 1,000.
Western blotting
Tissue samples were homogenized in lysis buffer
containing 0.25 M sucrose, 50 mM Tris-HCl (pH 7.4), 5 mM
MgCl2, 0.5% Triton X-100 and 1 mM PMSF. For each sample,
protein concentration was evaluated by Lowry protocol (27). The proteins (30 µg/well) were
separated by sodium dodecyl sulphate (SDS)-polyacrylamide gel
electrophoresis (7.5%) and transferred onto Immobilon-P membranes
(Millipore, Billerica, MA, USA) using the semi-dry system. The
membranes were incubated overnight at 4°C with primary antibodies
after prior blocking with 5% dry non-fat milk. Following extensive
washing with Tris-buffered saline with 0.1% Tween-20, the membranes
were incubated with horseradish peroxidase-conjugated (HRP)
secondary antibodies for 1 h and visualized with Novex HRP
Chromogenic Substrate-TMB (Invitrogen Inc., Carlsbad, CA, USA).
β-actin served as loading control. After TMB visualization, blots
were incubated at 50°C for 45 min in stripping buffer containing 2%
SDS, 62.5 mM Tris-HCl (pH 6.8), 0.8% β-mercaptoethanol, rinsed with
running water, and rehydrated with methanol. Immune-identification
of β-actin was carried out. Quantitative analysis was performed by
measuring IOD by GelProAnalyzer v. 3.0 for Windows™ software (Media
Cybernetics, Baltimore, MD, USA). The following commercially
available antibodies were applied: primary: rabbit polyclonal
anti-β-glycan antibodies (ab97459; Abcam, Cambridge, UK) against
protein fragment corresponding to a region within amino acids
88–274 of human β-glycan (dilution: 1:1,000), goat polyclonal
anti-actin antibodies (sc-1616, Santa Cruz Biotechnology, Santa
Cruz, CA, USA) against carboxy-terminus of actin of human origin
(dilution: 1:1,000); secondary: goat anti-rabbit polyclonal
antibodies (A9169, Sigma-Aldrich, Schnelldorf, Germany) against
whole molecule (dilution: 1/20,000), rabbit anti-goat polyclonal
antibodies (A8919, Sigma-Aldrich) against whole molecule (dilution:
1/5,000).
DNA isolation and PCR
Total genomic DNA was isolated according to the
phenol/chloroform protocol. Briefly, tissue was minced and
homogenized with denaturing solution consisting of 10 mM EDTA, 10
mM Tris/HCl (pH 8.0), 0.5% SDS. Following overnight incubation at
55°C with 20 µl of proteinase K (10 mg/ml), an equal volume
of phenol/chloroform/isoamyl alcohol (25:24:1) mixture was added to
the samples. After centrifugation, the aqueous phase was
precipitated with an equal volume of isopropanol. DNA pellet was
washed twice with 70% ethanol, re-suspended in TE buffer and stored
at −70°C for further analysis. The quality and quantity of DNA was
estimated spectrophotometrically with BioPhotometer Plus
(Eppendorf, Hamburg, Germany).
PCR was performed in Applied Biosystems 2720 thermal
cycler (Applied Biosystems, Foster City, CA, USA). Primers used in
the reaction were fluorescently labeled (Sigma-Aldrich). Table II presents the TGFBR3
microsatellite markers and PCR conditions applied. Briefly, the PCR
reaction was carried out in a total volume of 12.5 µl and
the mixture consisted of 1X PCR buffer [10 mM Tris/HCl (pH 8.3), 50
mM KCl, 1.5 mM MgCl2, 0.01% gelatin] dedicated for the
JumpStart™ Taq DNA polymerase (Sigma-Aldrich), 5 ng of
genomic DNA, 0.5 µM of each primer, 0.2 mM of each dNTP and
0.625 U of JumpStart™ Taq DNA polymerase. Sequences
of primers were adopted from the National Center for Biotechnology
Information database-NCBI (www.ncbi.nlm.nih.gov).
| Table IITGFBR3 microsatellite markers
and PCR conditions. |
Table II
TGFBR3 microsatellite markers
and PCR conditions.
| Marker | Chromosomal
localization | Repeat motif and
product sizes | Annealing
temperature | Primer
sequences | Dye |
|---|
| D1S188 | 1p31 | (CA)n
149–173 bp | 64°C | F:
5′-AACCAATCAAGGTGCCTGCA-3′ | – |
| | | R:
5′-TCCCCTAGTGTCCTGGCAG-3′ | FAM |
| D1S435 | 1p31 | (CA)n
157–177 bp | 62°C | F:
5′-GGTTATTAGGCATGATAAGGG-3′ | – |
| | | R:
5′-ACGCTGTCTCTGACAAGAAA-3′ | FAM |
| D1S1588 | 1p33-p32 | (AAT)n
118–139 bp | 58.5°C | F:
5′-CTGGTCCCATAGCTAGTAAACG-3′ | – |
| | | R:
5′-ATGAGGTCCCCATTTACCAT-3′ | TET |
LOH analysis
After PCR, the samples were mixed with solution
containing deionized formamide, GeneScan-350 TAMRA (Life
Technologies) dye size standard, and loading buffer (blue dextran,
EDTA). They were denatured, chilled on ice, and separated in 5%
Long Ranger (BioWhittaker Molecular Applications, Rockland, ME,
USA) containing 6 M urea. PCR products were analyzed using DNA
Sequencer ABI PRISM 377 (Applied Biosystems). Allele lengths were
determined with GeneScan v. 3.1.2 and Genotyper v. 2.5 software
(Applied Biosystems). Amplification of microsatellite markers
yielded one or two allele peaks, depending upon whether the
individual is homozygous (non-informative cases) or heterozygous
(informative cases) for that marker. Loss of heterozygosity (LOH)
was defined when one allelic band from tumor DNA disappeared
completely or when the signal intensity (allelic ratio ≤0.5) was
reduced <50% in the tumor DNA compared with the paired normal
DNA pattern. Allele ratios were calculated only for informative
cases according to the following formula:
T1:T2/N1:N2, where
T1 and N1 are the values for shorter length
allele product peak of tumor and normal sample, and T2
and N2 are the values for longer length allele product
peak of tumor and normal sample, respectively (28). All samples were analyzed in
replicates.
Statistical analysis
Statistical tests were performed applying GraphPad
Prism v. 5.00 software for Windows (GraphPad Software, Inc., La
Jolla, CA, USA). Data are presented as mean ± standard error of
mean (SEM). P-values were calculated using two-tailed paired
Student's t-test. The statistical analysis of allelic loss included
the assessment of the association between the prevalence of LOH and
clinical and pathological variables using Fisher's two-tailed exact
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
β-glycan expression pattern
Real-time PCR and western blotting were used for
β-glycan mRNA and protein quantification methods, respectively.
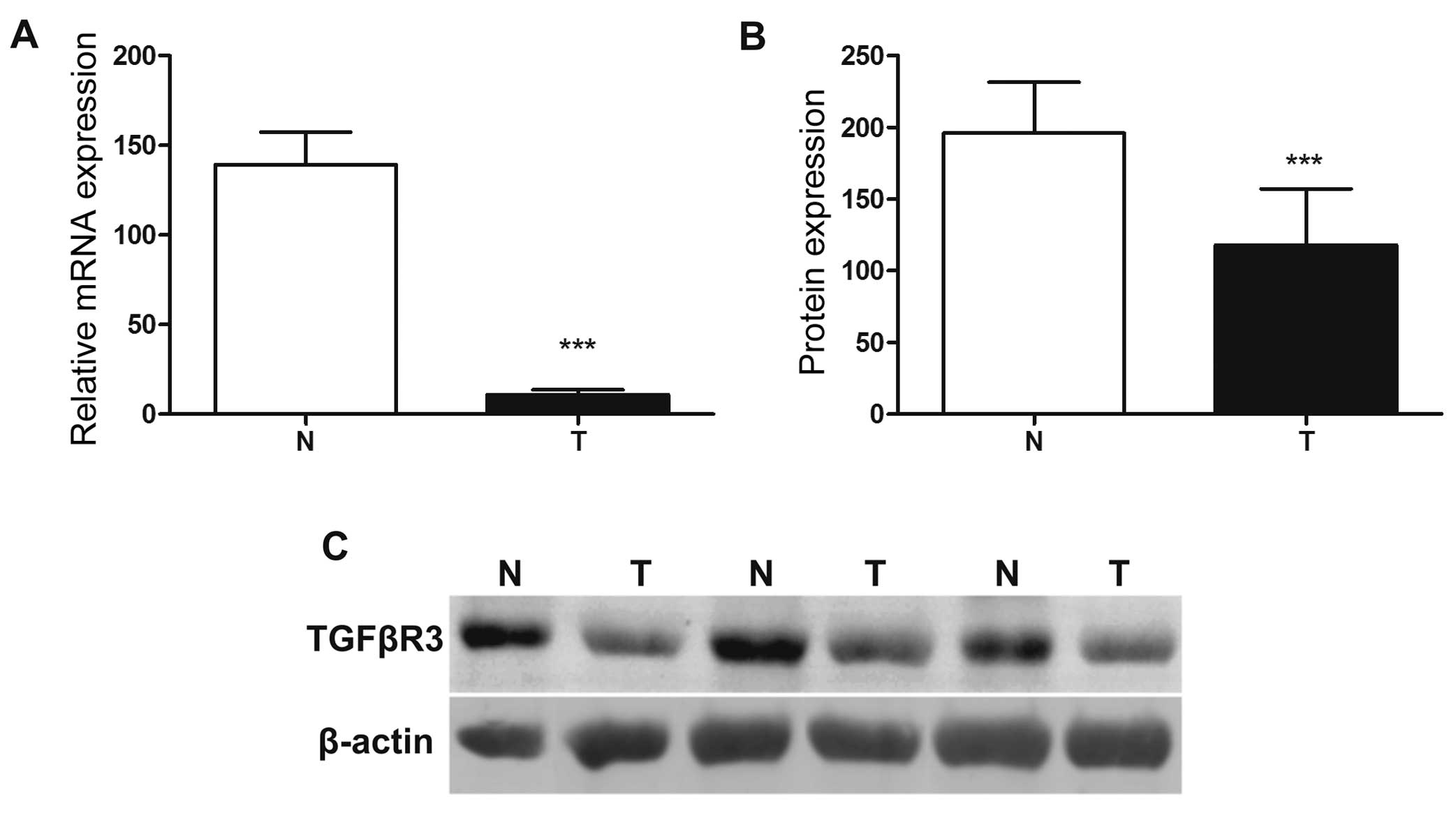
TGFBR3 expression was found to be significantly
downregulated in the EC samples compared to that noted in the
normal tissues (P<0.001) (Fig.
1A). Moreover, β-glycan mRNA decline corresponded to its
protein decrease, as demonstrated in all EC samples studied. The
decrease of β-glycan protein expression in ECs was highly
significant (P<0.001) (Fig. 1B).
Examples of immunoblots of β-glycan in normal and EC samples in
relation to actin (serving as an internal control) are shown in
Fig. 1C.
β-glycan allelic loss
To examine whether or not allelic loss is a bona
fide mechanism clearly responsible for decreased β-glycan
expression in human ECs, molecular analysis was performed with the
use of three different microsatellite markers: D1S188, D1S435 and
D1S1588. These markers are spanned within or in direct proximity to
the TGFBR3 locus (Table
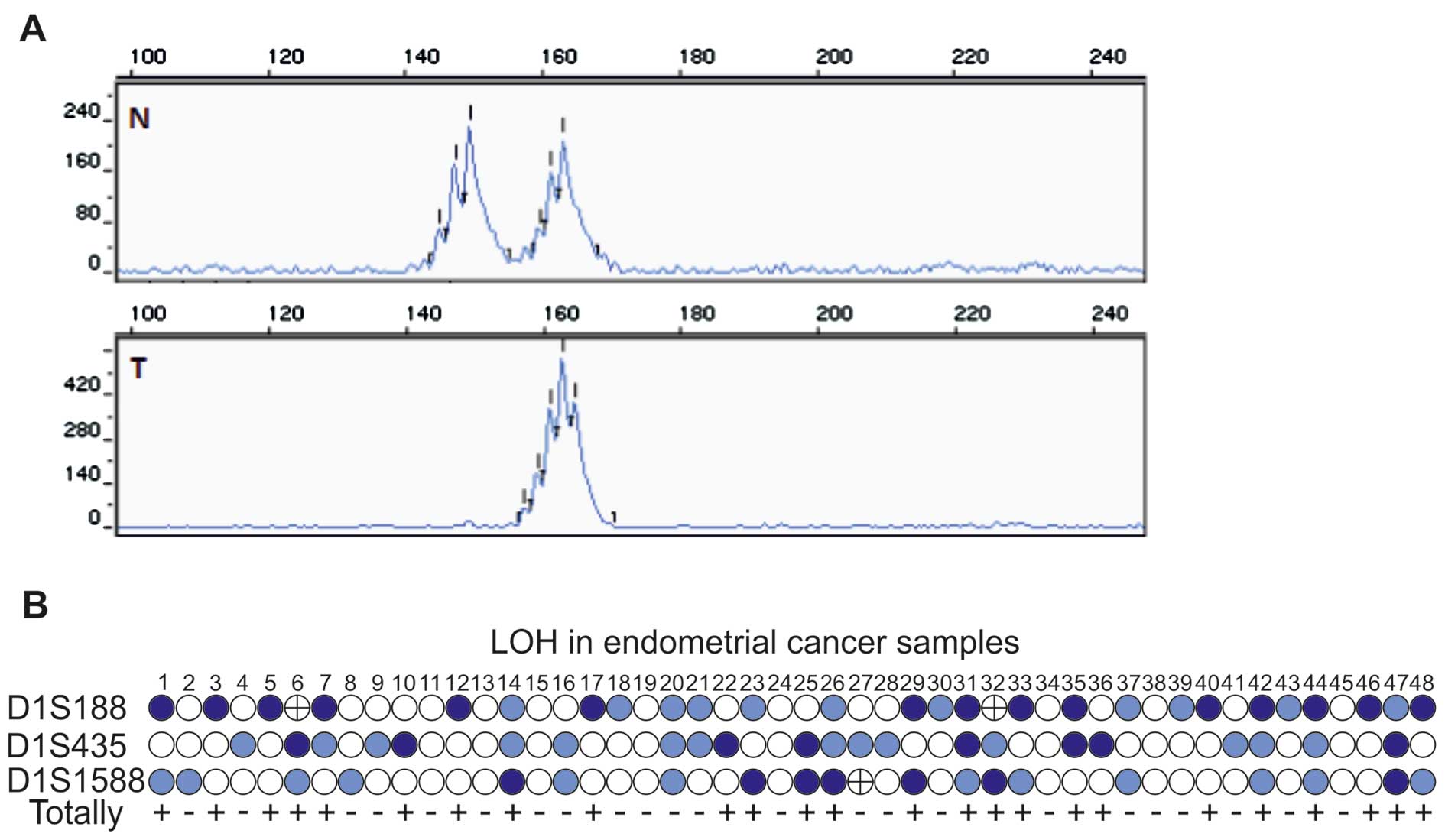
II). Loss of heterozygosity is a chromosomal event defined as a
direct loss of one allele or as an intensity reduction >50% in
EC samples compared to the corresponding normal tissue (Fig. 2A). It is worth pointing out that 25
of 39 (64%) informative cases and 25 of 48 (52%) uterine cancer
specimens studied revealed allelic imbalance in at least one
microsatellite marker evaluated. Altogether, 54% (15/28), 36%
(8/22) and 35% (7/20) of informative ECs revealed an allelic loss
in D1S188, D1S435 and D1S1588, respectively (Fig. 2B and Table III). It is worth pointing out that
5 out of 39 (13%) informative cases showed LOH at two
microsatellite markers. Microsatellite instability (MSI) was found
in only two markers, but to a very strictly limited extent (in two
cases - #6 and #32 and in case #27 in the D1S188 and D1S1588
microsatellite markers, respectively) (Fig. 2B).
| Table IIIFrequency of the loss of
heterozygosity (LOH) in the endometrial cancer samples. |
Table III
Frequency of the loss of
heterozygosity (LOH) in the endometrial cancer samples.
| Marker | Informative
cases/total cases | LOH-positive cases
(%) |
|---|
| D1S188 | 28/48 | 15 (54) |
| D1S435 | 22/48 | 8 (36) |
| D1S1588 | 20/48 | 7 (35) |
Correlation between β-glycan LOH and
clinicopathological variables of ECs
The correlation between LOH in the TGFBR3
locus and clinical and pathological variables of EC samples is
presented in Table IV. None of the
clinicoprognostic features, including clinical staging,
histological grading, myometrial invasion, VSI and age at
diagnosis, was found to be of significance. Moreover, additional
analysis of LOH occurrence in ECs in relation to combined
clinicopathological variables, i.e., FIGO stage/myometrial
invasion, histological grade/myometrial invasion, FIGO stage/age at
diagnosis, histological grade/age at diagnosis did not reveal any
significant differences (data not shown). Therefore, the presence
of LOH in the TGFBR3 locus did not appear to determine the
malignancy of the sporadic uterine neoplasms, and probably is an
early event during endometrial transformation in humans.
| Table IVLoss of heterozygosity (LOH) in
clinicopathological features of EC patients in relation to allelic
loss at the TGFBR3 locus using microsatellite markers
D1S188, D1S435 and D1S1588. |
Table IV
Loss of heterozygosity (LOH) in
clinicopathological features of EC patients in relation to allelic
loss at the TGFBR3 locus using microsatellite markers
D1S188, D1S435 and D1S1588.
| Clinicopathological
parameters | D1S188
| P-value | D1S435
| P-value | D1S1588
| P-value |
|---|
| I 28 | N 13 | LOH 15 | I 22 | N 14 | LOH 8 | I 20 | N 13 | LOH 7 |
|---|
| FIGO stage |
| I | 9 | 4 | 5 | 0.85 | 11 | 7 | 4 | 0.49 | 8 | 5 | 3 | 0.70 |
| II | 12 | 6 | 6 | | 6 | 5 | 1 | | 7 | 4 | 3 | |
| III | 4 | 1 | 3 | | 2 | 1 | 1 | | 3 | 3 | 0 | |
| IV | 3 | 2 | 1 | | 3 | 1 | 2 | | 2 | 1 | 1 | |
| Histological
grade |
| G1 | 6 | 2 | 4 | 0.41 | 6 | 5 | 1 | 0.58 | 3 | 1 | 2 | 0.33 |
| G2 | 20 | 9 | 11 | | 13 | 7 | 6 | | 15 | 11 | 4 | |
| G3 | 2 | 2 | 0 | | 3 | 2 | 1 | | 2 | 1 | 1 | |
| Myometrial
invasion |
| <1/2 | 11 | 5 | 6 | >0.99 | 12 | 7 | 5 | 0.67 | 9 | 6 | 3 | >0.99 |
| >1/2 | 17 | 8 | 9 | | 10 | 7 | 3 | | 11 | 7 | 4 | |
| Vascular space
invasion |
| Not present | 21 | 10 | 11 | >0.99 | 17 | 11 | 6 | 0.61 | 16 | 10 | 6 | >0.99 |
| Present | 7 | 3 | 4 | | 5 | 3 | 2 | | 4 | 3 | 1 | |
| Age (years) at
diagnosis |
| <60 | 11 | 5 | 6 | >0.99 | 9 | 7 | 2 | 0.38 | 7 | 6 | 1 | 0.33 |
| ≥60 | 17 | 8 | 9 | | 13 | 7 | 6 | | 13 | 7 | 6 | |
Discussion
EC is one of the leading causes of cancer-related
mortality among females with approximately 320,000 new cases and
76,000 deaths worldwide in 2012. In Poland, 5426 new cases were
diagnosed in 2011 (29). A
significantly higher incidence rate is observed in developed
countries in contrast to less developed ones. An increase in the EC
incidence rate should be expected in the near future due to the
intensified aging of human societies (30).
Other studies suggest that cancer development and
progression appears to be associated with alterations in the TGFβ
signaling cascade (31). Disturbed
signal mediation in the TGFβ pathway triggers its development from
a tumor suppressor, early in neoplastic transformation, to a
cancer-promoting and -metastatic agent in advanced clinical stages
of the disease (32). A particular
role in neoplastic transformation has been reported in the case of
β-glycan, which acts as a tumor inhibitory agent suppressing cancer
cell migration, invasion, proliferation and angiogenesis (6,11).
β-glycan downregulation is responsible for impaired ligand
presentation to TGFβ canonical receptors - TGFβRII and TGFβRI
(33). Loss of β-glycan expression
results in impaired signaling driven by TGFβ2 isoform, as TGFβ2
possesses the highest and exclusive affinity to β-glycan (33–35).
Besides TGFβ2 isoform signal mediation, β-glycan reduction may
favor development of an immunotolerant tumor microenvironment as an
immune suppressor and stimulator of Treg cells (36). Furthermore, involvement of β-glycan
in cancer development through non-canonical TGFβ signaling pathways
cannot be excluded. Significant alterations in β-glycan expression
have been reported in several human neoplasms originating from
different tissues, such as breast, endometrial, ovarian,
pancreatic, prostate, bladder, liver, lung and renal carcinomas
(24,37–46).
According to previous studies, chromosome 1p exhibits a meaningful
allelic imbalance in a number of primary human malignancies
(47).
Our current results are in line with those
previously published by Florio et al (48), who reported a significant
downregulation of β-glycan expression with concomitant decrease in
the inhibin α-subunit in the case of EC. Current results concerning
β-glycan protein expression are not in line with those published
previously by our group (24),
where ELISA assay was performed. Presently, protein expression was
analyzed by western blotting as a more specific and acknowledged
method. Moreover, we applied different primary antibodies which
corresponded to a region within amino acids 88–74 of the fully
processed and mature human form of β-glycan molecule. Previous
antibodies directed against β-glycan C-terminus may lead to
immunodetection of non-functionally synthesized β-glycan particles
with abrogated trafficking to the cell membrane and might also
cross-react with endoglin due to the sequence similarities.
The data of our study indicate LOH in the
TGFBR3 region located at chromosome 1p, and its association
with a decrease in β-glycan in sporadic human ECs. These results
suggest that allelic loss at the TGFBR3 region may be the
mechanism through which EC cells escape from TGFβ-mediated
suppression. Similar results have been achieved in studies on
allelic imbalance in different human neoplasms, in particular in
those derived from hormone-dependent tissues (38,39,41,42).
In breast carcinomas, the mechanisms responsible for
down-regulation of β-glycan included LOH in microsatellite markers
(D1S1588 and D1S188), in half of the samples analyzed (38). The study of the D1S1588, D1S2804 and
D1S435 microsatellite markers in prostate carcinomas showed LOH in
37.5% of samples studied (41).
Comparable results of allelic imbalance in the β-glycan locus have
been achieved in investigations of non-small cell lung carcinomas,
where LOH was reported in 38.5% of cases, at least in one
microsatellite marker (D1S1588, D1S188 or D1S2804) (42). Despite the high percentage of
LOH-positive cases in the TGFBR3 locus in our study (52% of
the samples investigated; 64% of informative cases), other
mechanisms causing mRNA and protein downregulation in ECs cannot be
excluded.
Studies on cancer cell lines revealed that loss of
β-glycan expression could be due to altered epigenetic regulation,
as observed in prostate and ovarian tumor cell lines, where
β-glycan expression was restored after treatment with
methyltransferase and histone deacethylase inhibitors (39,41).
Indirect epigenetic regulation was reported in bladder urothelioma
and renal cell carcinoma (23,49).
Β-glycan expression in these tumor types appeared to be positively
controlled by the GATA3 transcription factor (23,49).
Increased methylation of GATA3 was found to result in a decline in
β-glycan at the transcriptomic level (49). The molecular mechanisms determining
β-glycan expression alterations appear to involve other mechanisms
as well, since neither genetic aberrations nor allelic imbalance
were found to be associated with β-glycan expression in
hepatocellular carcinoma (44).
Transcriptional regulation is suggested to play an exclusive role.
Moreover, the lack of a statistical significance of LOH occurrence
and single or combined clinicopathological parameters of EC samples
strongly supports the hypothesis that β-glycan allelic imbalance
may be an early genomic event during endometrial neoplastic
transformation that does not determine cancer aggressiveness.
Acknowledgments
This study was supported by grant
UMO-2011/01/N/NZ4/01723 from the National Science Centre (NCN),
Craków, Poland.
References
|
1
|
Cheifetz S, Andres JL and Massagué J: The
transforming growth factor-beta receptor type III is a membrane
proteoglycan. Domain structure of the receptor. J Biol Chem.
263:16984–16991. 1988.PubMed/NCBI
|
|
2
|
Hempel N, How T, Dong M, Murphy SK, Fields
TA and Blobe GC: Loss of betaglycan expression in ovarian cancer:
Role in motility and invasion. Cancer Res. 67:5231–5238. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang XF, Lin HY, Ng-Eaton E, Downward J,
Lodish HF and Weinberg RA: Expression cloning and characterization
of the TGF-beta type III receptor. Cell. 67:797–805. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
López-Casillas F, Cheifetz S, Doody J,
Andres JL, Lane WS and Massagué J: Structure and expression of the
membrane proteoglycan betaglycan, a component of the TGF-beta
receptor system. Cell. 67:785–795. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Morén A, Ichijo H and Miyazono K:
Molecular cloning and characterization of the human and porcine
transforming growth factor-beta type III receptors. Biochem Biophys
Res Commun. 189:356–362. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gatza CE, Oh SY and Blobe GC: Roles for
the type III TGF-beta receptor in human cancer. Cell Signal.
22:1163–1174. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
López-Casillas F, Payne HM, Andres JL and
Massagué J: Betaglycan can act as a dual modulator of TGF-beta
access to signaling receptors: Mapping of ligand binding and GAG
attachment sites. J Cell Biol. 124:557–568. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Esparza-Lopez J, Montiel JL,
Vilchis-Landeros MM, Okadome T, Miyazono K and López-Casillas F:
Ligand binding and functional properties of betaglycan, a
co-receptor of the transforming growth factor-beta superfamily.
Specialized binding regions for transforming growth factor-beta and
inhibin A. J Biol Chem. 276:14588–14596. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kirkbride KC, Ray BN and Blobe GC:
Cell-surface co-receptors: Emerging roles in signaling and human
disease. Trends Biochem Sci. 30:611–621. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bernabeu C, Lopez-Novoa JM and Quintanilla
M: The emerging role of TGF-beta superfamily coreceptors in cancer.
Biochim Biophys Acta. 1792:954–973. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bilandzic M and Stenvers KL: Betaglycan: A
multifunctional accessory. Mol Cell Endocrinol. 339:180–189. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Andres JL, Stanley K, Cheifetz S and
Massagué J: Membrane-anchored and soluble forms of betaglycan, a
polymorphic proteoglycan that binds transforming growth
factor-beta. J Cell Biol. 109:3137–3145. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bandyopadhyay A, Zhu Y, Cibull ML, Bao L,
Chen C and Sun L: A soluble transforming growth factor β type III
receptor suppresses tumorigenicity and metastasis of human breast
cancer MDA-MB-231 cells. Cancer Res. 59:5041–5046. 1999.PubMed/NCBI
|
|
14
|
Cheung HK, Mei J and Xu RJ: Quantification
of soluble beta-glycan in porcine milk. Asia Pac J Clin Nutr.
12:S612003.
|
|
15
|
Velasco-Loyden G, Arribas J and
López-Casillas F: The shedding of betaglycan is regulated by
pervanadate and mediated by membrane type matrix metalloprotease-1.
J Biol Chem. 279:7721–7733. 2004. View Article : Google Scholar
|
|
16
|
Bandyopadhyay A, Wang L, López-Casillas F,
Mendoza V, Yeh IT and Sun L: Systemic administration of a soluble
beta-glycan suppresses tumor growth, angiogenesis, and matrix
metalloproteinase-9 expression in a human xenograft model of
prostate cancer. Prostate. 63:81–90. 2005. View Article : Google Scholar
|
|
17
|
Juárez P, Vilchis-Landeros MM, Ponce-Coria
J, Mendoza V, Hernández-Pando R, Bobadilla NA and López-Casillas F:
Soluble betaglycan reduces renal damage progression in db/db mice.
Am J Physiol Renal Physiol. 292:F321–F329. 2007. View Article : Google Scholar
|
|
18
|
Yan Z, Deng X and Friedman E: Oncogenic
Ki-ras confers a more aggressive colon cancer phenotype through
modification of transforming growth factor-beta receptor III. J
Biol Chem. 276:1555–1563. 2001. View Article : Google Scholar
|
|
19
|
Mythreye K and Blobe GC: The type III
TGF-beta receptor regulates epithelial and cancer cell migration
through beta-arrestin2-mediated activation of Cdc42. Proc Natl Acad
Sci USA. 106:8221–8226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eickelberg O, Centrella M, Reiss M,
Kashgarian M and Wells RG: Betaglycan inhibits TGF-beta signaling
by preventing type I-type II receptor complex formation.
Glycosaminoglycan modifications alter betaglycan function. J Biol
Chem. 277:823–829. 2002. View Article : Google Scholar
|
|
21
|
Gordon KJ, Dong M, Chislock EM, Fields TA
and Blobe GC: Loss of type III transforming growth factor beta
receptor expression increases motility and invasiveness associated
with epithelial to mesenchymal transition during pancreatic cancer
progression. Carcinogenesis. 29:252–262. 2008. View Article : Google Scholar
|
|
22
|
Antony ML, Nair R, Sebastian P and
Karunagaran D: Changes in expression, and/or mutations in TGF-β
receptors (TGF-β RI and TGF-β RII) and Smad 4 in human ovarian
tumors. J Cancer Res Clin Oncol. 136:351–361. 2010. View Article : Google Scholar
|
|
23
|
Liu XL, Xiao K, Xue B, Yang D, Lei Z, Shan
Y and Zhang HT: Dual role of TGFBR3 in bladder cancer. Oncol Rep.
30:130–138. 2013.
|
|
24
|
Zakrzewski PK, Mokrosiński J, Cygankiewicz
AI, Semczuk A, Rechberger T, Skomra D and Krajewska WM:
Dysregulation of betaglycan expression in primary human endometrial
carcinomas. Cancer Invest. 29:137–144. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Semczuk A, Zakrzewski PK, Forma E,
Cygankiewicz AI, Semczuk-Sikora A, Bryś M, Rechberger T and
Krajewska WM: TGFβ-pathway is down-regulated in a uterine
carcinosarcoma: A case study. Pathol Res Pract. 209:740–744. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pecorelli S: Revised FIGO staging for
carcinoma of the vulva, cervix, and endometrium. Int J Gynaecol
Obstet. 105:103–104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lowry OH, Rosebrough NJ, Farr AL and
Randall RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
28
|
Cawkwell L, Bell SM, Lewis FA, Dixon MF,
Taylor GR and Quirke P: Rapid detection of allele loss in
colorectal tumours using microsatellites and fluorescent DNA
technology. Br J Cancer. 67:1262–1267. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Didkowska J, Wojciechowska U and Zatoński
W: Cancer in Poland in 2011. National M. Sklodowska-Curie Cancer
Center; Warsaw: pp. 13–21. 2001
|
|
30
|
Ferlay J, Steliarova-Foucher E,
Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D and
Bray F: Cancer incidence and mortality patterns in Europe:
Estimates for 40 countries in 2012. Eur J Cancer. 49:1374–1403.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Drabsch Y and ten Dijke P: TGF-β
signalling and its role in cancer progression and metastasis.
Cancer Metastasis Rev. 31:553–568. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Principe DR, Doll JA, Bauer J, Jung B,
Munshi HG, Bartholin L, Pasche B, Lee C and Grippo PJ: TGF-β:
Duality of function between tumor prevention and carcinogenesis. J
Natl Cancer Inst. 106:djt3692014. View Article : Google Scholar
|
|
33
|
Blobe GC, Schiemann WP, Pepin MC,
Beauchemin M, Moustakas A, Lodish HF and O'Connor-McCourt MD:
Functional roles for the cytoplasmic domain of the type III
transforming growth factor β receptor in regulating transforming
growth factor β signaling. J Biol Chem. 276:24627–24637. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guo X and Wang XF: Signaling cross-talk
between TGF-β/BMP and other pathways. Cell Res. 19:71–88. 2009.
View Article : Google Scholar
|
|
35
|
Zhang YE: Non-Smad pathways in TGF-β
signaling. Cell Res. 19:128–139. 2009. View Article : Google Scholar :
|
|
36
|
Hanks BA, Holtzhausen A, Evans KS,
Jamieson R, Gimpel P, Campbell OM, Hector-Greene M, Sun L, Tewari
A, George A, et al: Type III TGF-β receptor downregulation
generates an immunotolerant tumor microenvironment. J Clin Invest.
123:3925–3940. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Copland JA, Luxon BA, Ajani L, Maity T,
Campagnaro E, Guo H, LeGrand SN, Tamboli P and Wood CG: Genomic
profiling identifies alterations in TGFbeta signaling through loss
of TGFbeta receptor expression in human renal cell carcinogenesis
and progression. Oncogene. 22:8053–8062. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dong M, How T, Kirkbride KC, Gordon KJ,
Lee JD, Hempel N, Kelly P, Moeller BJ, Marks JR and Blobe GC: The
type III TGF-beta receptor suppresses breast cancer progression. J
Clin Invest. 117:206–217. 2007. View
Article : Google Scholar
|
|
39
|
Hempel N, How T, Cooper SJ, Green TR, Dong
M, Copland JA, Wood CG and Blobe GC: Expression of the type III
TGF-beta receptor is negatively regulated by TGF-beta.
Carcinogenesis. 29:905–912. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sharifi N, Hurt EM, Kawasaki BT and Farrar
WL: TGFBR3 loss and consequences in prostate cancer. Prostate.
67:301–311. 2007. View Article : Google Scholar
|
|
41
|
Turley RS, Finger EC, Hempel N, How T,
Fields TA and Blobe GC: The type III transforming growth
factor-beta receptor as a novel tumor suppressor gene in prostate
cancer. Cancer Res. 67:1090–1098. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Finger EC, Turley RS, Dong M, How T,
Fields TA and Blobe GC: TbetaRIII suppresses non-small cell lung
cancer invasiveness and tumorigenicity. Carcinogenesis. 29:528–535.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gordon KJ and Blobe GC: Role of
transforming growth factor-beta superfamily signaling pathways in
human disease. Biochim Biophys Acta. 1782:197–228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bae HJ, Eun JW, Noh JH, Kim JK, Jung KH,
Xie HJ, Park WS, Lee JY and Nam SW: Downregulation of transforming
growth factor β receptor type III in hepatocellular carcinoma is
not directly associated with genetic alterations or loss of
heterozygosity. Oncol Rep. 22:475–480. 2009.PubMed/NCBI
|
|
45
|
Bilandzic M, Chu S, Farnworth PG, Harrison
C, Nicholls P, Wang Y, Escalona RM, Fuller PJ, Findlay JK and
Stenvers KL: Loss of betaglycan contributes to the malignant
properties of human granulosa tumor cells. Mol Endocrinol.
23:539–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu XL, Xue BX, Lei Z, Yang DR, Zhang QC,
Shan YX and Zhang HT: TGFBR3 co-downregulated with GATA3 is
associated with methylation of the GATA3 gene in bladder urothelial
carcinoma. Anat Rec (Hoboken). 296:1717–1723. 2013. View Article : Google Scholar
|
|
47
|
Ragnarsson G, Eiriksdottir G,
Johannsdottir JT, Jonasson JG, Egilsson V and Ingvarsson S: Loss of
heterozygosity at chromosome 1p in different solid human tumours:
Association with survival. Br J Cancer. 79:1468–1474. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Florio P, Ciarmela P, Reis FM, Toti P,
Galleri L, Santopietro R, Tiso E, Tosi P and Petraglia F: Inhibin
α-subunit and the inhibin coreceptor betaglycan are downregulated
in endometrial carcinoma. Eur J Endocrinol. 152:277–284. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cooper SJ, Zou H, Legrand SN, Marlow LA,
von Roemeling CA, Radisky DC, Wu KJ, Hempel N, Margulis V, Tun HW,
et al: Loss of type III transforming growth factor-beta receptor
expression is due to methylation silencing of the transcription
factor GATA3 in renal cell carcinoma. Oncogene. 29:2905–2915. 2010.
View Article : Google Scholar : PubMed/NCBI
|