Introduction
Members of the myristoylated alanine-rich C kinase
substrate (MARCKS) family, as specific protein kinase C (PKC)
substrates, modulate the PKC signaling pathway via its
phosphorylation. PKC has been implicated in cell proliferation, and
recent studies indicate that MARCKS may function as a cell growth
suppressor. For example, MARCKS and MARCKS-related protein (MRP)
are essential proteins implicated in various cellular events such
as cell motility, adhesion, secretion and phagocytosis in many cell
types (1,2). Myristoylated alanine-rich C kinase
substrate-like 1 (MARCKSL1) is part of the MARCKS family and an
essential cellular substrate for PKC as a membrane-bound protein.
MARCKSL1 (also known as MacMARCKS, MLP, MRP or F52) has been
associated with the coordination of membrane-cytoskeletal signaling
events such as regulation of integrin activation, cell adhesion,
cell spreading, cell migration and brain development, as well as
phagocytosis, mitogenesis, membrane trafficking, endocytosis and
exocytosis. It is expressed in a variety of tissues with the
highest levels found in the testis and uterus (3–8). Li
et al (9) reported that
MARCKS is a target of miR-21, a microRNA that promotes tumor
invasion and metastasis in a number of human cancers, suggesting
that it is an important tumor suppressor. Recent research suggests
that c-Jun N-terminal kinase (JNK) phosphorylation of MARCKSL1
regulates actin homeostasis, formation of lamellipodium and
filopodium, and neuronal migration under physiological conditions
in neurons and when ectopically expressed in prostate cancer cells
(8). Kim et al (10) reported that MARCKSL1 provides a
novel molecular mechanism for regulating cancer metastasis and can
serve as a therapeutic target for LOXL2-associated diseases,
including acting as a tumor suppressor.
Angiogenesis is a critical process in endothelial
cell development, movement, reproduction and degradation of the
extracellular matrix, as well as in wound repair. The complex
multi-step procedure of angiogenesis plays an important role in
cell division and metastasis in the majority of solid tumors. In
addition, angiogenic stimulators of endothelial cell functions are
associated with new blood vessel formation during embryonic
development, such as vascular endothelial growth factor (VEGF),
which plays a major role in the proliferation, migration and
invasion of vascular endothelial cells (11–13).
For example, VEGF plays a pivotal role in the progression of
ovarian and colon cancers by controlling cancer growth through
enhanced tumor angiogenesis. Recent studies have emphasized the
crucial role of the Wnt signaling pathway for embryo/organ-specific
endothelial cell differentiation in vasculogenesis (14–19).
In addition, VEGF upregulation has been associated with various
diseases. Solid tumors cannot grow beyond a certain size without an
adequate blood supply; however, tumors expressing VEGF are able to
grow and metastasize. VEGF acts via two specific binding receptor
tyrosine kinases, namely, VEGFR-1/flt-1 and VEGFR-2/KDR/flk-1, both
of which are expressed on normal vascular endothelial cells
(20). VEGFR-2 plays an important
role in the activation of essential downstream regulators
responsible for endothelial cell proliferation including migration,
invasion, survival and embryonic angiogenesis (21–23),
while VEGFR-1 is not associated with the proliferation of
endothelial cells (24). The
VEGF/VEGFR-2 signaling pathway is required for maintenance of
vascular homeostasis in various tissues and organs (25,26).
Physiological endothelial cell activity plays an
important role in regulating various vascular-related mechanisms
and vascular-associated diseases, including tumor growth and
maintenance. The detailed biological function of MARCKSL1 in
endothelial cells during ovarian tumor growth remains unclear.
Generally, the role of angiogenesis is examined based on cell
proliferation, migration and capillary-like tubule formation in
endothelial cells. In the present study, we investigated the direct
effect of MARCKSL1 on anti-angiogenic activity in tumor
angiogenesis using an in vitro human umbilical vein
endothelial cell (HUVEC) system. We also identified the molecular
regulators responsible for VEGFR-2/Akt/mammalian target of
rapamycin (mTOR)-dependent endothelial cell growth during
tumorigenesis. MARCKSL1 inhibited phosphorylation of signaling
modulators downstream of phosphatidylinositol-3′-kinase (PI3K) such
as Akt, phosphoinositide-dependent protein kinase 1 (PDK-1), mTOR
and tuberous sclerosis complex 2 (TSC-2) via direct interactions
with VEGFR-2. These interactions control powerful anti-angiogenic
activities. Therefore, our results strongly suggest that MARCKSL1
plays a major role in regulating cellular angiogenesis.
Materials and methods
Cell culture and antibodies
Human ovarian cancer cell line OVCAR-3 was seeded in
Dulbecco's modified Eagle's medium (DMEM) (Life Technologies,
Gaithersburg, MD, USA) supplemented with 10% heat-inactivated fetal
bovine serum (FBS), penicillin (100 U/ml)/streptomycin (100
µg/ml). Primary HUVECs (Clonetics, Walkersville, MD, USA)
were cultured on 0.3% gelatin-coated dishes (Sigma, St. Louis, MO,
USA) using EGM-2 BulletKit medium (Clonetics). All cells were
maintained in a humidified atmosphere containing 5% CO2
at 37°C. The following primary antibodies were used in the present
study: anti-mTOR, anti-phospho-mTOR, anti-p70S6K,
anti-phospho-p70S6K, anti-GSK-3β, anti-phospho-GSK-3β (Cell
Signaling, Beverly, MA, USA), anti-MARCKSL1, anti-Flag,
anti-VEGFR-2, anti-phospho-VEGFR-2, anti-HIF-1α, anti-PI3K,
anti-phospho-PI3K, anti-Akt, anti-phospho-Akt, anti-PDK-1,
anti-phospho-PDK-1, anti-TSC-2 and anti-phospho-TSC-2 (Santa Cruz
Biotechnology, Santa Cruz, CA, USA), anti-VEGF121 (Ab-1;
Oncogene, Cambridge, MA, USA) and β-actin (Sigma).
Migration and capillary tube formation
assay
Endothelial cell migration analyses were performed
according to a previously described protocol using Transwell
chambers (8-µm pore size; Costar) (27,28).
Briefly, the lower surface of the filter was coated with 10
µg gelatin. M199 containing 1% FBS with VEGF (10 ng/ml) was
placed in the lower wells. After 24 h, the cells were fixed with
99% methanol and stained with hematoxylin and eosin (H&E).
Cells were estimated as previously reported (27,28).
For the in vitro tube formation assays (27,28),
200 µl 10 mg/ml growth factor-reduced Matrigel was added
into a 24-well plate and polymerized at 37°C for 30 min.
Non-transfected (control/empty-inserted vector only), MARCKSL1 or
MARCKSL1 siRNA (siMARCKSL1)-transfected HUVECs (2.5×105)
were seeded on the surface of the Matrigel. Seeded whole cells were
then incubated for 48 h with or without 10 ng/ml VEGF in M199
containing 1% FBS. Tubular structure formation lengths were
measured using an inverted microscope equipped with a digital CCD
camera (Zeiss), and quantification was estimated with ImageLab
imaging software (MCM Design).
[3H]thymidine incorporation
assay
To calculate cell proliferation, HUVECs were seeded
at a density of 1.9×104/well in standard medium in
gelatinized plates on day 0. Next, the
[methyl-3H]thymidine incorporation rate was
evaluated according to a previously described protocol using a
liquid scintillation counter (Beckman Instrument) (27,28).
Immunoblot analysis
For whole protein extraction, the cultured cells
were harvested, washed with phosphate-buffered saline (PBS),
centrifuged and disrupted by adding ice-cold lysis buffer
containing protease inhibitors (50 mM Tris, pH 7.2, 150 mM NaCl, 1%
Triton X-100, 1 µg/ml leupeptin, 1 µg/ml pepstatin, 2
µg/ml aprotinin and 200 µg/ml phenylmethylsulfonyl
fluoride) for 1 h at 4°C. Proteins were subsequently separated by
8–12% SDS-PAGE and transferred to Immobilon-P membranes (Millipore
Corporation, Billerica, MA, USA). After blocking, the membranes
were incubated with the indicated primary antibodies at 4°C
overnight. The membranes were washed three times in TBST washing
buffer and incubated with horseradish peroxidase-conjugated
secondary antibodies. Protein bands were detected using the ECL
detection system (GE Healthcare, Little Chalfont, Buckinghamshire,
UK).
Yeast two-hybrid (Y2H) assay and
quantitation of interaction
For bait construction with human MARCKSL1, cDNA
encoding full-length human MARCKSL1 was subcloned into the
EcoRI and XhoI restriction enzyme sites of the
pGilda/LexA yeast shuttle cloning vector. The bait
pGilda/LexA-MARCKSL1 plasmid was introduced into a yeast strain
EGY48 [MATa, his3, trp1, ura3-52,
leu2::pLeu2-LexAop6/pSH18-34 (LexAop-lacZ reporter)]
using a modified lithium acetate method (29,30).
Human VEGFR-1 and VEGFR-2 were introduced, with each cDNA encoding
a full-length gene into the multi-cloning restriction enzyme sites
of the pJG4-5 plasmid vector, which included B42 fusion proteins
(Clontech, Palo Alto, CA, USA). The VEGFR-1 and VEGFR-2 cDNAs
encoding pJG4-5 fusion proteins were transformed into competent
yeast cells that already contained pGilda/LexA-MARCKSL1, while the
transformants were selected based on their tryptophan prototrophy
(plasmid marker) on a synthetic medium (Ura, His, Trp) containing
2% (w/v) glucose. The binding activity of the interaction was
estimated according to a previously described protocol (30).
Co-immunoprecipitation assay
For co-immunoprecipitation (co-IP), the cells were
trypsinized and then centrifuged. Cell pellets were rinsed with
ice-cold PBS and re-suspended in RIPA lysis buffer (50 mM Tris/HCl,
pH 7.2, 150 mM NaCl, 1% Triton X-100, protease inhibitor cocktail
containing 1 µg/ml leupeptin, 1 µg/ml pepstatin, 2
µg/ml aprotinin and 200 µg/ml PMSF). The cell lysates
were then incubated with anti-Flag antibody (Santa Cruz) and
precipitated with protein A-agarose (GE Healthcare). Approximately
20 µg of the precipitated proteins was separated by 10–12%
SDS-PAGE and transferred to Immobilon-P membranes. After blocking,
the membranes were incubated with the indicated primary antibodies,
including anti-MARCKSL1 and anti-VEGFR-2. Immunoreactive bands were
visualized using the ECL detection system (GE Healthcare).
Luciferase reporter-gene assay
In vitro VEGFR-2 promoter assays were
performed as previously described (31). Briefly, cells at 85% confluency were
transfected with a VEGFR-2 reporter expression vector. After lysis
in ice-cold RIPA buffer, whole cell lysates were cleared by
centrifugation for 15 min at 14,000 rpm, and cell extracts were
incubated at room temperature with the luciferase substrate reagent
for 30 min according to the manufacturer's instructions. Then, a
5-µl aliquot of each sample was examined using a MicroLumat
Plus LB96V luminometer.
Statistical analysis
All data are presented as means ± SD and were
assessed using the Student's t-test. Significant differences of 95%
confidence (P<0.05) are depicted by an asterisk (*)
on each graph.
Results
MARCKSL1 inhibits VEGF-induced
endothelial cell migration, capillary tube formation and cell
proliferation in vitro
During the multi-step process of angiogenesis, one
of the crucial stimulators during early development is VEGF, which
induces cell migration, invasion, proliferation and differentiation
of endothelial cells. Endothelial cell activity plays an important
role in controlling various vascular-associated functions and
diseases, including tumor cell growth and maintenance. To determine
whether MARCKSL1 controls the effects of VEGF on cell migration in
HUVECs, angiogenesis was examined based on cell migration,
capillary-like tubular structure formation and cell proliferation
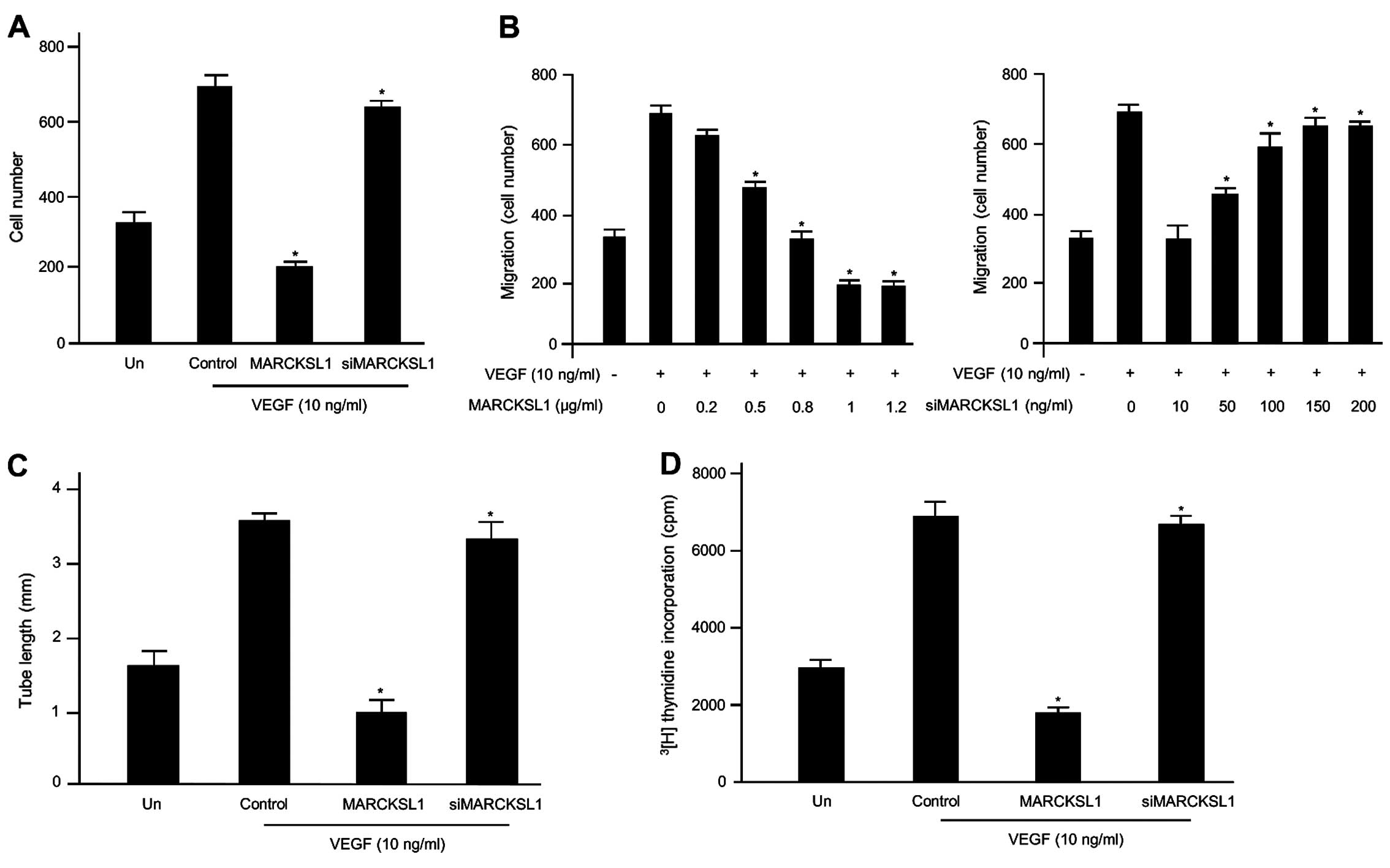
in endothelial cells. The inhibitory effects of MARCKSL1 on
VEGF-induced migration of endothelial cells were first assessed
using Transwell migration assays. As expected, VEGF promoted the
migration of the non-transfected cells and MARCKSL1-control
(empty-inserted vector)-transfected cells compared with
non-stimulated cells. Our results indicated that MARCKSL1
overexpression noticeably disrupted VEGF-induced cell migration,
while MARCKSL1-siRNA did not (Fig.
1A). We performed further experiments using various
concentrations of MARCKSL1 or siMARCKSL1 based on transient
transfection. As shown in Fig. 1B,
VEGF prompted the cell migration of the empty MARCKSL1-transfected
HUVECs compared with the unstimulated cells, as expected. However,
overexpression of MARCKSL1 gradually inhibited VEGF-induced cell
migration in a dose-dependent manner. Inhibition of MARCKSL1
expression via MARCKSL1-siRNA maintained the stimulatory effects of
VEGF on HUVEC migration. Therefore, overexpression of MARCKSL1
potently inhibits key events in the angiogenic process induced by
VEGF, such as the migration of endothelial cells in vitro.
Subsequently, we investigated the anti-angiogenic effects of
MARCKSL1 on the formation of VEGF-induced capillary-like tubular
structures on Matrigel using an in vitro angiogenesis model
system in HUVECs. As shown in Fig.
1C, untreated or control cells incubated with VEGF formed a
capillary-like structure on Matrigel. In contrast, the
overexpression of MARCKSL1 completely inhibited VEGF-induced
tubular structure formation. The inhibitory effect of MARCKSL1 on
VEGF-induced capillary tube formation was completely recovered by
siMARCKSL1 transient transfection. Finally, the inhibitory effects
on VEGF-induced proliferation of endothelial cells were assessed
using a [3H]thymidine incorporation assay system.
Generally, VEGF enhanced DNA synthesis of the untransfected HUVECs,
and the control (empty-inserted vector)-transfected HUVECs were
comparable to the non-induced cells (32). MARCKSL1 significantly suppressed
VEGF-induced DNA synthesis in HUVECs (Fig. 1D). Therefore, overexpression of
MARCKSL1 suppresses key events in VEGF-induced angiogenesis in
vitro, including endothelial cell migration and cell
proliferation. Collectively, these findings indicate that MARCKSL1
specifically controls VEGF-induced cell migration and tube
formation, as well as the proliferation of HUVECs.
MARCKSL1 tumor suppressor protein
inhibits VEGF-induced VEGFR-2 phosphorylation via the interaction
with VEGFR-2 protein
VEGFR-2 is an essential receptor involved in
angiogenesis and vasculogenesis and controls endothelial cell
migration, proliferation and mitogenesis. VEGFR-2 has been
implicated primarily in cancer metastasis and tumorigenesis. To
address the possible mechanism behind the inhibition of
angiogenesis by the tumor suppressor MARCKSL1, we used the Y2H
protein-protein interaction assay and the co-IP analysis system. We
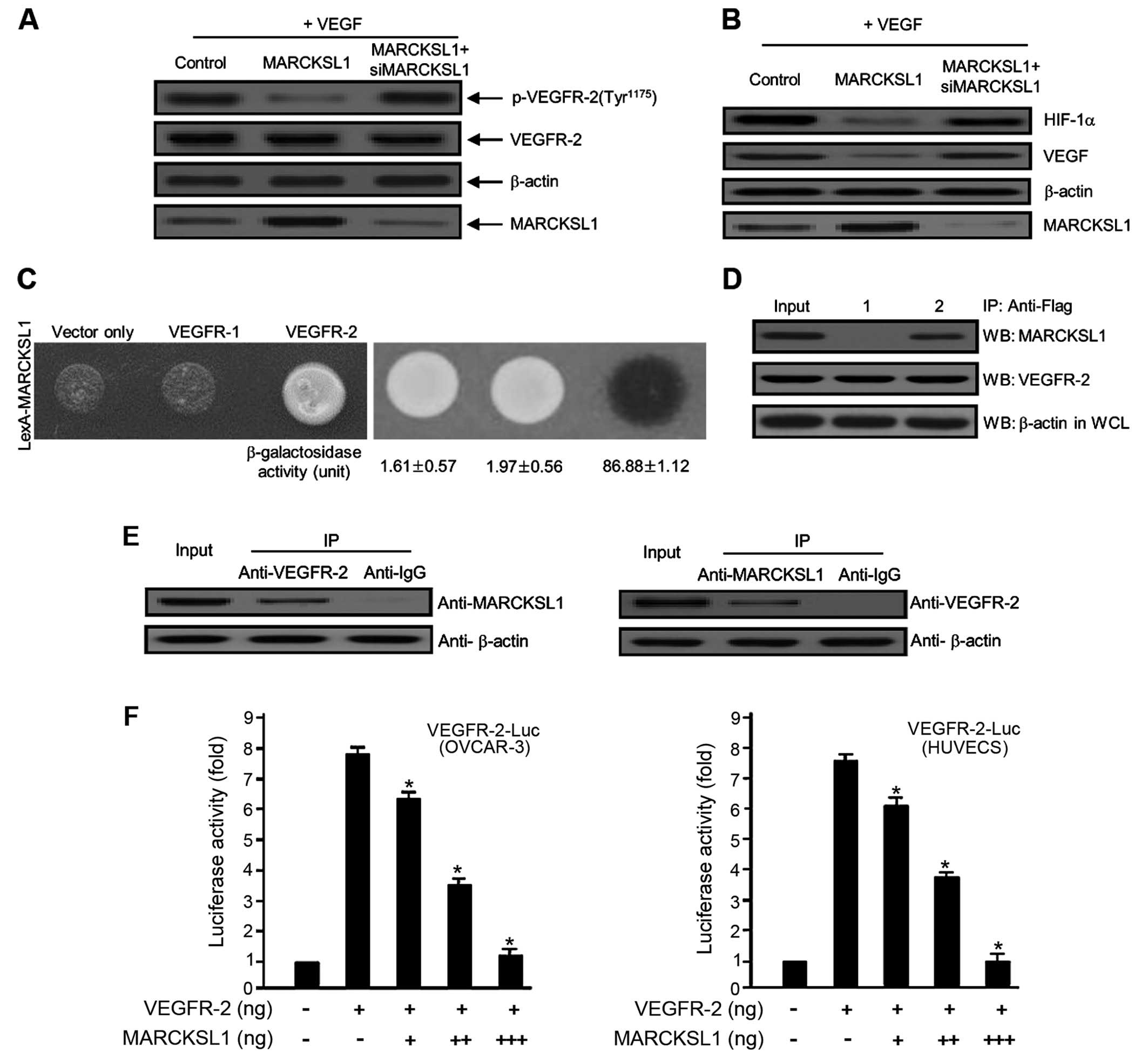
first investigated the inhibitory effect of MARCKSL1 on
VEGF-induced VEGFR-2 protein expression in ovarian carcinoma cells.
Overexpression of MARCKSL1 significantly reduced the
phosphorylation of VEGFR-2, whereas siMARCKSL1 completely reversed
the inhibitory effect (Fig. 2A).
VEGF rapidly activates angiogenesis in tumor metastasis as well as
tumorigenesis; hence, we evaluated the inhibitory effect of
MARCKSL1 on VEGF protein expression in OVCAR-3 tumor cells. The
overexpression of MARCKSL1 completely reduced VEGF expression,
whereas siMARCKSL1 reversed the inhibitory effect (Fig. 2B). We also demonstrated the effect
of MARCKSL1 on hypoxia-inducible factor 1α (HIF-1α) protein
expression. As a transcription factor, HIF-1α is an important
molecule for VEGF protein expression. As shown in Fig. 2B, MARCKSL1 markedly decreased the
expression of the HIF-1α protein. The inhibitory effect of MARCKSL1
was completely recovered by siMARCKSL1 transient transfection.
These observations indicate that VEGF-induced protein expression is
notably inhibited by the MARCKSL1 tumor suppressor. Direct
interactions among various proteins are critical for the majority
of cellular functions. For example, signaling cascades from the
exterior to the interior of a cell are regulated through
protein-protein interactions. Thus, signal transduction processes
play a pivotal role in cellular/physiological/biological processes,
as well as in aggressive solid tumors and various diseases.
Therefore, protein-protein interactions are important for the
majority of processes in a living cell microenvironment. Thus, we
investigated the cellular interaction between VEGFR-2 and MARCKSL1
protein in an in vivo and in vitro system. VEGFR-1
and the expression vector only (empty-inserted vector) were used as
negative controls. As shown in Fig.
2C, the β-galactosidase activity between MARCKSL1 and VEGFR-2
was fully activated (86.88±1.12), which was not the case with the
expression vector only (empty-inserted vector) (1.61±0.57) or
VEGFR-1 (1.97±0.56). To further confirm this direct interaction
between MARCKSL1 and VEGFR-2 observed in the Y2H assay system, we
explored this relationship using co-IP analysis. Bait constructs of
MARCKSL1 (pcDNA3.1/MARCKSL1) and VEGFR-2 (pcDNA3.1/Flag-VEGFR-2) or
pcDNA3.1/Flag-VEGFR-2 and expression plasmid vector only (pcDNA3.1)
were co-transfected into HEK293T cells. Continuously, IP was
performed using anti-Flag primary antibody with whole cell lysates
from both transfected cells. After IP, the precipitated proteins
were detected using either anti-MARCKSL1 or anti-VEGFR-2 primary
antibody. As shown in Fig. 2D,
VEGFR-2 co-immunoprecipitated with MARCKSL1 (lane 2 in the upper
panel). Next, we demonstrated the interaction between endogenous
MARCKSL1 and VEGFR-2. As shown in Fig.
2E, endogenous MARCKSL1 directly co-immunoprecipitated with
VEGFR-2. These results were indicative of the binding between
endogenous VEGFR-2 and MARCKSL1 in the cells. Finally, to determine
whether MARCKSL1 regulates VEGFR-2, the effect of MARCKSL1 on
VEGFR-2 transcription activity was estimated based on a luciferase
reporter gene assay using a construct fusing the VEGFR-2 promoter
to the luciferase gene. Luciferase activity was gradually reduced
by transient transfection of MARCKSL1 in a dose-dependent manner
(Fig. 2F), further supporting the
importance of MARCKSL1 for the regulation of VEGFR-2 activity. Our
results strongly indicate that overexpression of MARCKSL1 reduces
its transcriptional activity. Collectively, the results
demonstrated that MARCKSL1 disrupts the VEGFR-2 transcript levels
by inhibiting VEGFR-2 phosphorylation via the Akt/mTOR signaling
pathway.
Disrupted phosphorylation of downstream
components in the VEGFR-2/Akt/mTOR signaling pathways by the
MARCKSL1 tumor suppressor
PI3K/Akt phosphorylation is a crucial step in the
regulation of fundamental cellular processes involved in tumor
angiogenesis and tumor growth. Akt, as a pivotal downstream
regulator of PI3K, induces mTOR through a variety of cellular
functions, including phosphorylation and inactivation of apoptotic
cell death-associated proteins (33–35).
Therefore, to address the detailed functional mechanism underlying
its effects, we examined whether MARCKSL1 reduced PI3K and Akt
phosphorylation in OVCAR-3 tumor cells. As indicated in Fig. 3A, VEGF-induced PI3K and Akt
phosphorylation were significantly reduced by the MARCKSL1 tumor
suppressor. The inhibitory effect of MARCKSL1 on VEGF-induced
phosphorylation was nearly recovered by transient transfection of
siMARCKSL1. These results suggested that MARCKSL1 specifically
suppresses VEGF-stimulated PI3K/Akt phosphorylation in ovarian
tumor cells. We also investigated the phosphorylation effects on
upstream and downstream signaling components of the PI3K/Akt
pathway that control tumor endothelial cell function in the
inhibition of angiogenesis. For example, p70 ribosomal protein S6
kinase (p70S6K), a mitogen-activated serine/threonine kinase, is an
essential regulator of protein synthesis and plays an important
role in cell growth, proliferation, differentiation and survival.
As indicated in Fig. 3B, MARCKSL1
reduced VEGF-induced phosphorylation of the PI3K/Akt signaling
cascade components, including PDK-1, TSC-2, mTOR, p70S6K and
glycogen synthase kinase-3β (GSK-3β). Taken together, these results
suggest that MARCKSL1 has a novel biological function in activating
apoptosis, as well as inhibiting angiogenesis via concomitant
inactivation of VEGF-stimulated phosphorylation of the cascade
components in the VEGFR-2/Akt/mTOR signaling pathways.
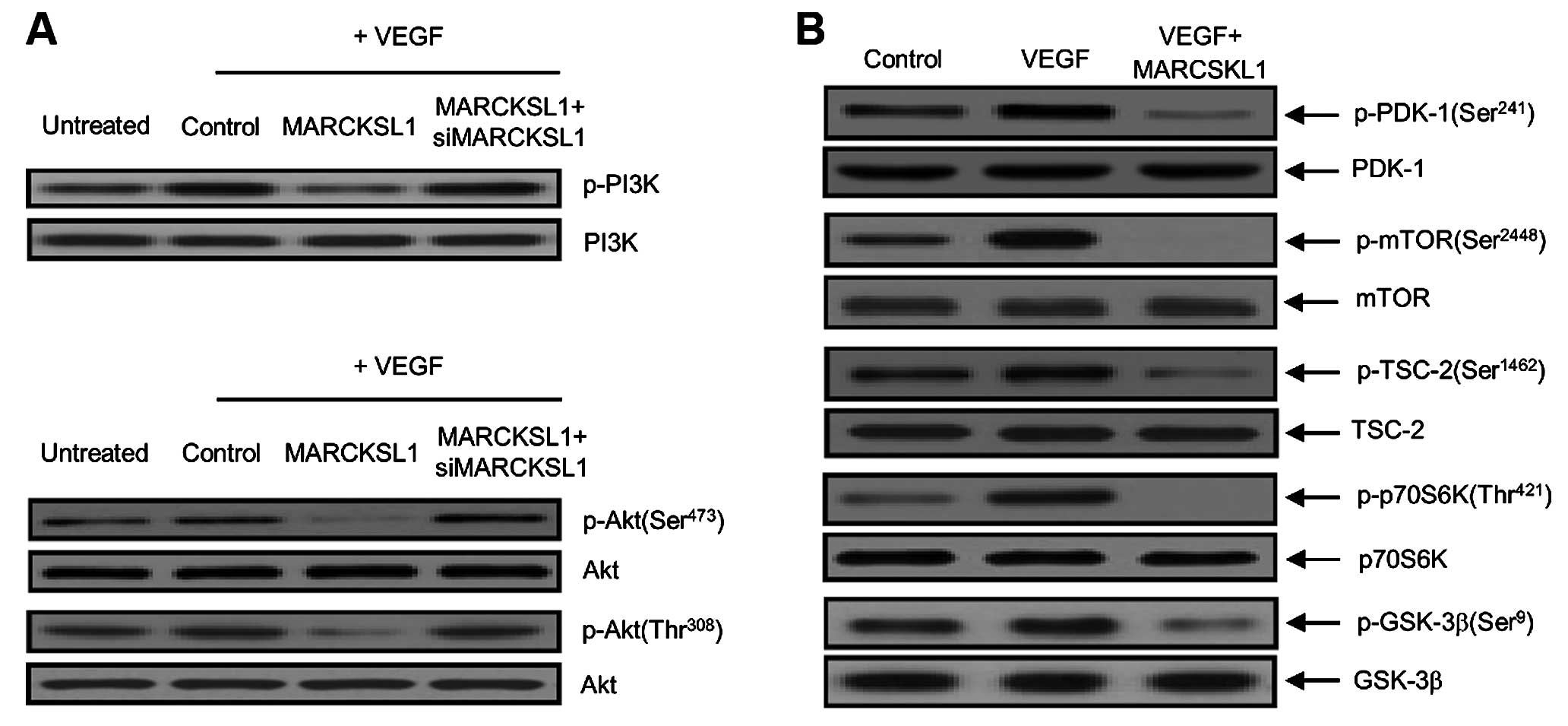 | Figure 3Disrupted phosphorylation of
downstream components in the VEGFR-2/Akt/mTOR signaling pathways by
the MARCKSL1 tumor suppressor. (A) Phospho-PI3K and phospho-Akt
suppression by MARCKSL1 disclosure in OVCAR-3 ovarian tumor cells.
Cells were incubated with 10 ng/ml VEGF and transfected with either
the control (empty-inserted vector only), MARCKSL1 or MARCKSL1 plus
siMARCKSL1. Phosphorylation of PI3K and Akt (Ser473 and Thr308)
were then developed by western blot analysis. Non-phosphorylated
PI3K and Akt were used as the loading control. Three independent
experiments were performed in triplicate. Protein levels were
measured based on densitometric analysis and normalized to levels
of the loading control (data not shown). (B) The phosphorylation
effects of MARCKSL1 on PDK-1 (Ser241), mTOR (Ser2448), TSC-2
(Ser1462), p70S6K (Thr421) and GSK-3β (Ser9) signaling components,
such as in the cases of upstream and downstream target regulators
of Akt. After transfection with MARCKSL1, equal amounts of cellular
total protein (20 µg) were separated by SDS-PAGE, followed
by immunoblotting with indicated primary antibodies specific to the
phosphorylated proteins (p-PDK-1, p-mTOR, p-TSC-2, p-p70S6K and
p-GSK-3β). Immunoblotting of non-phosphorylated PDK-1, mTOR, TSC-2,
p70S6K and GSK-3β were used to verify equal sample loading. All
experiments were performed at least three times with similar
results. |
Discussion
Previous research showed that LOXL2 recovers
MARCKSL1-induced apoptosis through inhibition of the FAK/Akt/mTOR
signaling pathway in carcinoma cells (10). According to this study, increased
LOXL2 expression activated cellular metastasis by suppressing
MARCKSL1-induced apoptotic cell death by regulating cell cycle- and
apoptosis-associated protein expression, including p21, cyclin D1,
p53 and Bcl-2 family genes. Second, LOXL2 was found to directly
bind to MARCKSL1, and MARCKSL1 overexpression reduced LOXL2
expression in carcinoma cells. Third, induction of the
LOXL2-mediated FAK/Akt/mTOR signaling pathways and tumor metastasis
were decreased by overexpression of MARCKSL1. In the present study,
we validated that MARCKSL1 had an inhibitory effect on VEGF-induced
angiogenesis in the endothelial cell system.
Angiogenesis plays a pivotal role in both early
development and progression of ovarian cancer, as well as in normal
ovarian function (36,37). As demonstrated via histologic
studies, ovarian tumors are highly neovascularized and
microvascular number and pathological/biological aggressiveness are
interrelated (38,39). As in other malignant tumors,
angiogenesis is an essential factor for ovarian carcinoma growth.
VEGF is associated with accelerated angiogenesis during the early
stages of ovarian carcinoma; thus, VEGF-induced angiogenesis is a
vital early regulator in ovarian carcinogenesis (36,40).
Generally, angiogenic signaling cascades are mediated by VEGF and
their receptors, VEGFRs (41). VEGF
expression is closely associated with the regulation of tumor
metastasis, growth, migration, invasion and aggression, as well as
poor survival (42–44). Notably, most human solid tumors show
upregulated expression of VEGF and HIF-1α, which enhance tumor
angiogenesis and tumor growth. Thus, suppression of tumor
angiogenesis by disturbed VEGF and HIF-1α expression is becoming a
pivotal approach for carcinoma treatment (45,46).
One study reported that maximal inhibition of tumor growth and
tumor angiogenesis can be accomplished by disrupting VEGF
circulation (47). Herein, we
demonstrated a novel cellular function for MARCKSL1 as a novel
potent angiogenic modulator that can disrupt VEGF and HIF-1α
expression in the VEGFR-2/Akt/mTOR signaling pathway using a cancer
model system. As indicated in Fig.
1, overexpression of MARCKSL1 considerably reduced the major
events of VEGF-induced angiogenesis in vitro, involving
endothelial cell migration and cell proliferation. At the same
time, overexpression of MARCKSL1 completely abrogated the
VEGF-induced capillary-like tubular structure network. In the
presence of MARCKSL1-siRNA, endothelial cells were not affected.
Taken together, these results indicate that MARCKSL1 specifically
regulates VEGF-induced HUVEC migration and tube formation. HIF-1α
enhances the transcript levels of many genes such as VEGF,
transferrin and endothelin-1 in tumor metastasis and
neovascularization (48–50). In addition, the HIF-1α protein is
commonly upregulated in various types of human tumors and in
regional and distant metastases (51). Therefore, its activation is closely
associated with gene expression, tumor angiogenesis, and tumor
growth (52). Fang et al
(53) reported that apigenin
significantly reduces the expression of VEGF and HIF-1α in ovarian
tumor cells. As mentioned above, HIF-1α, as a transcription factor,
is an essential mediator of VEGF protein expression. As indicated
in Fig. 2B, MARCKSL1 markedly
decreased the expression of VEGF and HIF-1α protein. In contrast,
the inhibitory effect of MARCKSL1 was completely restored by
siMARCKSL transfection. VEGFR-2 is phosphorylated through its
interaction with VEGF, and its downstream signaling cascade
activates PI3K/Akt phosphorylation, which is a potent cytokine in
the control of endothelial cell growth and survival of various
carcinomas, including ovarian tumors (46,54–57).
Akt phosphorylates mTOR and is regulated by mTOR through a negative
and positive feedback system (35),
and it also acts as a major modulator of cell proliferation,
promoting cell survival through various cellular mechanisms.
Despite these studies, the cellular mechanisms of ovarian tumor
angiogenesis remain unclear. Therefore, to address the detailed
functional mechanism underlying its effects, we investigated
whether MARCKSL1 downregulated PI3K and Akt phosphorylation in
OVCAR-3 tumor cells. Overexpression of MARCKSL1 notably inhibited
phosphorylation of VEGFR-2, whereas siMARCKSL1 completely reversed
the inhibitory effect (Fig. 2A). In
addition, VEGF-induced PI3K and Akt phosphorylation were
considerably reduced by the MARCKSL1 tumor suppressor. The
inhibitory effect of MARCKSL1 on VEGF-induced phosphorylation was
nearly recovered by MARCKSL1-siRNA transient transfection (Fig. 3A). Consistently, MARCKSL1 decreased
VEGF-induced phosphorylation of the PI3K/Akt signaling cascade
components, including PDK-1, TSC-2, mTOR, p70S6K and GSK-3β.
Collectively, these results suggest that MARCKSL1 disturbs VEGFR-2
transcriptional activity by restraining VEGFR-2 phosphorylation via
the Akt/PDK-1/mTOR signaling pathway.
In summary, the present study examined the
anti-angiogenic effects of the tumor suppressor MARCKSL1 in ovarian
tumorigenesis using an in vitro model system. In the present
study, we further revealed a novel biological/physiological
function and cellular molecular mechanism for MARCKSL1, which
serves as an anti-angiogenic factor that can mediate
phosphorylation of Akt/PDK-1/mTOR through inhibition of VEGF and
HIF-1α expression levels via interaction with VEGFR-2. Therefore,
combination therapy with MARCKSL1 and traditional anti-tumor
reagents may be a useful approach for more advanced ovarian cancer,
recurrent and certain other cancer patients.
Acknowledgments
This study was supported through a grant from the
National Cancer Center, Korea (NCC-1510050-1 and 1410312-2).
Abbreviations:
|
MARCKSL1
|
myristoylated alanine-rich C kinase
substrate-like 1
|
|
PKC
|
protein kinase C
|
|
MRP
|
MARCKS-related protein
|
|
VEGFR-2
|
vascular endothelial growth factor
receptor 2
|
|
HUVECs
|
human umbilical vein endothelial
cells
|
|
PDK-1
|
phosphoinositide-dependent protein
kinase 1
|
|
mTOR
|
mammalian target of rapamycin
|
|
TSC-2
|
tuberous sclerosis complex 2
|
|
p70S6K
|
p70 ribosomal protein S6 kinase
|
|
GSK-3β
|
glycogen synthase kinase 3β
|
References
|
1
|
Larsson C: Protein kinase C and the
regulation of the actin cytoskeleton. Cell Signal. 18:276–284.
2006. View Article : Google Scholar
|
|
2
|
Kalwa H and Michel T: The MARCKS protein
plays a critical role in phosphatidylinositol 4,5-bisphosphate
metabolism and directed cell movement in vascular endothelial
cells. J Biol Chem. 286:2320–2330. 2011. View Article : Google Scholar :
|
|
3
|
Aderem A: The MARCKS brothers: A family of
protein kinase C substrates. Cell. 71:713–716. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McNamara RK and Lenox RH: Distribution of
the protein kinase C substrates MARCKS and MRP in the postnatal
developing rat brain. J Comp Neurol. 397:337–356. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Underhill DM, Chen J, Allen LA and Aderem
A: MacMARCKS is not essential for phagocytosis in macrophages. J
Biol Chem. 273:33619–33623. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yue L, Lu S, Garces J, Jin T and Li J:
Protein kinase C-regulated dynamitin-macrophage-enriched
myristoylated alanine-rice C kinase substrate interaction is
involved in macrophage cell spreading. J Biol Chem.
275:23948–23956. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Arbuzova A, Schmitz AA and Vergères G:
Cross-talk unfolded: MARCKS proteins. Biochem J. 362:1–12. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Björkblom B, Padzik A, Mohammad H,
Westerlund N, Komulainen E, Hollos P, Parviainen L, Papageorgiou
AC, Iljin K, Kallioniemi O, et al: c-Jun N-terminal kinase
phosphorylation of MARCKSL1 determines actin stability and
migration in neurons and in cancer cells. Mol Cell Biol.
32:3513–3526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li T, Li D, Sha J, Sun P and Huang Y:
MicroRNA-21 directly targets MARCKS and promotes apoptosis
resistance and invasion in prostate cancer cells. Biochem Biophys
Res Commun. 383:280–285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim BR, Dong SM, Seo SH, Lee JH, Lee JM,
Lee SH and Rho SB: Lysyl oxidase-like 2 (LOXL2) controls
tumor-associated cell proliferation through the interaction with
MARCKSL1. Cell Signal. 26:1765–1773. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Folkman J and Shing Y: Angiogenesis. J
Biol Chem. 267:10931–10934. 1992.PubMed/NCBI
|
|
12
|
Risau W: Mechanisms of angiogenesis.
Nature. 386:671–674. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ferrara N: VEGF and the quest for tumour
angiogenesis factors. Nat Rev Cancer. 2:795–803. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van Gijn ME, Daemen MJ, Smits JF and
Blankesteijn WM: The wnt-frizzled cascade in cardiovascular
disease. Cardiovasc Res. 55:16–24. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cohen ED, Tian Y and Morrisey EE: Wnt
signaling: An essential regulator of cardiovascular
differentiation, morphogenesis and progenitor self-renewal.
Development. 135:789–798. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zerlin M, Julius MA and Kitajewski J:
Wnt/Frizzled signaling in angiogenesis. Angiogenesis. 11:63–69.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dejana E, Tournier-Lasserve E and
Weinstein BM: The control of vascular integrity by endothelial cell
junctions: Molecular basis and pathological implications. Dev Cell.
16:209–221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Franco CA, Liebner S and Gerhardt H:
Vascular morphogenesis: A Wnt for every vessel? Curr Opin Genet
Dev. 19:476–483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dejana E: The role of wnt signaling in
physiological and pathological angiogenesis. Circ Res. 107:943–952.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mustonen T and Alitalo K: Endothelial
receptor tyrosine kinases involved in angiogenesis. J Cell Biol.
129:895–898. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Breier G: Endothelial receptor tyrosine
kinases involved in blood vessel development and tumor
angiogenesis. Adv Exp Med Biol. 476:57–66. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ferrara N: Role of vascular endothelial
growth factor in regulation of physiological angiogenesis. Am J
Physiol Cell Physiol. 280:C1358–C1366. 2001.PubMed/NCBI
|
|
23
|
Meyer RD and Rahimi N: Comparative
structure-function analysis of VEGFR-1 and VEGFR-2: What have we
learned from chimeric systems? Ann NY Acad Sci. 995:200–207. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meyer RD, Singh A, Majnoun F, Latz C,
Lashkari K and Rahimi N: Substitution of C-terminus of VEGFR-2 with
VEGFR-1 promotes VEGFR-1 activation and endothelial cell
proliferation. Oncogene. 23:5523–5531. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang Y, Zhang Y, Cao Z, Ji H, Yang X,
Iwamoto H, Wahlberg E, Länne T, Sun B and Cao Y: Anti-VEGF- and
anti-VEGF receptor-induced vascular alteration in mouse healthy
tissues. Proc Natl Acad Sci USA. 110:12018–12023. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cao Y: VEGF-targeted cancer
therapeutics-paradoxical effects in endocrine organs. Nat Rev
Endocrinol. 10:530–539. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee OH, Kim YM, Lee YM, Moon EJ, Lee DJ,
Kim JH, Kim KW and Kwon YG: Sphingosine 1-phosphate induces
angiogenesis: Its angiogenic action and signaling mechanism in
human umbilical vein endothelial cells. Biochem Biophys Res Commun.
264:743–750. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee JH, Chun T, Park SY and Rho SB:
Interferon regulatory factor-1 (IRF-1) regulates VEGF-induced
angiogenesis in HUVECs. Biochim Biophys Acta. 1783:1654–1662. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rho SB, Lee KH, Kim JW, Shiba K, Jo YJ and
Kim S: Interaction between human tRNA synthetases involves repeated
sequence elements. Proc Natl Acad Sci USA. 93:10128–10133. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rho SB, Kim MJ, Lee JS, Seol W, Motegi H,
Kim S and Shiba K: Genetic dissection of protein-protein
interactions in multi-tRNA synthetase complex. Proc Natl Acad Sci
USA. 96:4488–4493. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rho SB, Song YJ, Lim MC, Lee SH, Kim BR
and Park SY: Programmed cell death 6 (PDCD6) inhibits angiogenesis
through PI3K/mTOR/p70S6K pathway by interacting of VEGFR-2. Cell
Signal. 24:131–139. 2012. View Article : Google Scholar
|
|
32
|
Plate KH, Breier G, Weich HA and Risau W:
Vascular endothelial growth factor is a potential tumour
angiogenesis factor in human gliomas in vivo. Nature. 359:845–848.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Downward J: Signal transduction. A target
for PI(3) kinase. Nature. 376:553–554. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Khwaja A: Akt is more than just a Bad
kinase. Nature. 401:33–34. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guertin DA and Sabatini DM: An expanding
role for mTOR in cancer. Trends Mol Med. 11:353–361. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ramakrishnan S, Subramanian IV, Yokoyama Y
and Geller M: Angiogenesis in normal and neoplastic ovaries.
Angiogenesis. 8:169–182. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kumaran GC, Jayson GC and Clamp AR:
Antiangiogenic drugs in ovarian cancer. Br J Cancer. 100:1–7. 2009.
View Article : Google Scholar :
|
|
38
|
Alvarez AA, Krigman HR, Whitaker RS, Dodge
RK and Rodriguez GC: The prognostic significance of angiogenesis in
epithelial ovarian carcinoma. Clin Cancer Res. 5:587–591.
1999.PubMed/NCBI
|
|
39
|
Hazelton D, Nicosia RF and Nicosia SV:
Vascular endothelial growth factor levels in ovarian cyst fluid
correlate with malignancy. Clin Cancer Res. 5:823–829.
1999.PubMed/NCBI
|
|
40
|
Ko YB, Kim BR, Yoon K, Choi EK, Seo SH,
Lee Y, Lee MA, Yang JB, Park MS and Rho SB: WIF1 can effectively
co-regulate pro-apoptotic activity through the combination with
DKK1. Cell Signal. 26:2562–2572. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ferrara N: Role of vascular endothelial
growth factor in physiologic and pathologic angiogenesis:
Therapeutic implications. Semin Oncol. 29(Suppl 16): S10–S14. 2002.
View Article : Google Scholar
|
|
42
|
Mu J, Abe Y, Tsutsui T, Yamamoto N, Tai
XG, Niwa O, Tsujimura T, Sato B, Terano H, Fujiwara H, et al:
Inhibition of growth and metastasis of ovarian carcinoma by
administering a drug capable of interfering with vascular
endothelial growth factor activity. Jpn J Cancer Res. 87:963–971.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hartenbach EM, Olson TA, Goswitz JJ,
Mohanraj D, Twiggs LB, Carson LF and Ramakrishnan S: Vascular
endothelial growth factor (VEGF) expression and survival in human
epithelial ovarian carcinomas. Cancer Lett. 121:169–175. 1997.
View Article : Google Scholar
|
|
44
|
Yamamoto S, Konishi I, Mandai M, Kuroda H,
Komatsu T, Nanbu K, Sakahara H and Mori T: Expression of vascular
endothelial growth factor (VEGF) in epithelial ovarian neoplasms:
Correlation with clinicopathology and patient survival, and
analysis of serum VEGF levels. Br J Cancer. 76:1221–1227. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ferrara N and Davis-Smyth T: The biology
of vascular endothelial growth factor. Endocr Rev. 18:4–25. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Park ST, Kim BR, Park SH, Lee JH, Lee EJ,
Lee SH and Rho SB: Suppression of VEGF expression through
interruption of the HIF-1α and Akt signaling cascade modulates the
anti-angiogenic activity of DAPK in ovarian carcinoma cells. Oncol
Rep. 31:1021–1029. 2014.
|
|
47
|
Gerber HP, Kowalski J, Sherman D, Eberhard
DA and Ferrara N: Complete inhibition of rhabdomyosarcoma xenograft
growth and neovascularization requires blockade of both tumor and
host vascular endothelial growth factor. Cancer Res. 60:6253–6258.
2000.PubMed/NCBI
|
|
48
|
Kerbel RS: New targets, drugs, and
approaches for the treatment of cancer: An overview. Cancer
Metastasis Rev. 17:145–147. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Semenza GL: Regulation of mammalian
O2 homeostasis by hypoxia-inducible factor 1. Annu Rev
Cell Dev Biol. 15:551–578. 1999. View Article : Google Scholar
|
|
50
|
Semenza GL: Hypoxia, clonal selection, and
the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol.
35:71–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhong H, De Marzo AM, Laughner E, Lim M,
Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL and Simons
JW: Overexpression of hypoxia-inducible factor 1alpha in common
human cancers and their metastases. Cancer Res. 59:5830–5835.
1999.PubMed/NCBI
|
|
52
|
Maxwell PH, Dachs GU, Gleadle JM, Nicholls
LG, Harris AL, Stratford IJ, Hankinson O, Pugh CW and Ratcliffe PJ:
Hypoxia-inducible factor-1 modulates gene expression in solid
tumors and influences both angiogenesis and tumor growth. Proc Natl
Acad Sci USA. 94:8104–8109. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fang J, Xia C, Cao Z, Zheng JZ, Reed E and
Jiang BH: Apigenin inhibits VEGF and HIF-1 expression via
PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 19:342–353. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Altomare DA, Wang HQ, Skele KL, De Rienzo
A, Klein-Szanto AJ, Godwin AK and Testa JR: AKT and mTOR
phosphorylation is frequently detected in ovarian cancer and can be
targeted to disrupt ovarian tumor cell growth. Oncogene.
23:5853–5857. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hanrahan AJ, Schultz N, Westfal ML, Sakr
RA, Giri DD, Scarperi S, Janakiraman M, Olvera N, Stevens EV, She
QB, et al: Genomic complexity and AKT dependence in serous ovarian
cancer. Cancer Discov. 2:56–67. 2012. View Article : Google Scholar : PubMed/NCBI
|