Introduction
Epigenetic mechanisms regulate gene expression and
establish cellular identity. Therefore, the absence of proper
epigenetic marks contributes to the development of diseases
including cancer (1). These
epigenetic marks are recognized by reader proteins that interpret
the chromatin information and signal other cellular components to
facilitate chromatin remodeling (2,3).
Proteins in the bromodomain and extra-terminal domain (BET) family,
including bromodomain-containing proteins (BRD)2, BRD3, BRD4, and
BRDT (testis-specific), are well-known readers of acetyl lysine
residues, which is not only the most abundant protein modification
in cells but also is critical to chromatin structure. BETs regulate
a variety of genes involved in the cell cycle, cell growth and
inflammation (4). Thus, the
targeting of these BETs has become an intense research issue in
diverse therapeutic areas.
The proto-oncogene MYC is a transcription factor
containing a basic helix-loop-helix (bHLH) domain that has been
studied for more than 30 years. The biology of MYC has been studied
extensively which has resolved various issues. MYC heterodimerizes
with the bHLH protein MAX and binds to CA(C/T)GTG of its target
genes. These genes encompass a broad spectrum of functions, from
cell cycle progression and cell growth to epithelial-mesenchymal
transition (EMT) (5). Activating
enhancer binding protein 4 (AP4) is considered a key mediator of
mitogenicity for MYC and EMT, as well as for further metastasis
(6,7). MYC directly binds to CACGTG motifs in
the first intron of the AP4 and functions as an activator (7). AP4 protein belongs to the bHLH
subfamily similar to MYC, although it recognizes symmetrical DNA
sequences such as CAGCTG and exclusively forms homodimers (7,8). The
role of the MYC-AP4 axis in cell cycle regulation and tumorigenesis
was only recently discovered (8).
Although a number of studies have reported MYC amplification and/or
overexpression in several types of cancers (9), targeting of the MYC or MYC-AP4 axis
remains a distant challenge.
Recently, a potent, selective, small-molecule
inhibitor of BET bromodomains, JQ1, was developed. The molecule
antagonized BET bromodomain proteins during MYC-dependent
transcription in several types of cancers, including multiple
myeloma, acute myeloid leukemia and mixed lineage leukemia
(10–12). However, a more recent study
suggested that the efficacy of BET inhibitors is not always
dependent on the downregulation of MYC (13). With characteristic targeting
epigenetic signaling molecules, BET inhibitors may function
differently in a cell context-dependent manner. Therefore, it is
important to define the main target and underlying mechanism of BET
inhibitors in different cellular contexts.
Breast cancer is the most frequent type of cancer
diagnosed in women and is a representative heterogeneous disease
that can be classified into several subtypes based on gene
expression profiling and tumor histology (14). Approximately 75% of patients are
hormone receptor-positive, and the treatment options for these
patients have relied on anti-hormonal strategies. While most
patients respond to endocrine agents and have shown improved
overall survival, eventually, the majority of such patients become
resistant to these agents (15).
Development of a therapeutic strategy to combat this resistance and
to effectively treat hormone receptor-negative breast cancer is
crucial. Recent studies have shown that BET inhibitors are valuable
candidates for overcoming resistance to endocrine agents by the
suppression of MYC and PI3K signaling (16,17).
In the present study, we determined whether JQ1, an
inhibitor of the epigenetic reader BRD4, suppresses the MYC-AP4
axis in breast cancer. We found that JQ1 suppressed the MYC-AP4
axis in ER-positive and -negative breast cancer cell lines. We
further studied the ER-negative breast cancer cell line MDA-MB-231
which is relative harder to target in the clinic. JQ1 downregulated
the MYC-AP4 axis by direct inhibition of BRD4 binding to the MYC
and AP4 promoters at early time-points and subsequently induced
antitumorigenic effects, including cell cycle arrest, reduced wound
healing, and soft agar colony formation. Using BRD4
loss-of-function experiments, we further demonstrated that MYC and
AP4 are the direct targets of BRD4 inhibition. We found that loss
of AP4 mimics almost all of the antitumorigenic effects of JQ1,
suggesting that AP4 is a more sensitive target for BRD4-mediated
inhibition of MDA-MB-231 cells. Of note, we demonstrated for the
first time that MYC is a downstream target of AP4; hence, there is
a bidirectional positive loop between MYC and AP4. Thus, inhibition
of the MYC-AP4 axis can be better amplified by JQ1. Altogether, the
results presented here demonstrate that the BET protein inhibitor
is effective against MYC-AP4 axis-activated cancers and other
diseases by targeting multiple points.
Materials and methods
RNA extraction and reverse transcription
PCR
Total RNA was extracted using TRIzol reagent,
digested with DNase I, and reverse transcribed using a High
Capacity cDNA reverse transcription kit (Applied Biosystems).
Amplification of cDNA was performed on a LightCycler®
480 II using the LightCycler® 480 SYBR Green I Master
(both from Roche), using the recommended conditions. cDNAs were
amplified using the following gene-specific primers: RT_MYC,
5′-CCCTGG TGCCGTGAAGC; 3′-TTGCTCGAGTTCTTTCTGCAGA; and RT_AP4,
5′-GCAGGCAATCCAGCACAT; 3′-GGAGGCGGTGTCAGAGGT; and RT_P21,
5′-GAGGCCGGGATGAGTTGGGAGGAG; 3′-CAGCCGGCGTTTGGAGTGGTAGAA and
RT_BRD4, 5′-AAGAAGCGCTTGGAAAACAA; 3′-CAGGTTTTGCTGTCCCTGTT and
RT_P53, 5′-CCCCTCCTGGCCCCTGTCATCTTC; 3′-GCAGCGCCTCACAACCTCCGTCAT;
and RT_GAPDH, 5′-GAGTCAACGGATTTGGTCGT; 3′-TGGAAGATGGTGATGGGATT.
Western blot analysis
Cells were lysed with RIPA buffer [150 mM NaCl, 1.0%
NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl (pH 8.0)
and protease inhibitors] and sonicated briefly (30% amplitude, 3
sec). Cell lysates were boiled in Laemmli sample buffer for 3 min,
and 30 μg of each protein was subjected to SDS-PAGE. The
protein concentration was measured using the Bradford protein
assay. Antibodies against MYC (cat no. 9402S; Cell Signaling
Technology or cat no. ab39688-100; Abcam), AP4 (cat no. HPA001912;
Sigma-Aldrich), P21 (cat no. sc-756; Santa Cruz Biotechnology),
BRD4 (cat no. 13440), and β-actin (cat no. 49675) (both from Cell
Signaling Technology) were used. Proteins were transferred to
polyvinylidene difluoride membranes; the membranes were blocked for
30 min in Tris-buffered saline (TBS) containing 0.1% Tween-20 and
5% (w/v) dry skim milk powder, and incubated overnight with the
primary antibodies (dilution ratio 1:1,000). The membranes were
then washed with TBS-0.1% Tween-20, incubated for 1 h with a
secondary antibody (dilution ratio 1:10,000), and visualized using
an enhanced chemiluminescence detection kit (Amersham Life
Sciences) after exposure on an LAS-3000 image detection system
(Fuji).
Chromatin immunoprecipitation assay
(ChIP)
ChiP assays were performed according to instructions
from Upstate Biotechnology. For each ChIP, 100 μg DNA,
sheared by sonication (the DNA fragment size was 200–500 bp), was
pre-cleared with protein A magnetic beads (cat no. 16-661; Upstate
Biotechnology), and then 40 μg of the DNA was precipitated
by BRD4 (cat no. 13440; Cell Signaling Technology) or by AP4 (cat
no. HPA001912; Sigma-Aldrich). After IP, the recovered chromatin
fragments were subjected to real-time PCR. IgG control experiments
were performed for all ChIPs and incorporated into the IP/Input
(1%) by presenting the results as (IP - IgG)/(Input - IgG). The
following primers were used for amplification of the chromatin
fragments by real-time PCR: ChIP_MYC promoter, 5′-ACACTAACATCC
CACGCTCTG; 3′-GATCAAGAGTCCCAGGGAGA and ChIP_MYC enhancer 1,
5′-TGCTAATTGTGCCTCTCCTGT; 3′-ACTCCCAGCAAATCAGCCTA; and ChIP_MYC
enhancer 2, 5′-GGTCGGACATTCCTGCTTTA; 3′-GATATGCGGTCCCTACTCCA and
ChIP_MYC_promoter_ AP4 binding motif, 5′-CACTCTCCCTGGGACTCTTG,
5′-CACTCTCCCTGGGACTCTTG; 3′-GCGCCTACCATTTTCTTTTG and ChIP_AP4
promoter, 5′-GGGCGCTGCAAATAGTCCTT; 3′-CCGGGCGTGTGTATGTGTGT and
ChIP_AP4 enhancer 1, 5′-CGCGACGTTTGTAAATTGC; 3′-CTCAGATCCCGAGGAAGGA
and ChIP_AP4 enhancer 2, 5′-GAGGTGGGCGTTCTACGG;
3′-GGTTGGGCAGGAGTGTCTAC.
Cell cycle analysis
Cell cycle assays were performed using the Cycletest
Plus DNA reagent kit (BD Biosciences), according to the
manufacturer's instructions. The cell cycle profile of the cells
was analyzed using a FACScan flow cytometer (BD Biosciences).
Soft agar colony-formation assay
The soft agar colony-formation assay was performed
in 6-well plates. A bottom layer of agar (0.5%) with enriched
Dulbecco's modified Eagle's medium (DMEM) (final 10% FBS) was
initially poured. After the bottom agar solidified, MDA-MB-231
cells (1.0×104) were seeded on the top agar (0.3%) with
enriched DMEM (final 10% FBS) and incubated at 37°C for 21 days.
The culture medium was replaced 1–2 times per week. Colonies were
visualized by staining for 1 h with 0.005% crystal violet.
Wound-healing assay
Cells were grown to confluency in culture dishes and
treated with 0.2 μM JQ1. After overnight starvation in
serum-free medium, the cell monolayers were scraped with a sterile
micropipette tip. Initial gap widths (0 h) and residual gap widths
at 6 and 24 h after wounding were determined by
photomicrographs.
shRNA infection
shBRD4 and shAP4 constructs were purchased from
Sigma-Aldrich. For lentiviral production, the Mission lentiviral
packaging mix was used. Infected derivative cells stably expressing
shRNA were selected in the presence of 1.25 μg/ml
puromycin.
Statistical analysis
Results are expressed as mean ± SEM. Most
statistical comparisons were calculated by one-way ANOVA followed
by Bonferroni's post hoc test using GraphPad Prism. A p-value
<0.05 was considered to indicate a significant result.
Results
JQ1 suppresses the MYC-AP4 axis in breast
cancer cell lines
To examine whether JQ1, a known BRD4 inhibitor,
targets the MYC-AP4 axis in breast cancer cells, we used
ER-negative MDA-MB-231 cells and ER-positive MCF7 cells as models.
MDA-MB-231 and MCF7 cells were treated for 24 h with the indicated
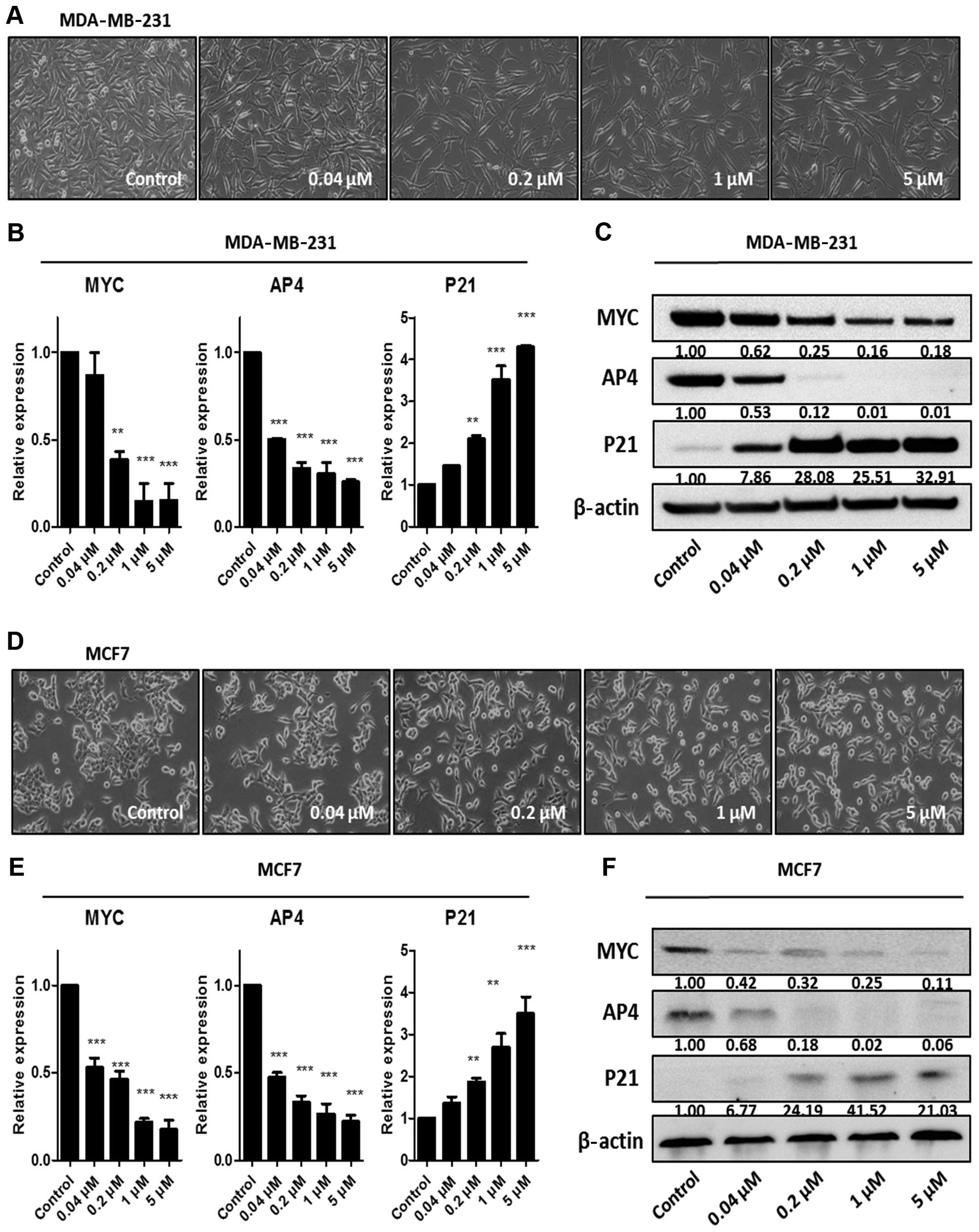
concentrations of JQ1 (Fig. 1). The
morphology of the MDA-MB-231 cells became thinner with protrusions
after treatment with increasing concentrations of JQ1 (Fig. 1A). In contrast, JQ1 had a modest
effect on the morphology of the MCF7 cells (Fig. 1D). JQ1 suppressed the MYC-AP4 axis
in a dose-dependent manner in the MDA-MB-231 and MCF7 cells,
although the sensitivities differed (Fig. 1B, C, E and F). MCF7 cells were more
sensitive than the MDA-MB-231 cells to suppression by JQ1; this
sensitivity may have been caused by a slightly higher expression of
MYC in the MDA-MB-231 cells (Fig.
1). Upregulation of P21 expression was accompanied by
downregulation of MYC and AP4 (Fig. 1B,
C, E and F), indicating that JQ1 targets the MYC-AP4 axis in
both ER-negative and -positive breast cancer cell lines.
JQ1 suppresses the tumorigenicity of
breast cancer cells
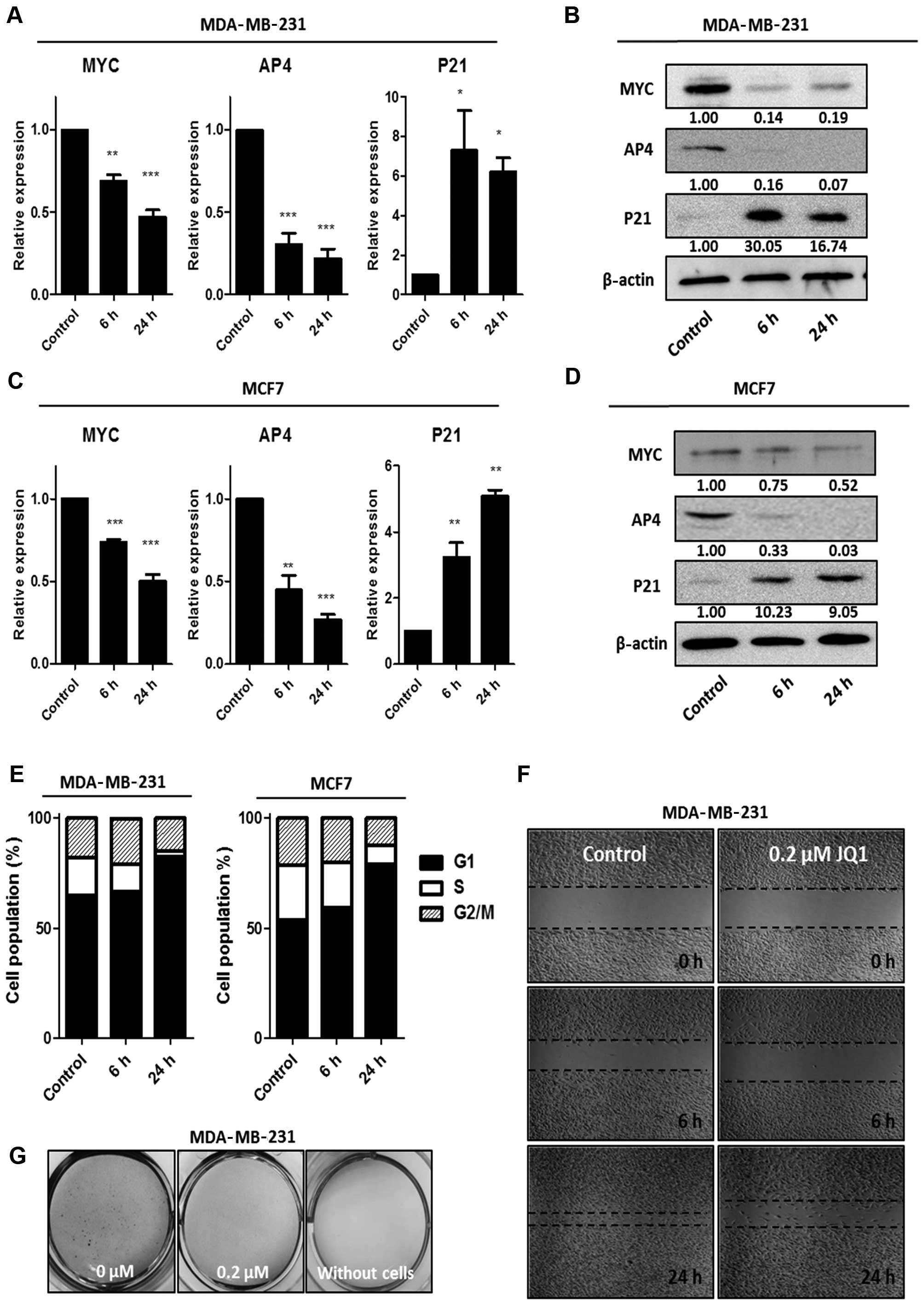
Based on a previous study showing that acute
treatment with JQ1 inhibits BRD4 (11), we measured the suppression of the
MYC-AP4 axis by JQ1 (0.2 μM) at an early time-point in the
MDA-MB-231 and MCF7 cells. Downregulation of MYC and AP4 was
observed after 6 h of treatment, and was accompanied by
upregulation of P21 (Fig. 2A–D).
Notably, the protein level of AP4 was almost completely abolished
at 6 h post-treatment in both breast cancer cell lines (Fig. 2B and D), suggesting that AP4 is a
highly sensitive and direct target of JQ1.
The MYC-AP4-P21 axis is responsible for G1 arrest in
several types of cancer cells (8);
therefore, we investigated the effect of JQ1 on the cell cycle. JQ1
treatment led to an increase in the percentage of cells in the G1
stage, from 64 to 85% in the MDA-MB-231 cells and from 55 to 86% in
the MCF7 cells (Fig. 2E). Next, to
assess additional antitumorigenic effects of JQ1, we performed a
scratch wound-healing assay and a soft agar colony-formation assay
using MDA-MB-231 cells. As shown in Fig. 2F and G, JQ1 efficiently reduced the
wound-healing capacity and soft agar colony formation of the
MDA-MB-231 cells, indicating that JQ1 has antitumor efficacy in
breast cancer cells.
JQ1 negatively regulates the MYC-AP4 axis
directly via suppression of BRD4 binding at the promoters of both
genes
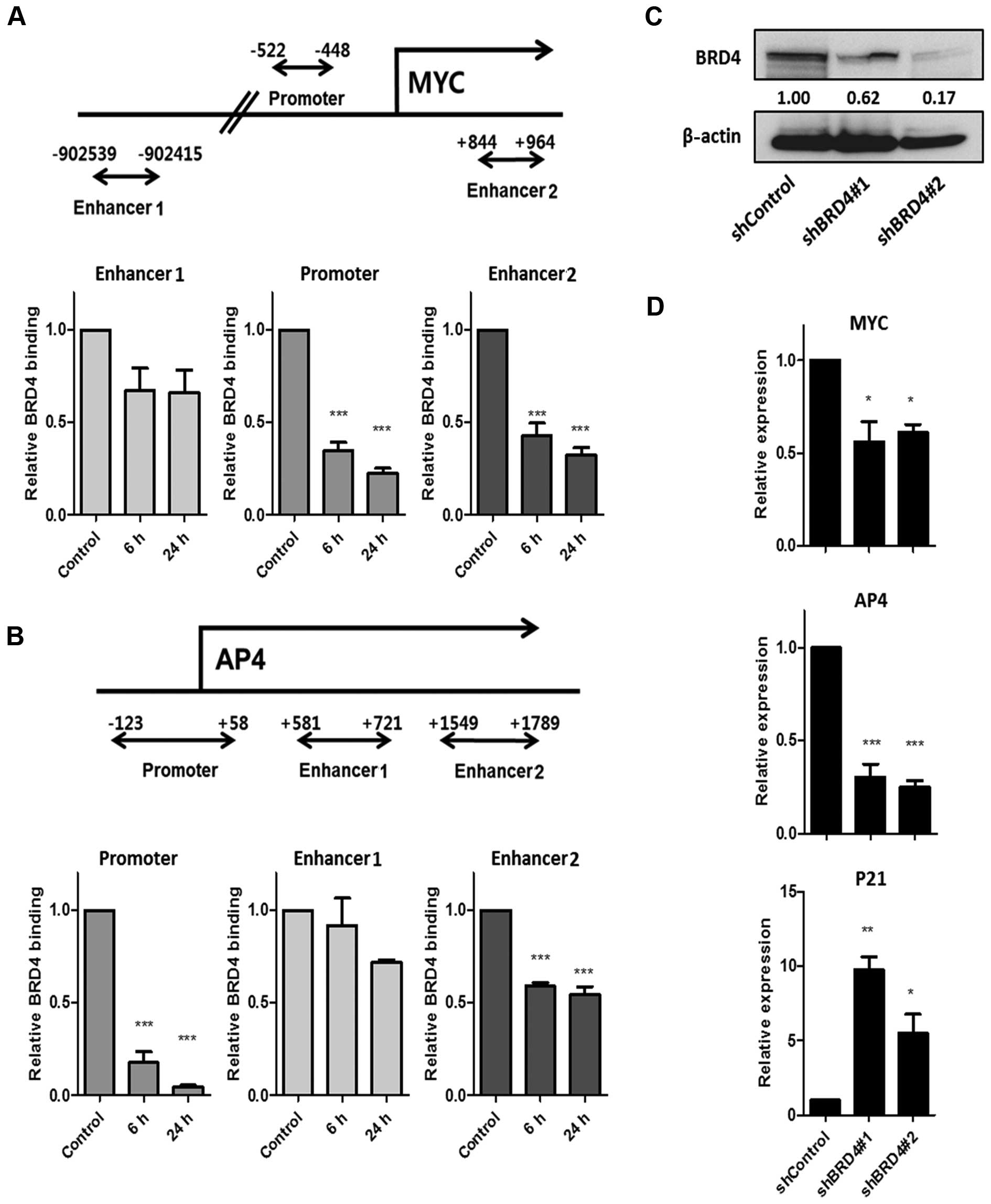
To determine whether downregulation of MYC and AP4
by JQ1 is associated with BRD4 binding, we performed chromatin
immunoprecipitation (ChIP) with a BRD4 antibody in MDA-MB-231 cells
(Fig. 3A and B). First, we measured
BRD4 binding to the previously identified promoter, enhancer 1 and
enhancer 2 of MYC (11). As shown
in Fig. 3A, we found that BRD4
binding decreased early at the promoter and at enhancer 2 upon
treatment with JQ1, but not at enhancer 1. Next, we measured BRD4
binding to the previously identified promoter, enhancer 1 and
enhancer 2 of AP4 (7). Reduced
binding by BRD4 was detected, mainly in the promoter, rather than
in the enhancers (Fig. 3B),
suggesting that suppression of the MYC-AP4 axis is through the
direct inhibition of BRD4 binding at the promoter of both
genes.
To further establish that BRD4 is required for
activation of the MYC-AP4 axis, we performed a loss-of-function
study by generating stable BRD4-knockdown cells (Fig. 3C). We detected reduced expression of
MYC and AP4 as well as an increase in P21 after BRD4 knockdown in
the MDA-MB-231 cells (Fig. 3D),
demonstrating that, in MDA-MB-231 cells, the MYC-AP4 axis is a
direct target of the epigenetic reader, BRD4.
Inhibition of MYC/MAX heterodimerization
suppresses AP4, but not as effectively as JQ1
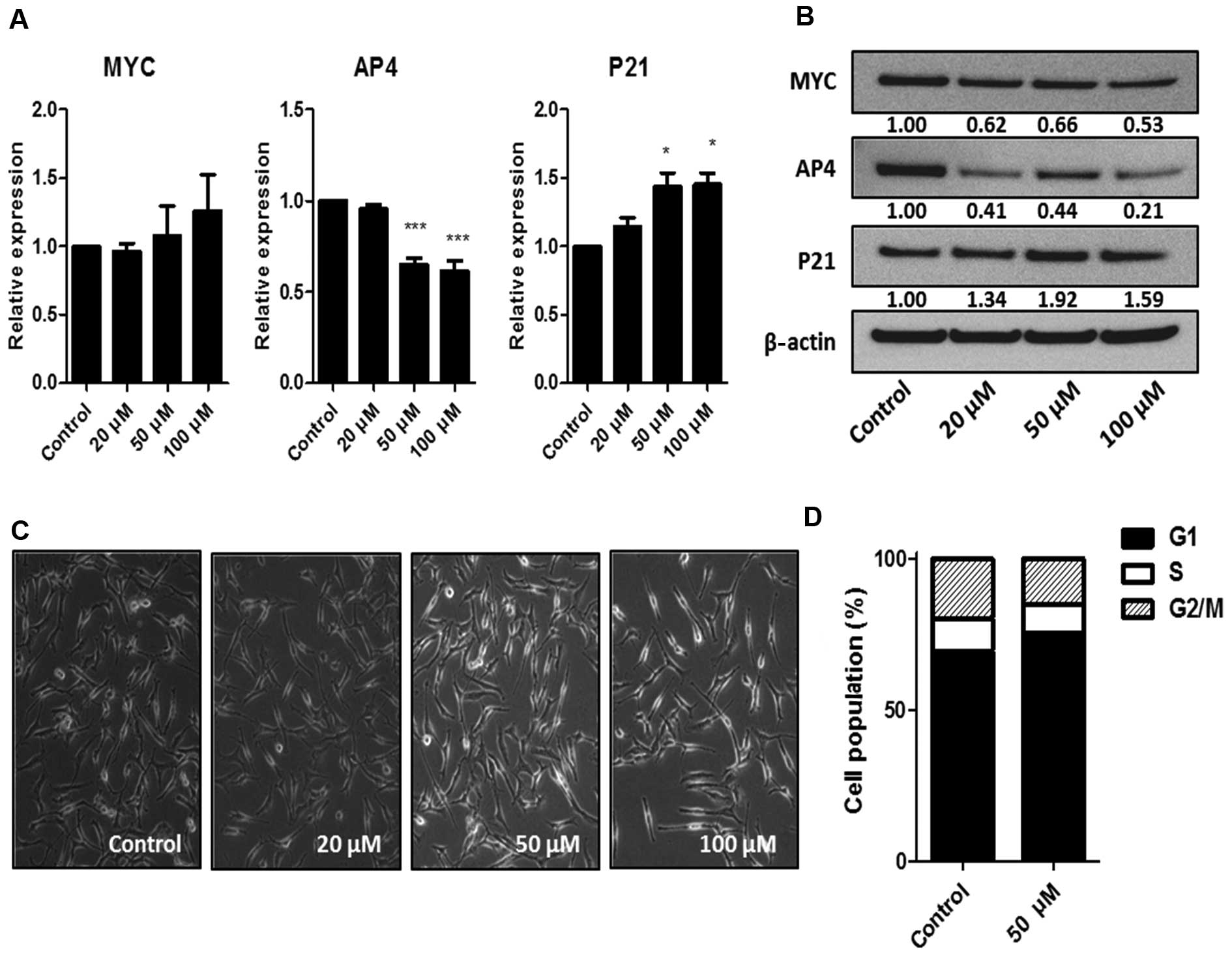
To determine whether the activation of the MYC-AP4
axis occurs through the action of the MYC/MAX dimer, we used the
small-molecule MYC inhibitor, 10058-F4. 10058-F4 prevents the
binding of MYC/MAX dimers to targets and inhibits MYC-driven
transformation (18,19). While 10058-F4 did not alter MYC mRNA
levels, a dose of 50 μM downregulated AP4 mRNA. This dose
was also suffi-cient to induce changes in cell morphology (Fig. 4A and C), suggesting that AP4 is a
target of the MYC/MAX dimer. There was a partial decrease in the
MYC and AP4 protein levels after treatment with 10058-F4 (Fig. 4B), and the effect of 10058-F4 on
cell cycle arrest was mild when compared to the effect of JQ1
(Fig. 4D). These results suggest
that inhibition of MYC/MAX dimerization in the activated MYC-AP4
axis is not as effective as BRD4 inhibition, presumably due to a
smaller downregulation of AP4.
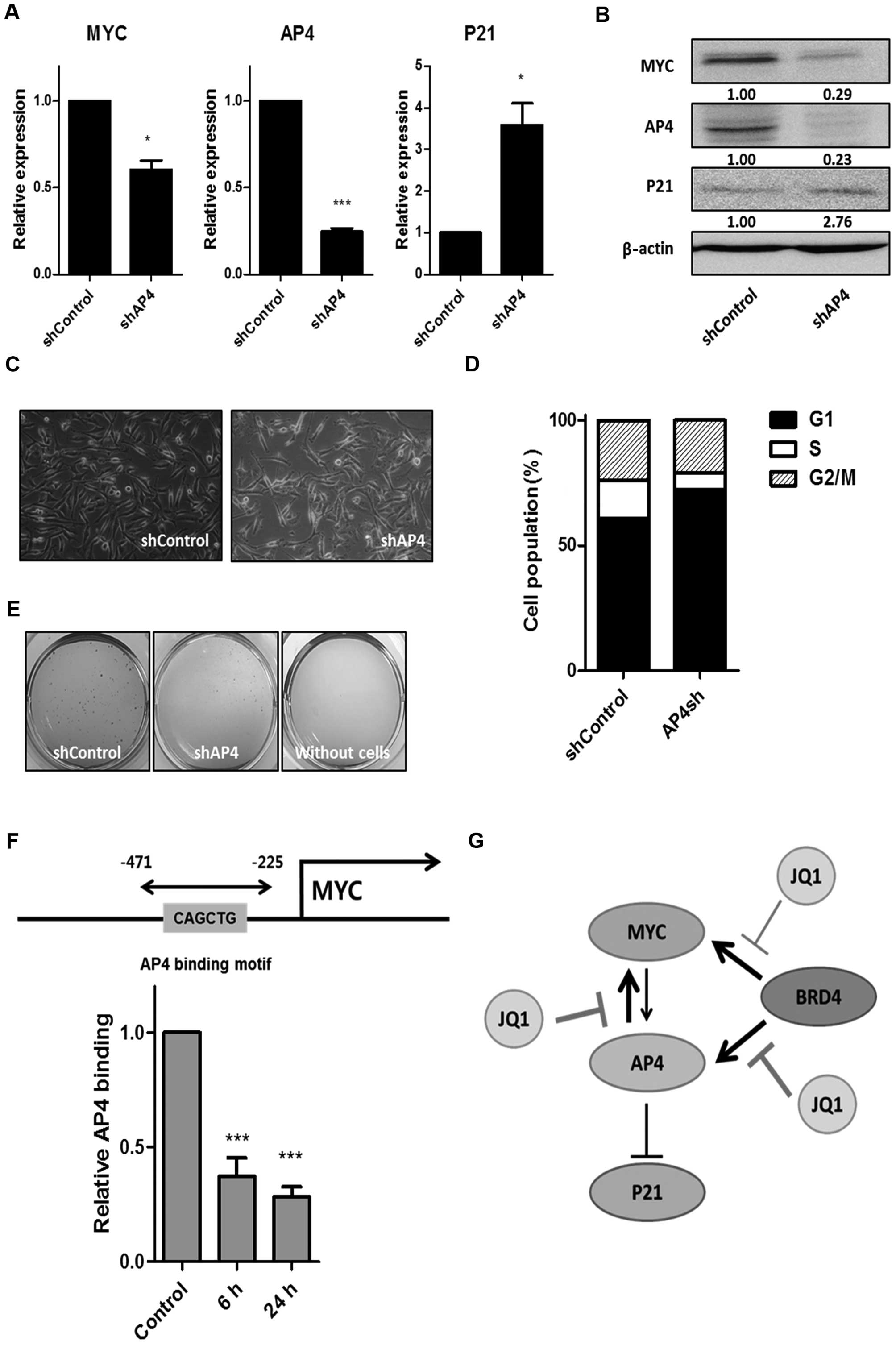
Knockdown of AP4 reveals a bidirectional,
positive loop between MYC and AP4 and the underlying mechanism by
which suppression of the MYC-AP4 axis is synergized after treatment
with JQ1
To determine whether AP4 is the major target of BRD4
inhibition, we generated a stable AP4-knockdown cell line using
shRNA in the MDA-MB-231 cells. We then analyzed the mRNA and
protein expression of AP4, MYC, and P21 (Fig. 5A and B). We verified complete loss
of AP4 by western blot analysis (Fig.
5B) and found that loss of AP4 mimicked most of the effects of
treatment with JQ1, including suppression of the MYC-AP4 axis and
sub-sequential induction of P21 (Fig.
5A and B), changes in cell morphology (Fig. 5C), cell cycle arrest (Fig. 5D), and reduced soft agar colony
formation (Fig. 5E). These results
suggest that AP4 is a major critical target of JQ1 in MDA-MB-231
cells. In addition, we confirmed that the expression of AP4 was
reduced by JQ1 treatment in other cancer cell lines, including cell
lines derived from the liver, colon and the esophagus (data not
shown), suggesting that AP4 may be a general target of JQ1.
Unexpectedly, we observed a decrease in MYC upon AP4
knockdown (Fig. 5A and B). AP4 is
known to be a direct target of MYC (6,7);
however, to the best of our knowledge, it has not been reported
that MYC is a downstream target of AP4. To confirm that MYC is a
downstream target of AP4 and that there is a bidirectional positive
loop between MYC and AP4, we performed a ChIP assay using the AP4
antibody following JQ1 treatment. We identified an AP4 binding
motif (CAGCTG) within the MYC promoter. We detected AP4 binding at
this site, and this binding decreased upon treatment with JQ1
(Fig. 5F), demonstrating that MYC
is a downstream target of AP4. Taken together, these data suggest
that suppression of the MYC-AP4 axis by JQ1 is synergized through
multiple mechanisms, including inhibition of BRD4 binding at the
MYC and AP4 promoters, followed by secondary inhibition of a
bidirectional loop between MYC and AP4.
Discussion
Here, for the first time, we demonstrated that the
inhibitor of the epigenetic reader BRD4, JQ1, effectively
suppressed the MYC-AP4 axis in breast cancer cells by targeting a
bidirectional, positive loop between MYC and AP4 (Fig. 5G). JQ1 inhibited BRD4 binding mainly
at the promoters for MYC and AP4 promoters, rather than at the
enhancer sites. We further demonstrated that MYC and AP4 are direct
targets of BRD4 by generating stable BRD4-knockdown breast cancer
cell lines. We found that the suppressive effect of an inhibitor of
MYC/MAX dimerization on the activated MYC-AP4 axis was not as
effective as inhibition of BRD4, which presumably acts through the
mild downregulation of AP4. Our AP4 loss-of-function study
demonstrated that AP4 is a major critical target of JQ1 and that
MYC is a novel target of AP4, which was further supported by an
anti-AP4 ChIP assay. Downregulation of MYC-AP4 by inhibition of
BRD4 induced antitumorigenic effects, including cell cycle arrest,
reduced wound healing and soft agar colony formation. Collectively,
our results suggest that the epigenetic reader BRD4 is a key
mediator of the overexpression of MYC and AP4. These findings have
important implications for the treatment of MYC-AP4 axis-activated
cancers.
Inhibitors of the epigenetic reader BET family,
including JQ1 and I-BET, have emerged as promising therapeutic
drugs for cancers, inflammation and obesity (20). The underlying mechanisms of the
effects of these small molecules often involve binding of these
drugs to the bromodomain of BET proteins, and the process is
completed upon lysine acetylation of histones. The molecules,
therefore, suppress target gene expression involved in
tumorigenesis or the inflammatory response (4,20). MYC
is the primary target in several cancers (11,12,21),
and more recently, the underlying mechanism was proposed to involve
inhibition by JQ1 of MYC by disrupting super-enhancers (22). Super-enhancers are defined as large
clusters of enhancers that determine cellular identity (23,24).
JQ1 was found to lead to preferential loss of BRD4 at
super-enhancers of MYC in multiple myeloma (22); however, it is not clear whether MYC
always has a super-enhancer in different cellular contexts. Indeed,
our results demonstrated that JQ1 inhibited BRD4 binding to the MYC
promoter and enhancer 2, but not to enhancer 1 (Fig. 3A). Thus, the detailed molecular
mechanism of BET protein inhibitors requires further
investigation.
AP4 is known to be a MYC-inducible repressor of P21
(7); however, it has not been
previously reported that MYC is a downstream target of AP4. Here,
we demonstrated, for the first time, that there is a bidirectional
positive loop between MYC and AP4 (Fig.
5). As shown in Fig. 5, AP4
binds to the MYC promoter. Binding by AP4 is reduced by JQ1
treatment, and knockdown of AP4 induces downregulation of MYC. This
may be the underlying mechanism by which suppression of the MYC-AP4
axis is amplified by the BET protein inhibitor. AP4 is also known
to contribute to several processes during cancer development,
including EMT and metastasis (6).
Our study showed that JQ1 downregulated AP4 at a very early time
point and at low concentrations in the ER-positive and -negative
cancer cell lines (Figs. 1 and
2). These results suggest that AP4
is the most sensitive and critical target of BET protein
inhibition. The clinical relevance of AP4 expression in cancers was
previously reported. Elevated expression of AP4 is associated with
an increased metastatic capacity in colorectal cancer (6) and AP4 predicts poor prognosis in
non-small cell lung cancer (25).
In addition, several studies have demonstrated the critical role of
AP4 in cancers and in immunology (26–28).
Therefore, this study contributes to our understanding of cancer
biology.
It is well known that epigenetic modifiers and
chromatin remodelers control gene expression and establish cellular
identities via the regulation of chromatin structure. However,
little is known concerning the specific targets of these components
of the epigenome and the underlying mechanisms of the effects of
the small molecules that target them. Our results showed, for the
first time, that the BET protein inhibitor suppressed the MYC-AP4
axis by targeting a bidirectional loop between MYC and AP4 to
induce antitumorigenic effects. Collectively, our results suggest
that a better understanding of epigenetic modifiers and chromatin
remodelers will facilitate the development of novel strategies for
treating many diseases caused by dysregulated epigenome
components.
Abbreviations:
|
BET
|
bromodomain and extra-terminal
domain
|
|
ChIP
|
chromatin immunoprecipitation
|
|
BRD
|
bromodomain-containing protein
|
|
bHLH
|
basic helix-loop-helix domain
|
|
AP4
|
activating enhancer binding protein
4
|
|
EMT
|
epithelial-mesenchymal transition
|
Acknowledgments
This research was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Science, ICT and Future Planning
(2013R1A1A1057575).
References
|
1
|
You JS and Jones PA: Cancer genetics and
epigenetics: Two sides of the same coin? Cancer Cell. 22:9–20.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arrowsmith CH, Bountra C, Fish PV, Lee K
and Schapira M: Epigenetic protein families: A new frontier for
drug discovery. Nat Rev Drug Discov. 11:384–400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
You JS and Han JH: Targeting components of
epigenome by small molecules. Arch Pharm Res. 37:1367–1374. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Filippakopoulos P and Knapp S: Targeting
bromodomains: Epigenetic readers of lysine acetylation. Nat Rev
Drug Discov. 13:337–356. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meyer N and Penn LZ: Reflecting on 25
years with MYC. Nat Rev Cancer. 8:976–990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jackstadt R, Röh S, Neumann J, Jung P,
Hoffmann R, Horst D, Berens C, Bornkamm GW, Kirchner T, Menssen A,
et al: AP4 is a mediator of epithelial-mesenchymal transition and
metastasis in colorectal cancer. J Exp Med. 210:1331–1350. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jung P, Menssen A, Mayr D and Hermeking H:
AP4 encodes a c-MYC-inducible repressor of p21. Proc Natl Acad Sci
USA. 105:15046–15051. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jung P and Hermeking H: The c-MYC-AP4-p21
cascade. Cell Cycle. 8:982–989. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Asangani IA, Dommeti VL, Wang X, Malik R,
Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S,
Engelke C, et al: Therapeutic targeting of BET bromodomain proteins
in castration-resistant prostate cancer. Nature. 510:278–282. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Delmore JE, Issa GC, Lemieux ME, Rahl PB,
Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et
al: BET bromo-domain inhibition as a therapeutic strategy to target
c-Myc. Cell. 146:904–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zuber J, Shi J, Wang E, Rappaport AR,
Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al:
RNAi screen identifies Brd4 as a therapeutic target in acute
myeloid leukaemia. Nature. 478:524–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lockwood WW, Zejnullahu K, Bradner JE and
Varmus H: Sensitivity of human lung adenocarcinoma cell lines to
targeted inhibition of BET epigenetic signaling proteins. Proc Natl
Acad Sci USA. 109:19408–19413. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rakha EA, Reis-Filho JS and Ellis IO:
Basal-like breast cancer: A critical review. J Clin Oncol.
26:2568–2581. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ignatiadis M and Sotiriou C: Luminal
breast cancer: From biology to treatment. Nat Rev Clin Oncol.
10:494–506. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bihani T, Ezell SA, Ladd B, Grosskurth SE,
Mazzola AM, Pietras M, Reimer C, Zinda M, Fawell S and D'Cruz CM:
Resistance to everolimus driven by epigenetic regulation of MYC in
ER+ breast cancers. Oncotarget. 6:2407–2420. 2015.
View Article : Google Scholar
|
|
17
|
Stratikopoulos EE, Dendy M, Szabolcs M,
Khaykin AJ, Lefebvre C, Zhou MM and Parsons R: Kinase and BET
inhibitors together clamp inhibition of PI3K signaling and overcome
resistance to therapy. Cancer Cell. 27:837–851. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang MJ, Cheng YC, Liu CR, Lin S and Liu
HE: A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle
arrest, apoptosis, and myeloid differentiation of human acute
myeloid leukemia. Exp Hematol. 34:1480–1489. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mo H and Henriksson M: Identification of
small molecules that induce apoptosis in a Myc-dependent manner and
inhibit Myc-driven transformation. Proc Natl Acad Sci USA.
103:6344–6349. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Belkina AC and Denis GV: BET domain
co-regulators in obesity, inflammation and cancer. Nat Rev Cancer.
12:465–477. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Filippakopoulos P, Qi J, Picaud S, Shen Y,
Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et
al: Selective inhibition of BET bromodomains. Nature.
468:1067–1073. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lovén J, Hoke HA, Lin CY, Lau A, Orlando
DA, Vakoc CR, Bradner JE, Lee TI and Young RA: Selective inhibition
of tumor oncogenes by disruption of super-enhancers. Cell.
153:320–334. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hnisz D, Abraham BJ, Lee TI, Lau A,
Saint-André V, Sigova AA, Hoke HA and Young RA: Super-enhancers in
the control of cell identity and disease. Cell. 155:934–947. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hnisz D, Schuijers J, Lin CY, Weintraub
AS, Abraham BJ, Lee TI, Bradner JE and Young RA: Convergence of
developmental and oncogenic signaling pathways at transcriptional
super-enhancers. Mol Cell. 58:362–370. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gong H, Han S, Yao H, Zhao H and Wang Y:
AP-4 predicts poor prognosis in non small cell lung cancer. Mol Med
Rep. 10:336–340. 2014.PubMed/NCBI
|
|
26
|
Chou C, Pinto AK, Curtis JD, Persaud SP,
Cella M, Lin CC, Edelson BT, Allen PM, Colonna M, Pearce EL, et al:
c-Myc-induced transcription factor AP4 is required for host
protection mediated by CD8+ T cells. Nat Immunol.
15:884–893. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jackstadt R and Hermeking H: AP4 is
required for mitogen- and c-MYC-induced cell cycle progression.
Oncotarget. 5:7316–7327. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jackstadt R, Jung P and Hermeking H: AP4
directly downregulates p16 and p21 to suppress senescence and
mediate transformation. Cell Death Dis. 4. pp. e7752013, View Article : Google Scholar
|