Introduction
Breast cancer is one of the most common malignant
cancers, and is also the leading cause of cancer-related deaths
among women due to the metastatic spread of the cancer to vital
organs, such as the lung and liver (1,2). For
patients with early breast cancer, surgery is the primary treatment
which effectively improves patient long-term survival. However, it
is ineffective for individuals with advanced disease, and the
systemic chemotherapy is considered as an alternative option when
tumor resection is not feasible (3,4).
Unfortunately, chemotherapy is mostly ineffective due to the
development of chemoresistance in cancer patients (5). Doxorubicin (DOX) is a widely used
antitumor antibiotic for the treatment of multiple types of cancers
including breast cancer. However, high-dose DOX shows
cardiotoxicity as well as killing normal self-reborn cells
(6,7). Therefore, efforts have been made to
reduce the dose of DOX by reversing chemoresistance (8,9).
MicroRNAs (miRNAs) are a class of small non-coding
RNAs, typically 19–25 nucleotides in length. They function as gene
regulators by downregulating the expression of specific target
genes (10,11). Approximately 50% of miRNA genes are
located in tumor-associated genomic regions, and more than 30% of
all human protein-coding genes may be regulated by miRNAs,
including a wide range of genes involved in tumorigenesis (12,13).
Therefore, it is well acknowledged that miRNAs are important for
cancer development and progression, which can act as either
oncogenes or tumor suppressors by regulating their respective
target genes (14,15). Studies have indicated that a
systematic characterization of miRNAs could enable their
identification as biomarkers for the diagnosis of breast cancer,
and many miRNAs may be chosen as therapeutic targets for the
treatment of breast cancer (16,17).
However, the role and the mechanism of miRNAs in tumorigenesis and
cancer chemotherapy remain largely unknown.
In the present study, we sought to determine the
role of miR-181b in the growth and migration of breast cancer
cells. We demonstrated that the expression of miR-181b is
upregulated in patients with breast cancer, and is involved in the
development and metastasis of breast cancer cells. More
importantly, we also present evidence for miR-181b upregulation as
a mechanism for DOX resistance, and provide the first link between
miR-181b and the intrinsic apoptotic pathway activated by DOX in
breast cancer.
Materials and methods
Clinical samples and cell culture
Blood samples were obtained from 30 healthy controls
and 30 breast cancer patients at Zhejiang Cancer Hospital
(Hangzhou, China). The use of the blood for the present study was
approved by the Hospital's Protection of Human Subjects Committee.
MCF-10A, T-47D, MCF-7, MDA-MB-231 and MDA-MB-435 cell lines were
provided by the Institute of Biochemistry and Cell Biology of the
Chinese Academy of Science (Shanghai, China). The breast cancer
cell lines (including T-47D, MCF-7, MDA-MB-231 and MDA-MB-435) were
cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented
with 10% fetal bovine serum (both from Gibco, USA), 100 IU/ml
penicillin and 100 µg/ml streptomycin sulfate. The MCF-10A
cell line was cultured in DMEM/F12 media supplemented with 5% horse
serum, 10 µg/ml insulin (all from Gibco), 100 ng/ml cholera
toxin, 20 ng/ml EGF and 0.5 µg/ml hydrocortisone (all from
Sigma-Aldrich, USA) at 37°C in a humidified incubator with 5%
CO2. To study the role of miR-181b in chemoresistance,
we established a doxorubicin-resistant T-47D cell line (T-47D-R) by
stepwise exposure of T-47D cells to increasing concentrations of
DOX (Sigma-Aldrich). Briefly, the T-47D cells were initially
cultured with 0.1 µg/ml DOX for 8 weeks, and then the DOX
concentration was increased every 4 weeks by 0.02 µg/ml up
to a final concentration of 0.3 µg/ml. The T-47D-R cells
were exposed to DOX over a time period of 12 months. Before the
following experiments were performed, the T-47D-R cells were
cultured in DOX-free DMEM for 2 weeks.
RNAs and transfection
Human miR-181b mimics, 2′-omethyl modified miR-181b
inhibitors, negative control oligonucle-otides (NCO) and Bim siRNA
were purchased from GenePharma Co. (China). The sequences of RNAs
were as follows: miR-181b mimics, 5′-AACAUUCAUUGCUGUCGGUGGGU-3′;
miR-181b inhibitors; 5′-ACCCACCGACAGCAAUGAAUGUU-3′; NCO,
5′-AUCCCAUGGUGGGUUACAUGGUU-3′; and Bim siRNA,
5′-GACCGAGAAGGUAGACAAUUU-3′. The RNAs were transfected into cells
with Lipofectamine 2000 (Invitrogen, USA) at the final
concentration of 50 nM according to the manufacturer's
protocols.
Quantitative real-time PCR (qRT-PCR)
The expression levels of miR-181b and Bim were
measured by quantitative RT-PCR (qRT-PCR), using TaqMan MicroRNA
assays kit and supplies (Applied Biosystems, USA) according to the
manufacturer's instructions. The expression of miR-181b was
normalized to U6 snRNA, and the Bim level was normalized to GAPDH.
Relative quantities of miR-181b and Bim were calculated using the
2−ΔΔCt method (18).
Luciferase reporter assay
The Bim 3′-UTR was cloned into the pMIR-REPORT™
miRNA Expression Reporter Vector (Life Technologies, USA) to
generate the wild-type constructs. The mutant plasmid was created
by mutating the seed regions of the miR-181b-binding sites (UGAAUGU
to UGAUAGU) using the Site-Directed Mutagenesis kit (Takara, Japan)
based on the wild-type constructs. Then, the Dual-Luciferase
Reporter assay (Promega, Madison, WI, USA) was conducted to
investigate the interaction between miR-181b and its predicted
target gene Bim. Briefly, the T-47D-R cells were co-transfected
with the miR-181b mimics or NCO and Bim 3′UTR or Bim 3′UTR-mutant.
Forty-eight hours later, the firefly and Renilla luciferase
activities were detected according to the manufacturer's protocol.
The firefly luciferase activity was normalized to the
Renilla luciferase activity.
Western blot analysis
The whole cells were lysed with RIPA buffer (Cell
Signaling Technology, USA) containing 2 mM of phenylmethanesulfonyl
fluoride. The samples were then subjected to SDS-PAGE and
transferred to nitrocellulose membranes. The membranes were probed
with primary antibodies at 4°C overnight, and were then incubated
with appropriate horseradish peroxidase-conjugated secondary
antibodies for 2 h. Blots were developed using an enhanced
chemiluminescence detection kit (Pierce, USA). The primary
antibodies against Bim, caspase-3, caspase-9, PARP and β-actin were
purchased from Cell Signaling Technology.
Cell viability and proliferation
Breast cancer cells were transfected with miR-181b
mimics or miR-181b inhibitors at a final concentration of 50
µM. Forty-eight hours after transfection, the medium was
replaced with fresh medium containing DOX at different
concentrations. After culturing for 48 h, the cells were subjected
to cell viability assay. DOX concentrations leading to 50% cell
death (IC50) were calculated by the viability curve
determined by the MTT assay. To determine cell proliferation, the
3H thymidine incorporation assay was used to determine
the cell proliferation during the last 6 h of incubation as
previously described (19).
Cell migration in vitro
Breast cancer cells were trans-fected with miR-181b
mimics or inhibitors for 48 h. Before being seeded, the
undersurface of the upper chamber of the Transwell was coated with
collagen I overnight at 4°C, and then 1×105 cells were
seeded in serum-free media on the upper chambers of Transwells with
a porous transparent polyethylene terephthalate membrane having a
pore size of 8-micron (Corning Costar Corporation, USA), and
hydroxyurea (Sigma-Aldrich) was added to stop cellular
proliferation. After 24 h, cells on the undersurface of the upper
units were fixed, stained and then counted under a phase-contrast
microscope.
Apoptosis and mitochondrial membrane
potential (MMP, ΔΨm) analysis
Apoptosis was assessed using the Annexin V-FITC
apoptosis detection kit (Sigma-Aldrich) according to the
manufacturer's protocol. After transfection and DOX treatment,
T-47D-R cells were harvested and rinsed in cold phosphate-buffered
saline (PBS), followed by resuspension in 1X Annexin binding buffer
at 1×106 cells/ml. Annexin V (5 µl) and 0.1
µg of propidium iodide (PI) were then added to the cells.
Samples were incubated at room temperature for 15 min in the dark
and were analyzed using flow cytometry (Becton-Dickinson, USA).
ΔΨm was detected using
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbo-cyanine
iodide (JC-1; Molecular Probes, USA) as an indicator (20). After treatment, the T-47D-R cells
were collected and resuspended with PBS containing JC-1 at a final
concentration of 5 µM. Following a 20-min incubation period
at 37°C in the dark, MMP was determined by flow cytometric
analysis.
Statistical analysis
Data are represented as mean ± SE of three
independent experiments. The Student's t-test was conducted with
SPSS 14.0 software to assess the statistical significance between
treatments. p<0.05 was considered to indicate a statistically
significant result.
Results
miR-181b is upregulated in breast cancer
cell lines and patient serum
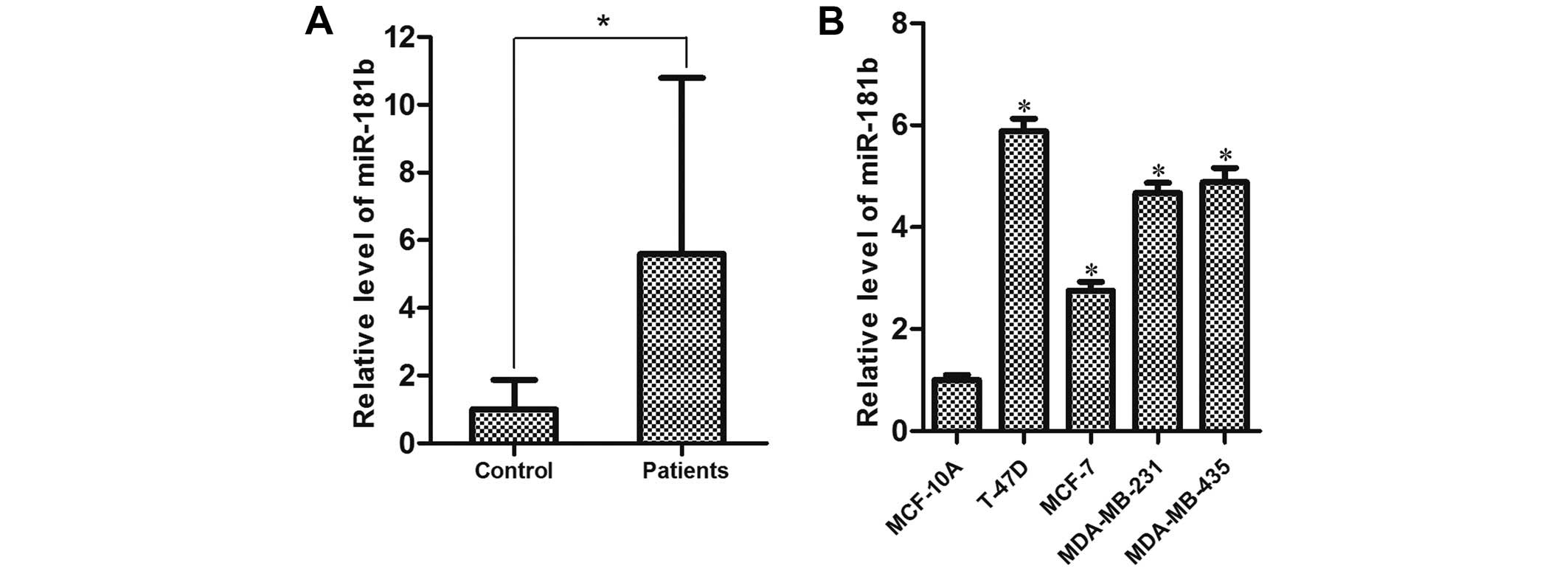
The expression of miR-181b was analyzed in the serum
of breast cancer patients and the healthy controls using RT-qPCR.
miR-181b was significantly upregulated in the cancer patient serum
compared with that in the normal controls (Fig. 1A). Furthermore, we found that the
expression level of miR-181b was significantly higher in all of the
four breast cancer cell lines compared with the level in the normal
breast cell line MCF-10A (21)
(Fig. 1B). These results suggest
that miR-181b may function as a tumor promoter in breast
cancer.
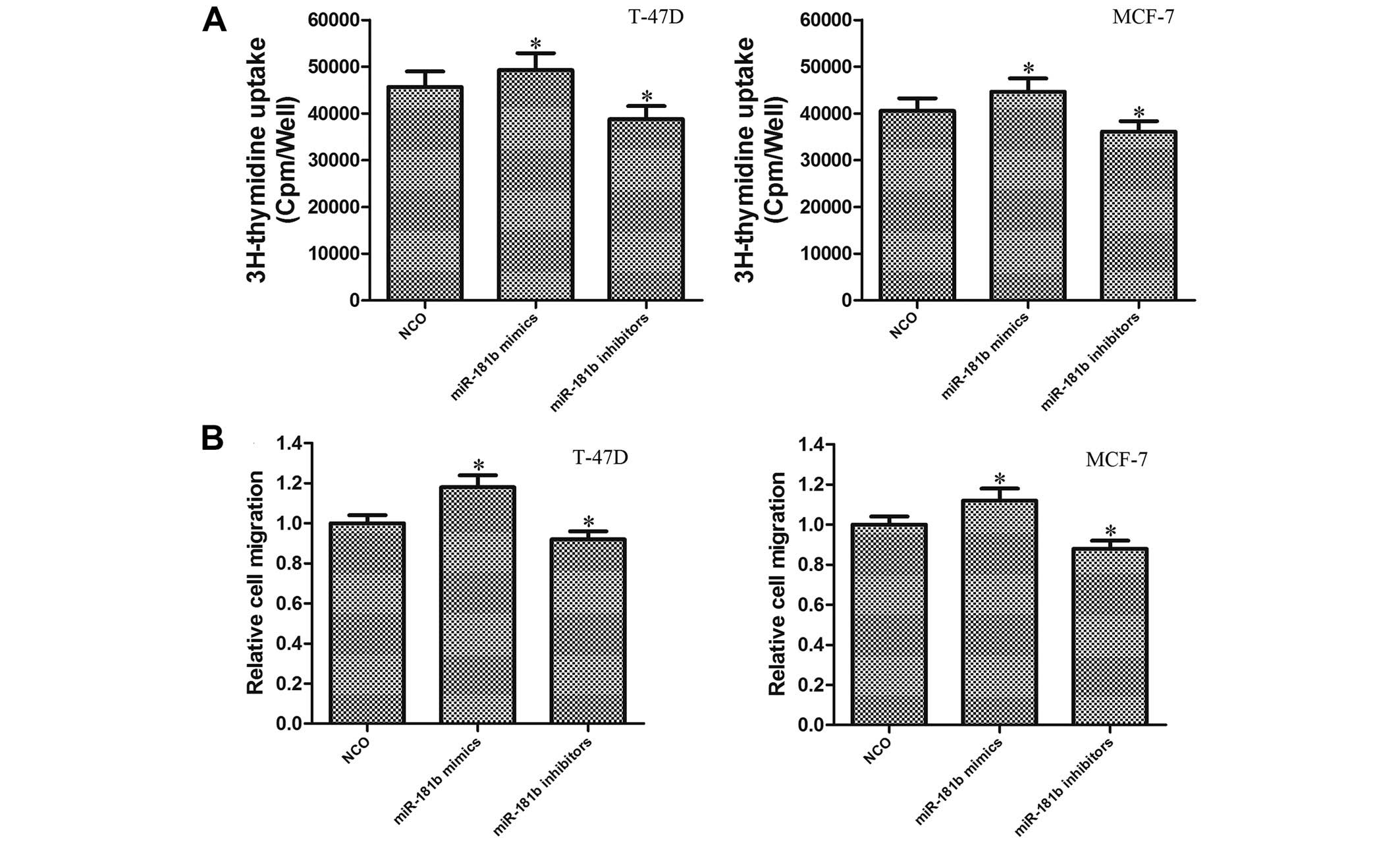
miR-181b promotes cell proliferation and
migration in breast cancer cells
To study the role of miR-181b in the tumor
progression of breast cancer cells, miR-181b mimics or inhibitors
were transfected to change the level of miR-181b in the T-47D and
MCF-7 cells. Briefly, the miR-181b level was increased ~10.22-fold
after the miR-181b mimics were introduced, and the miR-181b level
was decreased ~4.27-fold after the miR-181b inhibitors were
introduced (data not shown). Results of 3H thymidine
incorporation assays demonstrated that the proliferation of both
T-47D and MCF-7 cells was significantly enhanced in the
miR-181b-overexpressing group compared with the control group. In
contrast, the proliferation was significantly impaired in the
miR-181b-knockdown cells (Fig. 2A).
Furthermore, we evaluated the effect of miR-181b on cell migration
using a Transwell system. We observed that the overexpression of
miR-181b significantly promoted the migration in breast cancer
cells, which could be obviously inhibited by knockdown of miR-181b
(Fig. 2B). These results suggest
that miR-181b may function as a novel oncogene in breast
cancer.
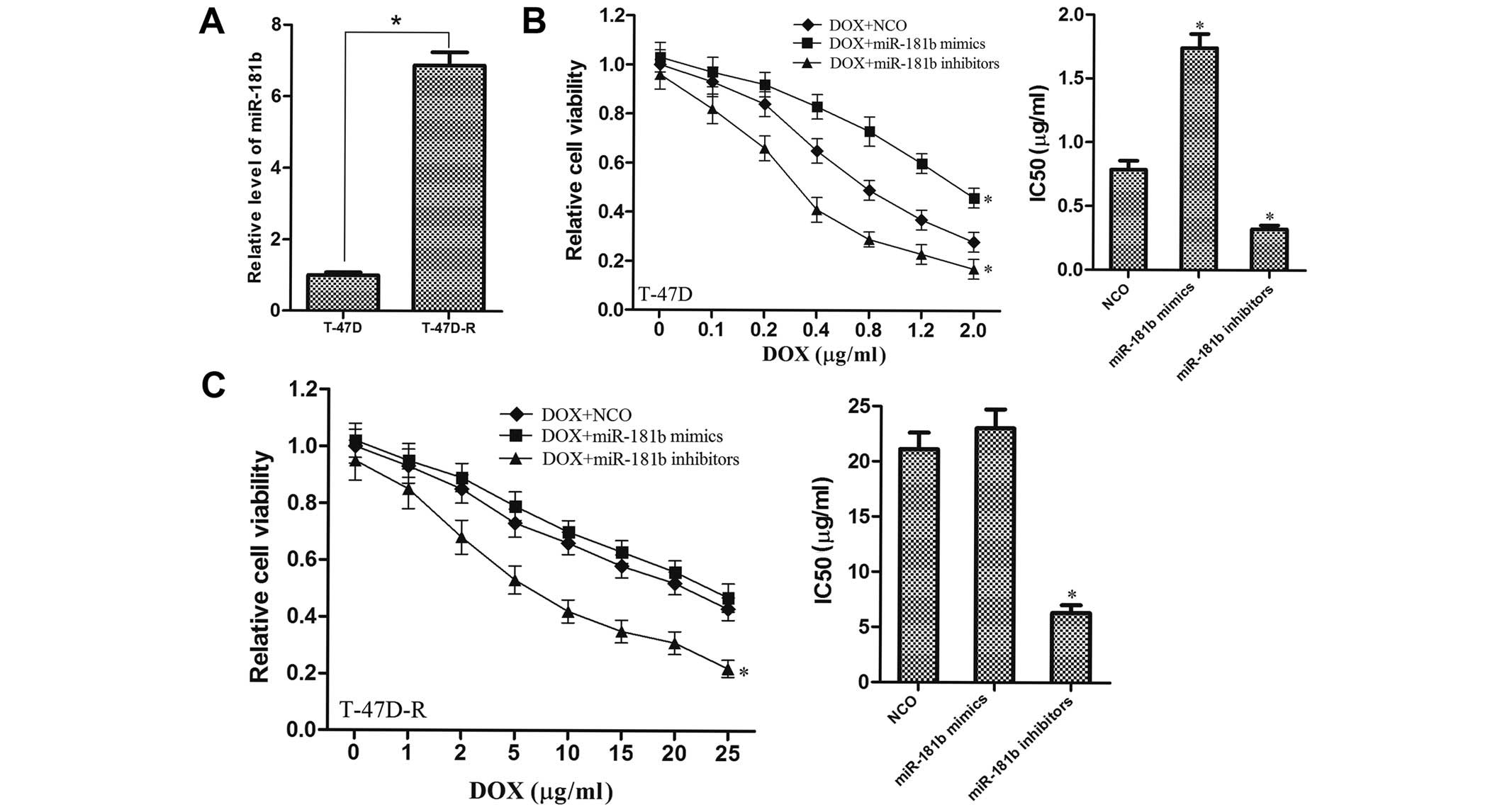
miR-181b is associated with the
resistance of breast cancer cells to DOX
To investigate whether miR-181b can modulate the
sensitivity of breast cancer cells to DOX which is a potent
anticancer drug, we stepwisely exposed the T-47D cells to
increasing concentrations of DOX to establish a DOX-resistant T-47D
cell line (T-47D-R). As shown in Fig.
3A, the expression level of miR-181b in the T-47D-R cells was
significantly upregulated compared with that in the parental T-47D
cells, suggesting that miR-181b promotes the chemoresistance in
breast cancer. To reveal the effects of miR-181b on the efficacy of
chemotherapy, T-47D cells were simultaneously treated with miR-181b
and DOX. It was found that the miR-181b mimics significantly
increased the IC50 of DOX, which was obviously decreased
by the transfection of miR-181b inhibitors compared with the
controls (Fig. 3B). This indicated
that the downregulation of miR-181b sensitized breast cancer cells
to DOX resistance. Furthermore, knockdown of miR-181b re-sensitized
the T-47D-R (DOX-resistant T-47D) cells to DOX, although the
antitumor effect of DOX was not influenced by the transfection of
miR-181b mimics (Fig. 3C). Taken
together, these results indicated that miR-181b may play an
important role in chemoresistance in breast cancer, and that
miR-181b silencing reverses the resistance to DOX treatment.
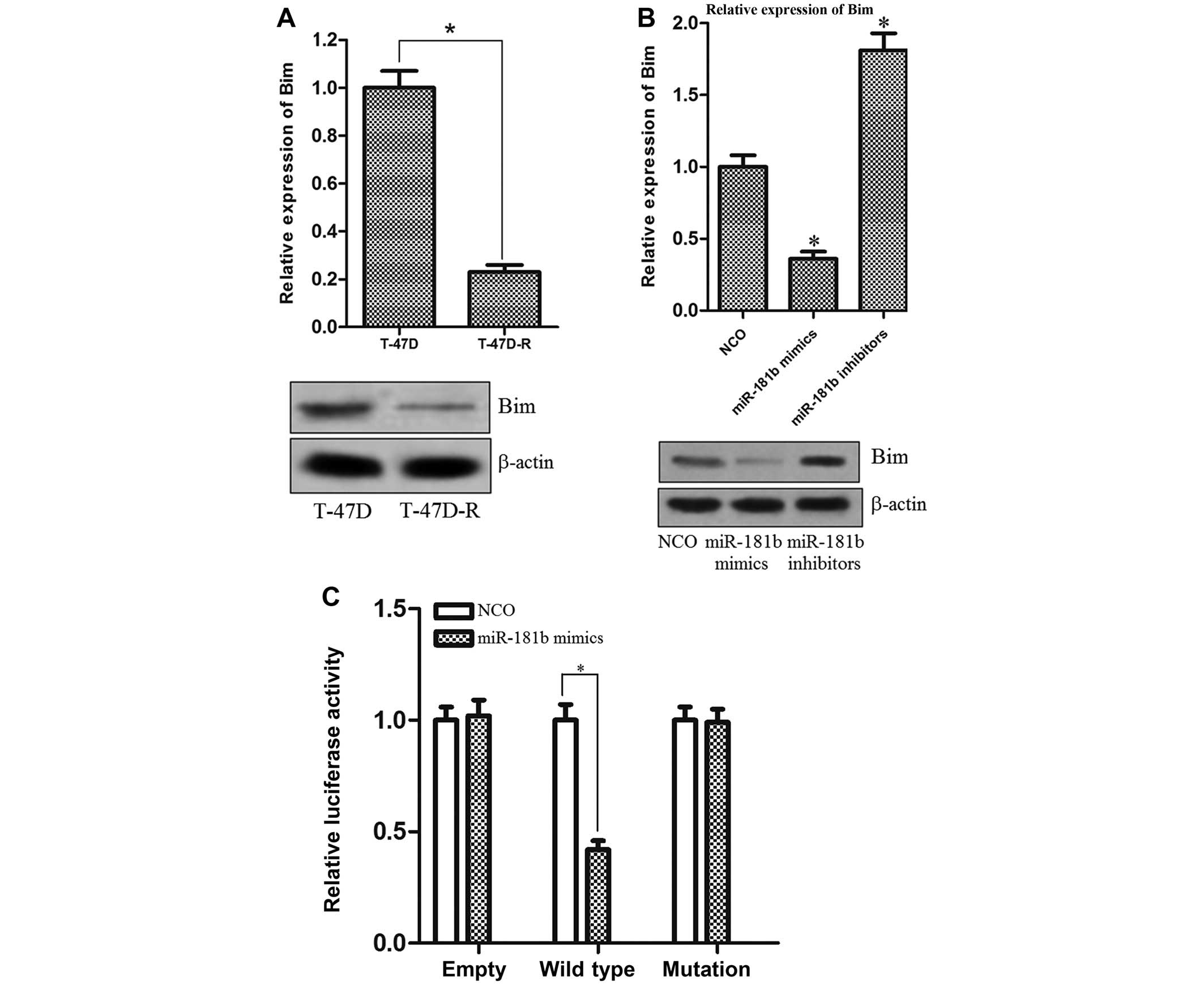
miR-181b in T-47D-R cells regulates the
expression of Bim
In order to understand the underlying mechanism for
the promotion of miR-181b to DOX resistance, we used TargetScan
database (http://www.targetscan.org/) and found
that Bim was a putative target of miR-181b. Notably, the position
474–480 of Bim 3′-UTR was observed to be a complementary site
(5′-…..UGAAUGU….-3′) for the seed region of miR-181b. Furthermore,
we observed that the expression level of Bim was significantly
lower in the T-47D-R cells compared with the parental T-47D cells
(Fig. 4A). Therefore, we inferred
that the T-47D cells became DOX resistant by downregulating Bim
expression, which is a target of miR-181b. To confirm this
speculation, T-47D-R cells were transfected with miR-181b mimics,
miR-181b inhibitors or NCO to alter the miR-181b level. As
expected, a pronounced decrease in both Bim mRNA and protein levels
was observed in the T-47D-R cells by transfection of the miR-181b
mimics. On the contrary, the level of Bim was significantly
upregulated when the miR-181b inhibitors were introduced into the
T-47D-R cells (Fig. 4B). To further
investigate whether Bim is directly targeted by miR-181b, a
luciferase reporter vector was constructed, containing the putative
miR-181b binding sites within the Bim 3′-UTR. The results showed
that the relative luciferase activity of the reporter which
contained wild-type 3′UTR of Bim was significantly inhibited in the
miR-181b group compared with the control group. However, the
mutations in the miR-181b binding site from the Bim 3′-UTR
abolished this effect (Fig. 4C).
Taken together, these results suggested that the expression of Bim
was negatively regulated by miR-181b, which may play an essential
role in the DOX resistance of breast cancer cells.
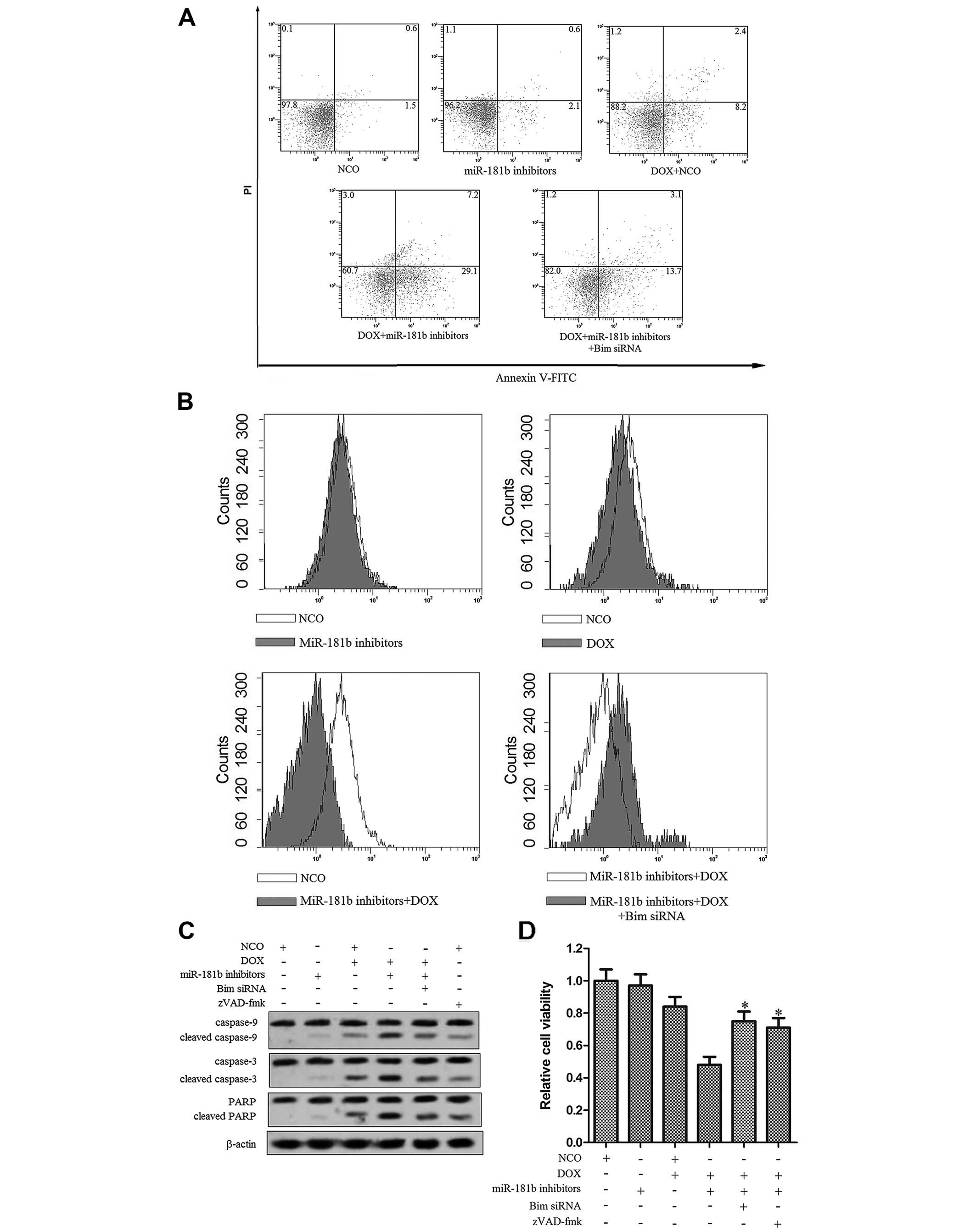
miR-181b inhibitors reverse DOX
resistance through the miR-181b-Bim-MMP-caspase pathway
Our preceding results showed that miR-181b
inhibitors reversed the DOX resistance in T-47D-R cells and that
the Bim gene was the direct target of miR-181b. We, therefore,
investigated the pathway and the relationship between Bim
regulation and the antitumor effect of the treatment with DOX plus
miR-181b inhibitors in T-47D-R cells. As Bim is the key member of
the pro-apoptotic Bcl-2 family proteins (22), we found that the downregulation of
miR-181b by its specific inhibitors significantly increased the
apoptotic rate of the T-47D-R cells treated with DOX, and this
synergistic effect of miR-181b inhibitors was abolished when Bim
siRNA (after transfecion, the expression of Bim was reduced
~5.12-fold, data not shown) was co-transfected (Fig. 5A). The results indicated that
miR-181b regulated the DOX resistance via targeting the Bim gene.
We next observed that although miR-181b inhibitors alone did not
influence the MMP of the T-47D-R cells, they significantly promoted
DOX to damage the mitochondria of the T-47D-R cells. Notably, the
mitochondrial dysfunction caused by miR-181b inhibitors plus DOX
was inhibited by Bim siRNA (Fig.
5B). Since a previous study indicated that intrinsic apoptosis
is activated by mitochondrial dysfunction, mitochondrial-derived
apoptogenic proteins are then released from mitochondria, leading
to the activation of caspase-9 which finally activates caspase-3
(23). Our results demonstrated
that the apoptosis induced by miR-181b inhibitors plus DOX was
caspase-dependent (Fig. 5C).
Finally, using MTT assay, we indicated that both the Bim siRNA and
zVAD-fmk significantly inhibited the cell death induced by DOX
combined with the miR-181b inhibitors in the T-47D-R cells
(Fig. 5D). Our data strongly
suggest the important role of the Bim pathway in reversing the DOX
resistance in breast cancer.
Discussion
Studies have demonstrated that miR-181b may act as
an oncogene in multiple types of cancer. For instance, the
upregulation of the miR-181 family promoted the growth, clonogenic
survival, migration and invasion in hepatocellular carcinoma cells
by targeting TIMP3 (24). It was
also reported that expression levels of the miR-181 family were
elevated in breast, colon and pancreatic cancer (25–27).
Furthermore, miR-181b was found to contribute to the drug
resistance of tamoxifen which is the irreplaceable drug for the
treatment of breast cancer (28).
Although the previous studies indicated that the miR-181b level is
commonly upregulated in many types of tumor, the functions and
targets concerning miR-181b remain unknown. In the present study,
we showed that miR-181b was significantly upregulated in the the
blood of breast cancer patients and in breast cancer cell lines.
Moreover, we also found that the expression level of miR-181b in
breast cancer cells was positively related with cell proliferation
and migration in vitro, suggesting that miR-181b acts as a
tumor promoter in breast cancer.
Chemoresistance is the major limitation for
achieving a satisfactory chemotherapeutic effect, and much effort
has been made to prove that treatment using various miRNAs may
improve the antitumor effect of chemotherapeutic drugs in multiple
tumor types. For instance, the anti-miR-21 oligonucleotide
significantly enhanced the chemosensitivity of glioblastoma cells
to DOX by inducing apoptosis (29).
Enforced expression of miR-26b improved the curative effect of
hepatocellular carcinoma cells to TRAIL by downregulating the
anti-apoptotic gene myeloid cell leukemia-1 (Mcl-1) (30). miR-193b was proven to act as a drug
sensitizer, and promoted the induction of apoptosis following
cisplatin treatment in hepatocellular carcinoma (31). In this context, it was of interest
to examine whether or not miR-181b is associated with
chemoresistance. We found that knockdown of miR-181b re-sensitized
the T-47D-R cells to DOX, significantly decreasing the
IC50 value of DOX. We further found the miR-181b mimics
did not further impair the antitumor effect of DOX. We explained
that the expression of miR-181b in the T-47D-R cells is high enough
to have a biological effect so that the transfection of miR-181b
mimics became unnecessary.
Bcl-2 interacting mediator of cell death [Bim, also
known as B-cell chronic lymphocytic leukemia-lymphoma-like 11
(BCL2L11)], is a member of the Bcl-2 family genes and belongs to
the BH3-only subfamily of Bcl-2 family proteins (32). Bim has emerged as a crucial
regulator of the mitochondrial (intrinsic) apoptotic pathway, which
directly activates the pro-apoptotic function, and binds to all of
the pro-apoptotic Bcl-2 family members to promote cell apoptosis
(33,34). Recently, accumulating evidence shows
that Bim deletion in cancers is associated with a poorer response
to targeted therapy treatment (35,36).
Bim may act as a biomarker to predict the survival of cancer
patients (37). According to our
data, miR-181b inhibitors re-sensitized T-47D-R cells to DOX by
targeting Bim, depending on the upregulation of the cellular level
of Bim in chemoresistant cancer cells. To further explore the
mechanisms, we tested the mitochondrial membrane potential which is
downstream of the Bim pathway, which controls mitochondrial
(intrinsic) apoptosis (38). We
found that regain of Bim mediated by miR-181b inhibitors in T-47D-R
cells significantly promoted DOX to damage the mitochondria,
leading to a decrease in the mitochondrial membrane potential. As a
result, caspase-9 which is a biomarker of intrinsic apoptosis was
activated, following the cleavage of caspase-3 (39).
In conclusion, the present study provides evidence
that miR-181b is a novel oncogene and one important factor for
developing the chemoresistance in breast cancer. The results
suggest that the miR-181b-Bim-MMP-caspase pathway may be a
potential therapeutic target for the chemotherapy of breast cancer
in the future.
Acknowledgments
The present study was supported by the Natural
Science Foundation of Zhejiang Province (no. LY12H29004). The study
was approved by the Ethics Committee of Zhejiang Cancer
Hospital.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weigelt B, Peterse JL and van 't Veer LJ:
Breast cancer metastasis: Markers and models. Nat Rev Cancer.
5:591–602. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jerusalem G, Rorive A and Collignon J:
Chemotherapy options for patients suffering from heavily pretreated
metastatic breast cancer. Future Oncol. 11:1775–1789. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakamura S, Yagata H, Ohno S, Yamaguchi H,
Iwata H, Tsunoda N, Ito Y, Tokudome N, Toi M, Kuroi K, et al:
Multicenter study evaluating circulating tumor cells as a surrogate
for response to treatment and overall survival in metastatic breast
cancer. Breast Cancer. 17:199–204. 2010. View Article : Google Scholar
|
|
5
|
Yao YS, Qiu WS, Yao RY, Zhang Q, Zhuang
LK, Zhou F, Sun LB and Yue L: miR-141 confers docetaxel
chemoresistance of breast cancer cells via regulation of EIF4E
expression. Oncol Rep. 33:2504–2512. 2015.PubMed/NCBI
|
|
6
|
Xu F, Wang F, Yang T, Sheng Y, Zhong T and
Chen Y: Differential drug resistance acquisition to doxorubicin and
paclitaxel in breast cancer cells. Cancer Cell Int. 14:5382014.
View Article : Google Scholar :
|
|
7
|
Shuhendler AJ, Prasad P, Zhang RX, Amini
MA, Sun M, Liu PP, Bristow RG, Rauth AM and Wu XY: Synergistic
nanoparticulate drug combination overcomes multidrug resistance,
increases efficacy, and reduces cardiotoxicity in a
nonimmunocompromised breast tumor model. Mol Pharm. 11:2659–2674.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mohell N, Alfredsson J, Fransson Å,
Uustalu M, Byström S, Gullbo J, Hallberg A, Bykov VJ, Björklund U
and Wiman KG: APR-246 overcomes resistance to DOX and doxorubicin
in ovarian cancer cells. Cell Death Dis. 6:e17942015. View Article : Google Scholar
|
|
9
|
Gao JH, Chen FH, Wang L, Wei H and Meng
SL: YM155 inhibits tumor growth and enhances chemosensitivity to
cisplatin in osteosarcoma. Eur Rev Med Pharmacol Sci. 19:2062–2069.
2015.PubMed/NCBI
|
|
10
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sevignani C, Calin GA, Siracusa LD and
Croce CM: Mammalian microRNAs: A small world for fine-tuning gene
expression. Mamm Genome. 17:189–202. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ambros V: MicroRNA pathways in flies and
worms: Growth, death, fat, stress, and timing. Cell. 113:673–676.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Calin GA, Sevignani C, Dumitru CD, Hyslop
T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M,
et al: Human microRNA genes are frequently located at fragile sites
and genomic regions involved in cancers. Proc Natl Acad Sci USA.
101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qiu F, Xiong JP, Deng J and Xiang XJ:
TRIM29 functions as an oncogene in gastric cancer and is regulated
by miR-185. Int J Clin Exp Pathol. 8:5053–5061. 2015.PubMed/NCBI
|
|
15
|
Tian X, Xu L and Wang P: MiR-191 inhibits
TNF-α induced apoptosis of ovarian endometriosis and endometrioid
carcinoma cells by targeting DAPK1. Int J Clin Exp Pathol.
8:4933–4942. 2015.
|
|
16
|
Toraih EA, Mohammed EA, Farrag S, Ramsis N
and Hosny S: Pilot study of serum nicroRNA-21 as a diagnostic and
prognostic biomarker in Egyptian breast cancer patients. Mol Diagn
Ther. 19:179–190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kaboli PJ, Rahmat A, Ismail P and Ling KH:
MicroRNA-based therapy and breast cancer: A comprehensive review of
novel therapeutic strategies from diagnosis to treatment. Pharmacol
Res. 97:104–121. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
19
|
Czeczuga-Semeniuk E, Bielawski T,
Lemancewicz D, Rusak M and Wołczyński S: Vitamin A family
compounds, estradiol, and docetaxel in proliferation, apoptosis and
immunocytochemical profile of human ovary endometrioid cancer cell
line CRL-11731. Folia Histochem Cytobiol. 47:S127–S135. 2009.
|
|
20
|
Prathapan A, Vineetha VP and Raghu KG:
Protective effect of Boerhaavia diffusa L. against mitochondrial
dysfunction in angiotensin II induced hypertrophy in H9c2
cardiomyoblast cells. PLoS One. 9:e962202014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zahedifard M, Faraj FL, Paydar M, Yeng
Looi C, Hajrezaei M, Hasanpourghadi M, Kamalidehghan B, Abdul Majid
N, Mohd Ali H and Ameen Abdulla M: Synthesis, characterization and
apoptotic activity of quinazolinone Schiff base derivatives toward
MCF-7 cells via intrinsic and extrinsic apoptosis pathways. Sci
Rep. 5:115442015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Frank DO, Dengjel J, Wilfling F,
Kozjak-Pavlovic V, Häcker G and Weber A: The pro-apoptotic BH3-only
protein Bim interacts with components of the translocase of the
outer mitochondrial membrane (TOM). PLoS One. 10:e01233412015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Geserick P, Wang J, Feoktistova M and
Leverkus M: The ratio of Mcl-1 and Noxa determines ABT737
resistance in squamous cell carcinoma of the skin. Cell Death Dis.
5:e14122014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang B, Hsu SH, Majumder S, Kutay H, Huang
W, Jacob ST and Ghoshal K: TGFbeta-mediated upregulation of hepatic
miR-181b promotes hepatocarcinogenesis by targeting TIMP3.
Oncogene. 29:1787–1797. 2010. View Article : Google Scholar :
|
|
25
|
Yan LX, Huang XF, Shao Q, Huang MY, Deng
L, Wu QL, Zeng YX and Shao JY: MicroRNA miR-21 overexpression in
human breast cancer is associated with advanced clinical stage,
lymph node metastasis and patient poor prognosis. RNA.
14:2348–2360. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nakajima G, Hayashi K, Xi Y, Kudo K,
Uchida K, Takasaki K, Yamamoto M and Ju J: Non-coding MicroRNAs
hsa-let-7g and hsa-miR-181b are associated with chemoresponse to
S-1 in colon cancer. Cancer Genomics Proteomics. 3:317–324.
2006.
|
|
27
|
Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner
MR, Frankel WL, Morgan DL, Postier RG, Brackett DJ and Schmittgen
TD: Expression profiling identifies microRNA signature in
pancreatic cancer. Int J Cancer. 120:1046–1054. 2007. View Article : Google Scholar
|
|
28
|
Miller TE, Ghoshal K, Ramaswamy B, Roy S,
Datta J, Shapiro CL, Jacob S and Majumder S: MicroRNA-221/222
confers tamoxifen resistance in breast cancer by targeting
p27Kip1. J Biol Chem. 283:29897–29903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Giunti L, da Ros M, Vinci S, Gelmini S,
Iorio AL, Buccoliero AM, Cardellicchio S, Castiglione F, Genitori
L, de Martino M, et al: Anti-miR21 oligonucleotide enhances
chemosensitivity of T98G cell line to doxorubicin by inducing
apoptosis. Am J Cancer Res. 5:231–242. 2015.PubMed/NCBI
|
|
30
|
Jiang C, Long J, Liu B, Xie X and Kuang M:
Mcl-1 Is a novel target of miR-26b that is associated with the
apoptosis induced by TRAIL in HCC cells. Biomed Res Int.
2015:5727382015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yin W, Nie Y, Zhang Z, Xie L and He X:
miR-193b acts as a cisplatin sensitizer via the caspase-3-dependent
pathway in HCC chemotherapy. Oncol Rep. 34:368–374. 2015.PubMed/NCBI
|
|
32
|
Kivits RA and Furneaux C: BIM: Enabling
sustainability and asset management through knowledge management.
Scientific World Journal. 2013:9837212013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar
|
|
34
|
O'Connor L, Strasser A, O'Reilly LA,
Hausmann G, Adams JM, Cory S and Huang DC: Bim: A novel member of
the Bcl-2 family that promotes apoptosis. EMBO J. 17:384–395. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Faber AC, Corcoran RB, Ebi H, Sequist LV,
Waltman BA, Chung E, Incio J, Digumarthy SR, Pollack SF, Song Y, et
al: BIM expression in treatment-naive cancers predicts
responsiveness to kinase inhibitors. Cancer Discov. 1:352–365.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shao YY, Chang YL, Huang CY, Hsu CH and
Cheng AL: The germline BIM deletion polymorphism is not associated
with the treatment efficacy of sorafenib in patients with advanced
hepatocellular carcinoma. Oncology. 85:312–316. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee JH, Lin YL, Hsu WH, Chen HY, Chang YC,
Yu CJ, Shih JY, Lin CC, Chen KY, Ho CC, et al: Bcl-2-like protein
11 deletion polymorphism predicts survival in advanced
non-small-cell lung cancer. J Thorac Oncol. 9:1385–1392. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Weber K, Harper N, Schwabe J and Cohen GM:
BIM-mediated membrane insertion of the BAK pore domain is an
essential requirement for apoptosis. Cell Reports. 5:409–420. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Park C, Hong SH, Kim GY and Choi YH:
So-Cheong-Ryong-Tang induces apoptosis through activation of the
intrinsic and extrinsic apoptosis pathways, and inhibition of the
PI3K/Akt signaling pathway in non-small-cell lung cancer A549
cells. BMC Complement Altern Med. 15:1132015. View Article : Google Scholar : PubMed/NCBI
|