Introduction
Cellular senescence is irreversible cell cycle
arrest, a process that was originally identified in normal human
fibroblasts (1). It has been
recently shown that various stresses, such as DNA damage, lack of
nutrients, oxidative stress, improper cell contacts and oncogene
activation, could trigger cellular senescence in normal and cancer
cells (2). Thus, cellular
senescence has been accepted as a general biological program in the
last decade.
Senescent cells are metabolically active and exhibit
characteristic flattened, enlarged morphologies with
senescence-associated β-galactosidase activity. Senescent cells
also undergo many molecular changes in gene expression, protein
processing and chromatin organization (3,4). Since
a variety of DNA-damaging agents, including ionizing radiation (IR)
and chemotherapeutic drugs, can prematurely induce cancer cell
senescence, premature senescence might serve as a tumor-suppressing
mechanism as potent as apoptosis both in vitro and in
vivo (5). Senescent cells are
detected in early-stage premalignant lesions in the tissues of
mouse tumor models and human cancer patients, supporting the notion
that cellular senescence is an anticancer barrier in early human
tumorigenesis (6–8).
As described above, mounting evidence supports that
cellular senescence is a strong tumor-suppressing mechanism.
However, senescent cancer cells have multiple facets facilitated by
their altered secretory profiles. Collectively, these changes are
referred to as the senescence-associated secretory phenotype (SASP)
(9). The SASP is comprised of three
major classes of secretory proteins, including chemokines and
cytokines, matrix-remodeling proteases and growth factors (9). Senescent tumor cells promote cancer
progression, stimulate angiogenesis, induce
epithelial-to-mesenchymal transitions, facilitate tissue repair and
contribute to cellular senescence (10,11).
The SASP could also trigger specific innate immune responses, which
ultimately contribute to the elimination of senescent cancer cells
(12,13). Thus, the SASP could have both
beneficial and detrimental effects on the tumor
microenvironment.
Radiotherapy is one of the major regimens used to
treat cancer patients. We previously reported that premature
senescence is efficiently induced by IR treatment in a variety of
carcinoma cell lines and in a human tumor xenograft mouse model
(14–16). To minimize the side effects of
radiotherapy, we must expand our knowledge of the SASP brought on
by IR-induced senescent tumor cells. In the present study, we
listed the top 20 cytokines that were increased in IR-induced
senescent MCF7 cells using cytokine microarray analysis. From this
list, we demonstrated that osteoprotegerin (OPG), midkine (MDK) and
apolipoprotein E3 (ApoE3) can influence the tumor microenvironment
and that OPG is likely a promising target capable of reducing the
side effects associated with radiation therapy.
Materials and methods
Reagents and antibodies
Recombinant human proteins of OPG, MDK and ApoE3
were purchased from Biovision (Milpitas, CA, USA). Phospho-pRb
antibody was purchased from Cell Signaling Technology (Danvers, MA,
USA). p53 antibody was purchased from Leica Biosystems (Wetzlar,
Germany). p21 antibody was purchased from Santa Cruz Biotechnology
(CA, USA). Actin antibody was purchased from ABM (Richmond, BC,
Canada). The neutralizing OPG antibody was purchased from R&D
Systems (Minneapolis, MN, USA). The Matrigel matrix and Transwell
upper chambers were purchased from Corning (Cambridge, MA, USA).
The Ultracel-3K filter was purchased from Millipore (Billerica, MA,
USA). Crystal violet dye was purchased from Sigma-Aldrich (St.
Louis, MO, USA).
Cell culture and irradiation
MCF-7 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) (Welgene, Inc., Daegu, Korea), MDA-MB-231
cells were cultured in RPMI-1640 media (Welgene, Inc.), and human
umbilical vein endothelial cells (HUVECs) were grown in EBM-2 media
(Lonza, Walkersville, MD, USA) supplemented with 10% fetal bovine
serum (FBS) and 1% penicillin and streptomycin (all from Welgene,
Inc.) at 37°C in a 5% CO2 incubator. For irradiation,
cells were exposed to γ-rays with a 137Cs γ-ray source
(Modle 68; JL Shepherd and Associates, Glenwood, CA, USA) at a dose
rate of 200–300 cGy/min.
Senescence-associated β-galactosidase
staining
Cells were washed in phosphate-buffered saline
(PBS), fixed at room temperature (RT) for 3–5 min in 3.7%
formaldehyde, washed with PBS, and incubated at 37°C in 0%
CO2 with fresh staining solution consisting of 1 mg/ml
5-bromo-4-chloro-3-indolyl β-D-galactoside (X-Gal, from a stock
solution of 20 mg/ml in dimethylformamide) in 40 mM citric
acid/sodium phosphate, pH 6.0, 5 mM potassium ferrocyanide, 5 mM
potassium ferricyanide, 150 mM NaCl and 2 mM MgCl2.
Staining was maximal at 12–16 h.
Collection of conditioned media (CM)
MCF7 cells were seeded at 5×104 cells/ml
in 100 mm culture dishes prior to overnight culture and exposed to
either 0 or 6 Gy radiation. Cells were kept in a CO2
incubator for 3 days, washed with PBS three times and incubated in
5 ml of fresh serum-free media without antibiotics. CM were
collected after an additional 24 h of incubation, centrifuged to
remove any residual cells, and filtered with a syringe filter with
a pore size of 0.2 µm. Filtered CM were concentrated 5- or
10-fold using a Centricon-10 concentrator (Millipore) at 4°C. After
collecting CM, the number of cells on the dish was counted and
normalized to the volume of CM used in each experiment.
Sample preparation for cytokine
microarray analysis
Proteins from CM were obtained using a gel matrix
column that was included in the antibody array assay kit (Full Moon
Biosystems, Sunnyvale, CA, USA). The column was vortexed for 5 sec
and hydration-treated for 60 min at RT. After hydration, the column
was centrifuged at 750 × g for 2 min. After centrifugation, the
column was placed into a collection tube, and 100 µl of the
protein sample was transferred into the column and centrifuged at
750 × g for 2 min. The concentration of the purified sample was
measured with the bicinchoninic acid assay (BCA) protein assay kit
(Pierce, Rockford, IL, USA) using a NanoPhotometer™ (Implen, UK),
and the purity of the purified sample was confirmed with an
ultraviolet spectrophotometer.
Cytokine microarray analysis
For each sample, 75 µl of labeling buffer was
added to a 50-µg sample of protein. Subsequently, the sample
was treated with 3 µl of 10 µg/µl biotin/DMF
solution and incubated at RT for 1 h with mixing. To stop the
reaction, the sample was treated with 35 µl of stop reagent
and incubated at RT for 30 min with mixing. The cytokine-profiling
antibody array slide (Full Moon Biosystems) was treated with 30 ml
of blocking solution in a Petri dish and incubated at 55 rpm for 1
h at RT with shaking. After blocking, the slide was rinsed with
Milli-Q grade water. The labeled sample was mixed with 6 ml of
coupling solution. The blocked array slide was incubated in
coupling mixture at 60 rpm for 2 h at RT with shaking in a coupling
dish. After coupling, the slide was washed six times with 30 ml of
washing solution in a Petri dish on a shaker at 55 rpm for 5 min
and rinsed with Milli-Q grade water. Thirty microliters of 0.5
mg/ml Cy3-streptavidin (GE Healthcare, Chalfont St. Giles, UK) was
mixed with 30 ml of detection buffer. The coupled array slide was
incubated with detection mixture in a Petri dish at 55 rpm for 20
min at RT with shaking, washed six times with 30 ml of washing
solution in a Petri dish at 55 rpm for 5 min with shaking and
rinsed with Milli-Q grade water.
Cytokine microarray data acquisition and
analysis
We scanned the slide using a GenePix 4000B scanner
(Molecular Devices, Sunnyvale, CA, USA). The slides were completely
dried and scanned within 24–48 h after drying. The slides were
scanned at 10-µm resolution, with optimal laser power and
PMT. Using GenePix Software (Molecular Devices), grids were applied
to the scanned images, and the pixels were quantified. The obtained
numerical data were analyzed using Genowiz 4.0™ (Ocimum
Biosolutions, Indianapolis, IN, USA). Following analysis, the
protein data were annotated using UniProt DB (www.uniprot.org).
Western blot analysis
To prepare samples for western blot analysis, CM was
collected and boiled with sodium dodecyl sulfate (SDS) sample
buffer [250 mM Tris-HCl, 8% (w/v) SDS, 40% (v/v) glycerol, 8% (v/v)
β-mercapto-ethanol, and 0.02% (w/v) bromophenol blue] and subjected
to SDS-polyacryamide gel electrophoresis. Thereafter, the gel was
transferred to a nitrocellulose membrane for immunoblotting.
Membranes were blocked with 5% (w/v) skim milk in Tris-buffered
saline with 0.1% (v/v) Tween-20 (TBST) solution and incubated with
the appropriate primary and secondary antibodies for 1 h at RT.
After washing, the membranes were subjected to enhanced
chemiluminescence assays (Thermo Fisher Scientific, Waltham, MA,
USA) and exposed to X-ray film (Agfa Gevaert NV, Mortsel, Antwerp,
Belgium) to illuminate the protein bands.
In vitro cell migration and invasion
assays
The Transwell migration and Matrigel invasion assays
were conducted using the methods described by the manufacturers
using modified Boyden chambers with 8-µm pore filter inserts
for 24-well plates (Costar, Cambridge, MA, USA). For the invasion
assay, filters were pre-coated with 10 µl ice cold 0.1%
Matrigel (BD Biosciences, San Jose, CA, USA) in DPBS. Cells
(1×105) in 200 µl of serum-free media were added
to the upper chamber. For the negative and positive control groups,
cells were incubated in serum-free media and media containing 1%
serum, respectively. For the experimental groups, the lower chamber
was filled with 600 µl of either serum-free media containing
1 µg/ml of protein (OPG, MDK, or ApoE3) or CM containing 20
µg/ml of neutralizing OPG antibody. After 16 h of
incubation, the membranes were incubated with 3.7% formaldehyde
solution for 20 min to fix the cells and stained with crystal
violet solution for 30 min. Subsequently the non-invasive cells in
the inserts were removed with cotton swabs. The invasive cells on
the underside of the membrane were counted using a light microscope
(Olympus CKX41; Olympus, Shinjuku, Tokyo, Japan) and
photographed.
Wound-healing assay
MDA-MB-231 cells were dispensed into 12-well plates
at a high density and incubated in complete media in a
CO2 incubator for 16 h. Then, the media were replaced
with serum-free media for an additional 16 h. After scraping the
cell monolayer with a sterile 200-µl pipette tip, the debris
was removed by washing with DPBS, and the wells were refilled with
1 ml of fresh serum-free media. For the negative and positive
control groups, cells were incubated in serum-free media and media
containing 10% serum for 30 h, respectively. For the experimental
groups, cells were incubated in either serum-free media containing
1 µg/ml of recombinant protein (OPG, MDK, or ApoE3) or CM
containing 20 µg/ml of neutralizing OPG antibody for 30 h.
Cells were washed with DPBS, fixed with 3.7% paraformaldehyde for
30 min, and stained with 0.5% (w/v) crystal violet (Sigma-Aldrich).
After washing with water, cells were air dried and inspected with a
light microscope (Olympus CKX41; Olympus). Data are expressed as
percentage of wound healing (WH %) calculated by dividing the
migrated distance by the scratched distance.
Tube formation assay
Each well of a 48-well plate was coated with 100
µl of Matrigel (BD Biosciences) and incubated at 37°C for 30
min. For the negative and positive control groups, gels were
overlaid with HUVECs (4×104) suspended in serum-free
media and media containing 0.5% serum and growth factors (VEGF,
EGF, FGF and IGF), respectively. For the experimental groups, gels
were overlaid with HUVECs (4×104) suspended in media
containing 0.5% serum, growth factors (VEGF, EGF, FGF and IGF), and
1 µg/ml of recombinant protein (OPG, MDK, or ApoE3). After
16 h of incubation, the area of tube growth was photographed under
a microscope. To quantify HUVEC tube formation, the numbers of
branch points and branches were counted, and the tube area was
quantified using Fuji Multi Gauge V2.3 software (Fuji, Tokyo,
Japan).
Statistical analysis
Data are presented as mean ± SD from at least three
separate experiments. Results were considered significant when
P≤0.05 was obtained. All the statistical analyses were performed
using SPSS 19 (IBM® SPSS® Statistics, IBM
Corporation, Somers, NY, USA).
Results
Identification of cytokines from
IR-induced senescent MCF7 cells
To examine cytokines differentially secreted from
IR-induced senescent cancer cells, we exposed MCF7 breast carcinoma
cells to 6 Gy of IR. IR-exposed MCF7 cells exhibited specific
characteristics of cellular senescence, such as enlarged, flattened
morphological changes and positive senescence-associated
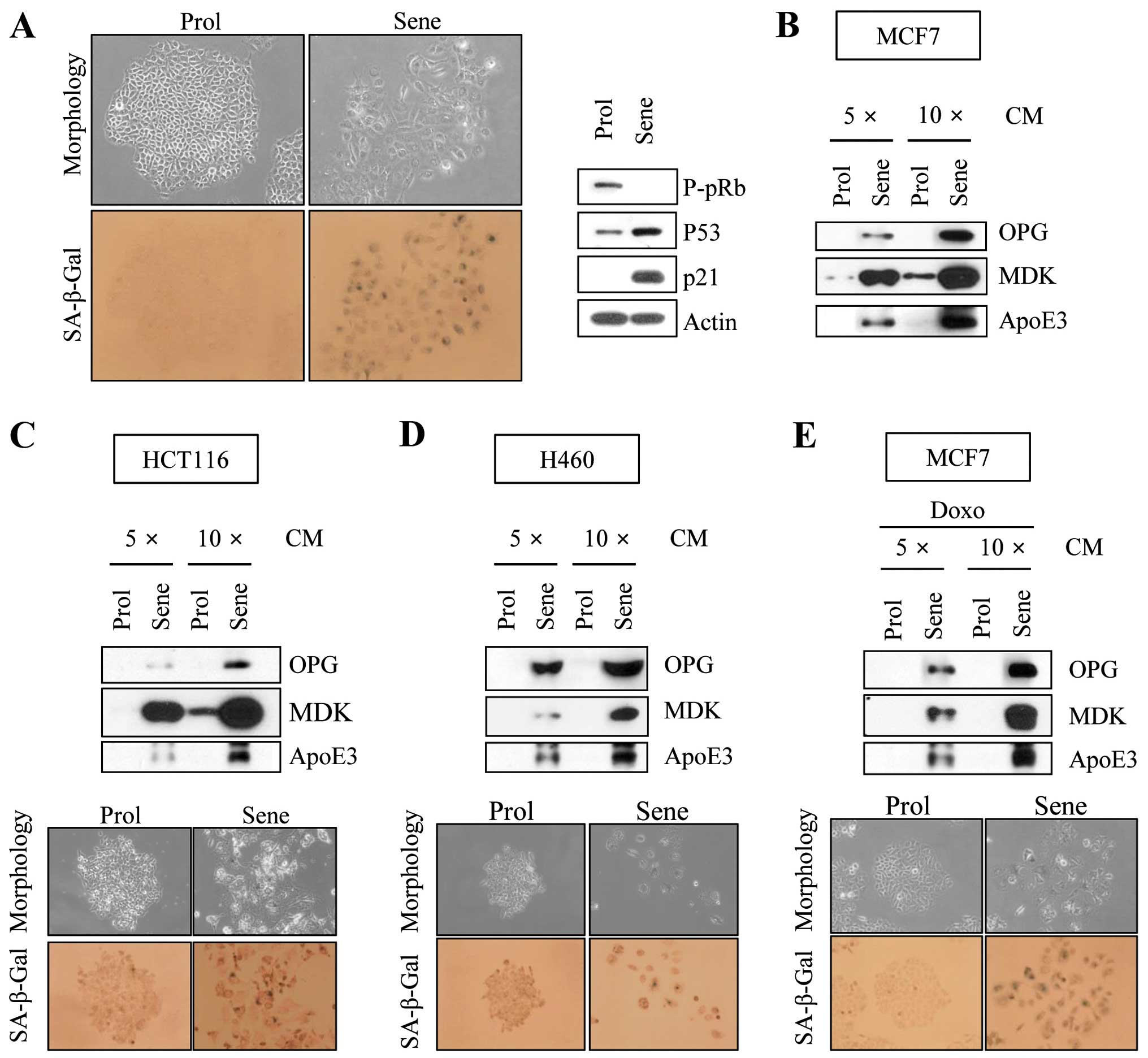
β-galactosidase staining (Fig. 1A).
These cells also showed pRb hypophosphorylation, p53 accumulation
and p21 induction in western blot analyses (Fig. 1A). Together, these results indicate
that 6 Gy of IR efficiently induced premature senescence in MCF7
cells.
To identity cytokines differentially secreted in
proliferating and senescent MCF7 cells, we collected CM from
proliferating and senescent cells and analyzed their respective
cytokine profiles using a cytokine-profiling array. Table I shows the top 20 cytokines that
were increased in IR-induced senescent MCF7 cells. From this list
of cytokines, we selected MDK and ApoE3 for further analysis since
they have not previously been identified as factors that
demonstrate increased secretion in senescent tumor cells. We also
selected OPG, which is reported to increase in secretion in
senescent human fibroblasts and epithelial cells (17). To confirm that their secretion
levels increased in IR-induced senescent MCF7 cells, we performed
western blot analysis after normalizing the volume of CM with the
number of cells on the dish following CM collection. The levels of
MDK, ApoE3 and OPG were elevated in senescent CM compared to
proliferating CM (Fig. 1B). When we
examined the secretion of MDK, ApoE3 and OPG in other types of
cancer cell lines, such as HCT116 colon carcinoma and H460 lung
carcinoma cells, under IR-induced senescence conditions, we
observed elevated levels of these proteins (Fig. 1C and D). Furthermore, we found that
OPG, MDK and ApoE3 were secreted, under anticancer drug,
doxorubicin-induced senescence condition (Fig. 1E).
 | Table IList of the top 20 cytokines with
increased levels of secretion following IR-induced senescence in
MCF7 cells. |
Table I
List of the top 20 cytokines with
increased levels of secretion following IR-induced senescence in
MCF7 cells.
| List of
cytokines | Fold-change
(senescent cells/proliferating cells) | Gene ID |
|---|
| Serpin peptidase
inhibitor clade E member 1 (Serpine1) | 5.15 | P05121 |
| Midkine (MDK) | 4.56 | P21741 |
| Apolipoprotein E3
(ApoE3) | 3.36 | Q13791 |
| Transforming growth
factor β1 (TGF-β1) | 3.16 | P01137 |
| Galectin-3 | 2.99 | P17931 |
| Galectin-1 | 2.61 | P09382 |
| Osteoprotegerin
(OPG) | 2.55 | O00300 |
| Receptor activator
of nuclear factor κB ligand (RANKL) | 2.49 | O14788 |
| Ferritin heavy
polypeptide 1 (FTH1) | 2.14 | P02794 |
| Nicotinamide
phosphoribosyltransferase (NAMPT) | 1.98 | P43490 |
| Insulin-like growth
factor binding protein 5 (IGFBP5) | 1.94 | P24593 |
| Aminoacyl tRNA
synthetase complex-interacting multifunctional protein 1
(AIMP1) | 1.88 | Q12904 |
| Chemokine (C-X3-C
motif) ligand 1 (CX3CL1) | 1.83 | P78423 |
| Fibroblast growth
factor 5 (FGF5) | 1.67 | P12034 |
| Kallikrein-related
peptidase 3 (KLK3) | 1.57 | P07288 |
| Nephroblastoma
overexpressed (NOV) | 1.51 | P48745 |
| Thyroglobulin
(TG) | 1.49 | P01266 |
| Adenomatous
polyposis coli (APC) | 1.48 | P25054 |
| Betacellulin
(BTC) | 1.47 | Q86UF5 |
| Myostatin
(MSTN) | 1.46 | O14793 |
Effects of OPG, MDK, and ApoE3 on
migration, invasion and wound healing in MDA-MB-231 cells
To test the effects of secreted MDK, ApoE3 and OPG
on the tumor microenvironment, we treated MDA-MB-231 breast
carcinoma, MCF-10A breast epithelial and HUVEC endothelial cells
with recombinant human MDK, ApoE3 and OPG proteins and observed
phenotypes related to cell motility, invasiveness and angiogenesis.
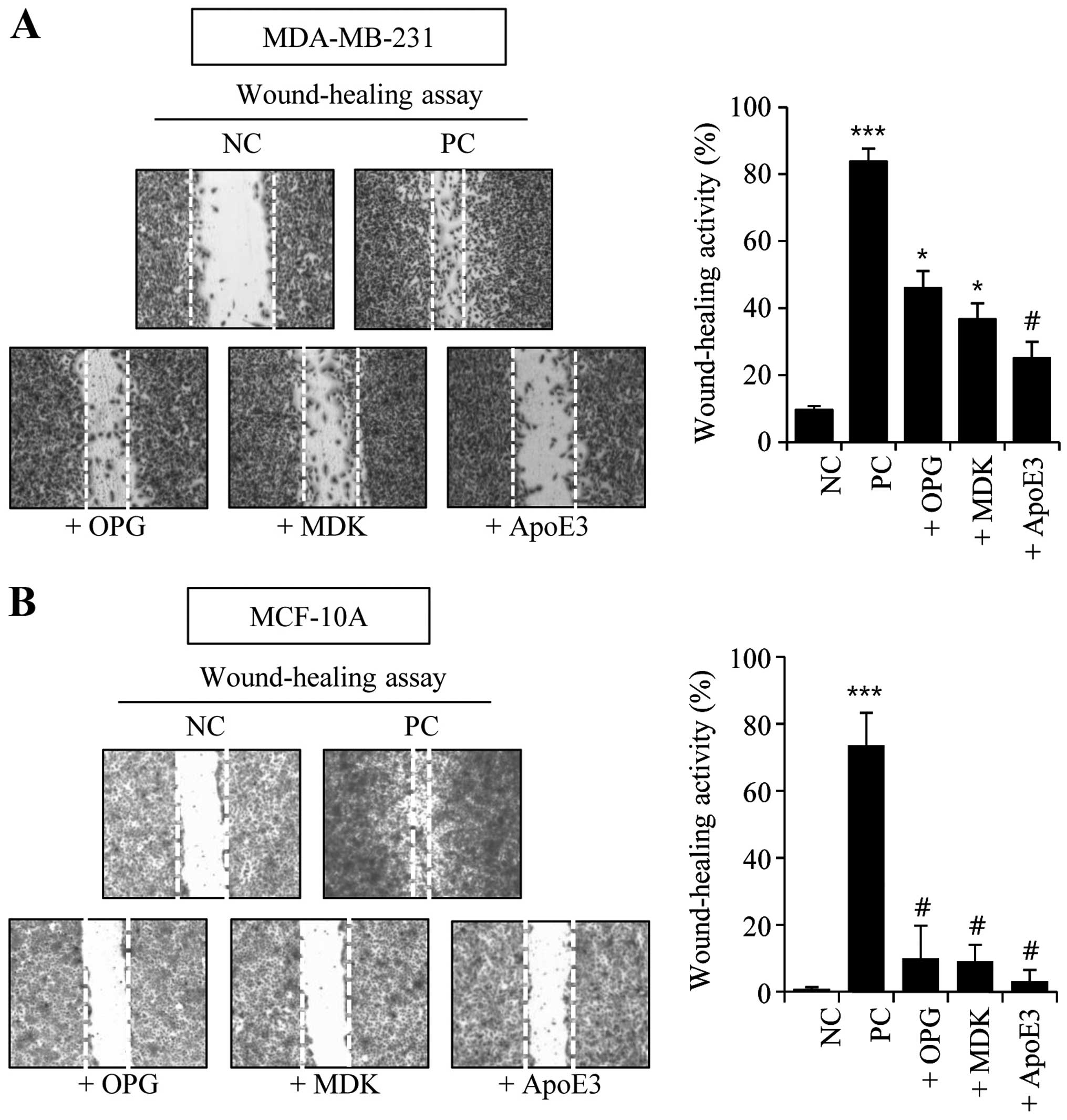
We first examined the effects of these proteins on wound-healing
activity. For the negative and positive control groups, MDA-MB-231
and MCF7 cells were incubated in serum-free media and media
containing 10% serum, respectively. Simultaneously, additional
batches of cells were incubated in serum-free media containing 1
µg/ml of OPG, MDK, or ApoE3 recombinant proteins. OPG-, MDK-
and ApoE3-treated groups clearly showed increased wound-healing
activity in the MDA-MB-231 cells (Fig.
2A). However, the wound-healing activity in OPG-, MDK-, or
ApoE3-treated MCF-10A cells was not significantly changed compared
to the negative control cells (Fig.
2B).
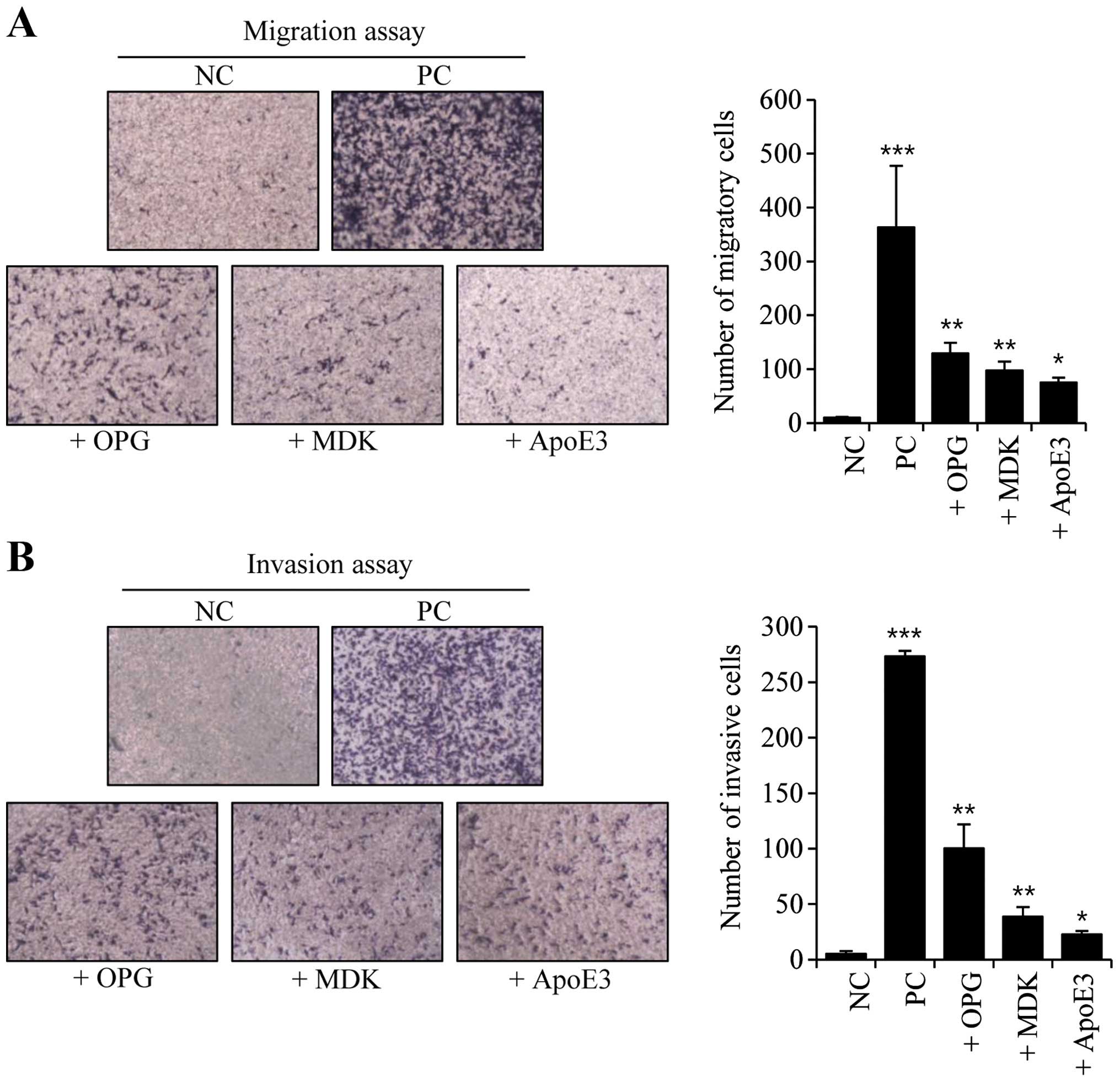
Next, we conducted migration and invasion assays in
OPG-, MDK- and ApoE3-treated MDA-MB-231 cells. Both the migratory
and invasive activities were increased following treatment with
OPG, MDK, or ApoE3 (Fig. 3). These
activities were increased the most in OPG-treated cells, whereas
ApoE3-treated cells had the least impact on migration and
invasiveness (Fig. 3).
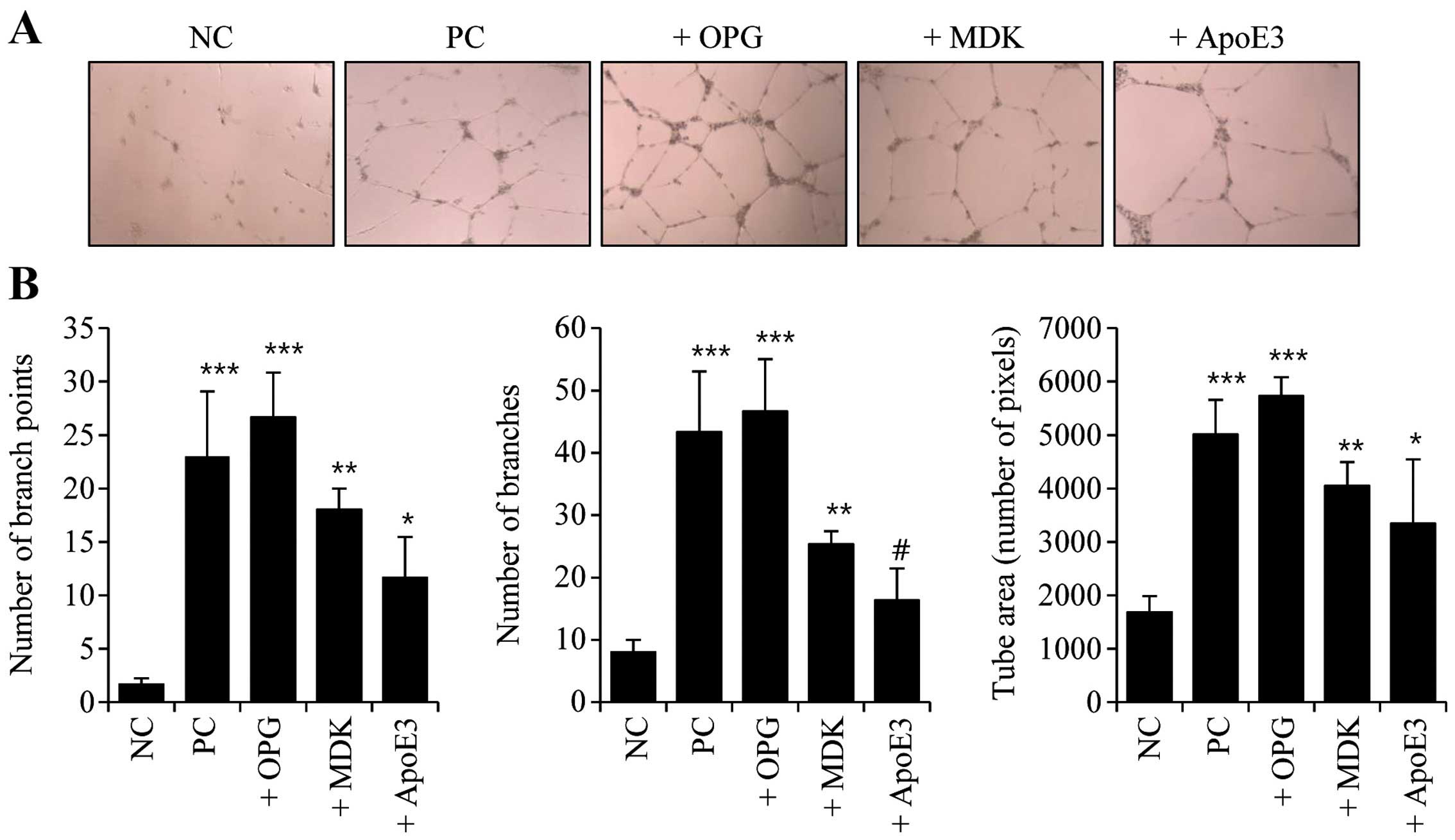
Effects of OPG, MDK and ApoE3 on tube
formation in HUVECs
To identify the possible effects of secreted OPG,
MDK and ApoE3 on angiogenesis in the tumor microenvironment, we
assayed tube formation. For the negative and positive control
groups, gels were overlaid with HUVECs (4×104) suspended
in serum-free media and media containing 0.5% serum and growth
factors (VEGF, EGF, FGF and IGF), respectively. For the OPG-, MDK-,
and ApoE3-treated groups, gels were overlaid with HUVECs
(4×104) suspended in media containing 0.5% serum, growth
factors (VEGF, EGF, FGF and IGF), and 1 µg/ml of OPG, MDK,
or ApoE3 recombinant proteins. Tube-forming activity clearly
increased following treatment with OPG, MDK and ApoE3 (Fig. 4A). The graphs in Fig. 4B show the quantifications of
tube-forming activity in the OPG-, MDK- and ApoE3-treated HUVECs.
OPG-treated cells exhibited the greatest increases in branch point
number, branch number and tube area (Fig. 4). These data revealed that OPG, MDK
and ApoE3 secreted from senescent cancer cells likely affect
angiogenic activity in neighboring endothelial cells.
Effects of the neutralizing OPG antibody
on migration and wound healing in MDA-MB-231 cells
Since OPG was the most effective at increasing wound
healing, invasion and migration in MDA-MB-231 carcinoma cells as
well as tube formation in HUVECs, we hypothesized that OPG is one
of the most detrimental factors to the tumor microenvironment.
Thus, we examined the role of OPG in senescence using CM and a
neutralizing antibody against OPG (Neut OPG Ab). MDA-MB-231 cells
were incubated in CM in both the absence and presence of 20
µg/ml Neut OPG Ab. For the negative and positive control
groups, cells were incubated in serum-free media and media
containing 1% serum, respectively. When we assessed the migratory
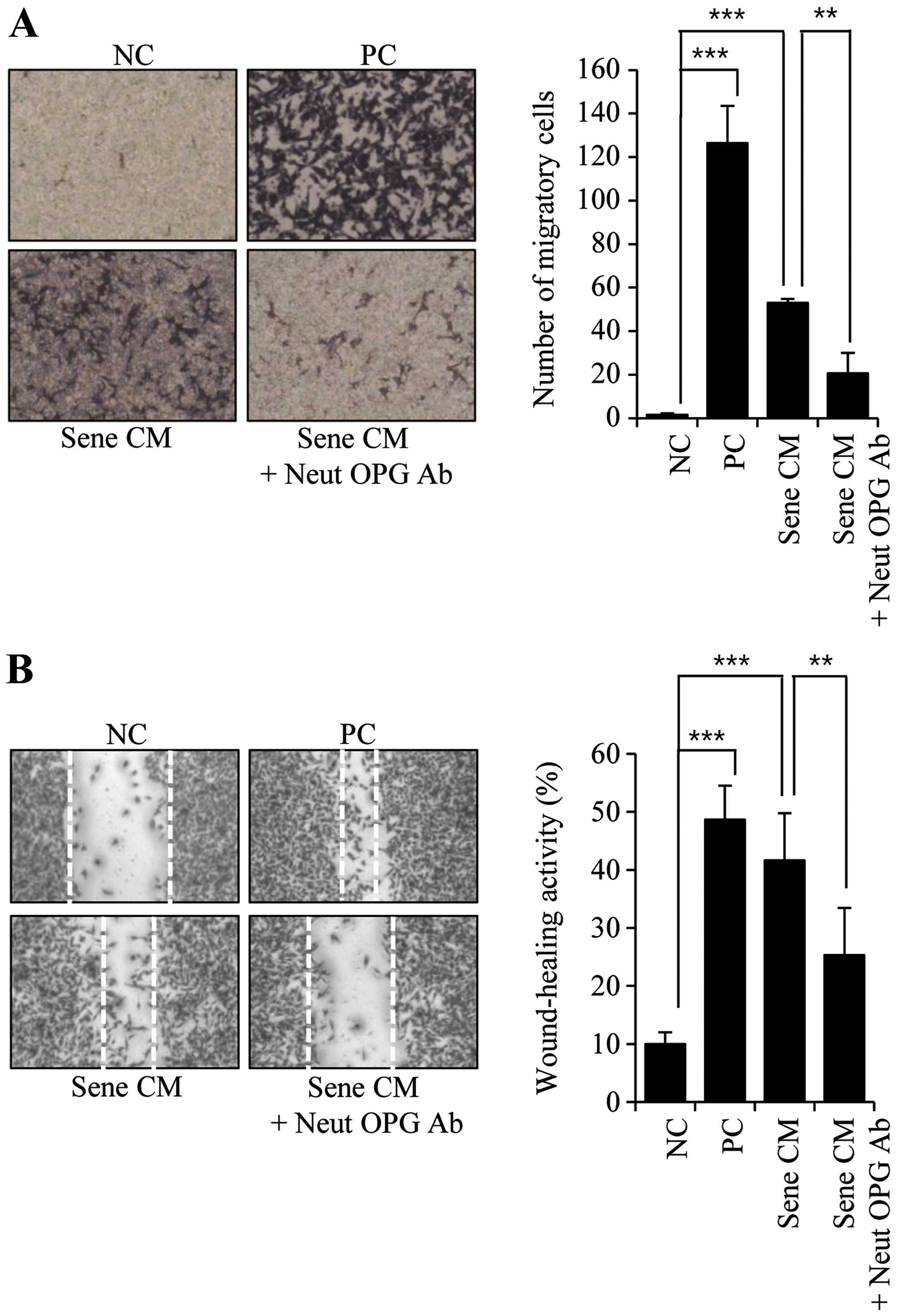
activity of MDA-MB-231 cells in OPG-depleted CM,
senescence-associated migratory activity was diminished (Fig. 5A). Additionally, the presence of the
Neut OPG Ab also diminished senescence-associated wound-healing
activity (Fig. 5B).
Discussion
The development of cancer is not dictated solely by
cell growth. The tumor microenvironment is an indispensable
participant in the neoplastic process, fostering cell proliferation
and survival (18,19). Normal cells can be recruited to the
tumor microenvironment to aid malignant progression (20). Additionally, tumor cells produce
various cytokines and chemokines that stimulate the innate immune
response and other receptors necessary for invasion, migration and
metastasis (18). Multiple
mechanisms of intercellular communication in the tumor
microenvironment can contribute both positive and negative signals
to the tumor (20). Therefore,
multiple therapeutic approaches to target different cell types in
the tumor microenvironment are needed.
Cellular senescence, which is an extremely stable
form of cell cycle arrest, could be triggered by exposures to
various stresses, including telomere attrition, DNA damage,
oxidative stress, lack of nutrition and improper cell contacts
(21). Given that neoplastic
transformation involves events that inhibit senescence, tumor cells
are believed to lose the ability to senesce. However, tumor cells
can be triggered to undergo cellular senescence following
treatments with anticancer drugs, radiation, DNA-damaging agents,
or genetic manipulation (22).
Currently, cellular senescence is recognized as a tumor-suppressing
mechanism. Accumulating evidence has shown that cellular senescence
is as potent as apoptosis at depriving the self-renewal capacity of
cancer cells after radiotherapy and chemotherapy (23–25).
Thus, triggering senescence is considered a key component of
therapeutic interventions necessary for treating cancer, and
potential senescence-inducing small molecules are currently in
development (26).
Senescent cells undergo a variety of characteristic
changes in cellular morphology, cell size, gene expression pattern,
granularity, chromatin configuration and enzyme activity.
Additionally, despite the importance of cellular senescence in
cancer treatments, senescent cells paradoxically secrete factors
that alter the tissue microenvironment and favor tumor growth
(27–30). The SASP allows tumor cells to
proliferate inappropriately, invade surrounding tissues, and
reinforce senescence in cell-autonomous and -nonautonomous manners
(10,27,31–33).
The SASP also cell-nonautonomously affects angiogenesis and the
infiltration of immune cells (23,34,35).
Since a variety of inflammatory cytokines comprise the SASP, there
are at least four major processes by which cellular senescence is
thought to contribute to tumor suppression, aging, tumor promotion
and tissue repair (10,11).
Currently, exposure to IR is being increased not
only for cancer treatments, but also for diagnostic and
occupational reasons. Epithelial, mesenchymal, and tumor cells
derived from theses tissues primarily respond to IR by initiating
senescence (14,36,37). A
long-standing question regarding the bystander effects exhibited by
unirradiated cells raises the possibility that the observed
irradiation effects might be attributed to the SASP mediated by
IR-induced senescent tumor cells (37,38).
The major factors secreted by senescent cells are
inflammatory cytokines, chemokines, growth factors, matrix
metalloproteinases, and their inhibitors, such as IL-1, IL-6, IL-8,
GRO, MCP-1, VEGF, MMP-1, MMP-3, MMP-10 and PAI (34,36).
However, secreted factors from IR-induced senescent cancer cells
and their functions in the tumor micro-environment have not yet
been fully elucidated. We previously identified novel proteins
secreted from IR-induced senescent MCF7 cells using proteomic
techniques and elucidated their roles in the tumor microenvironment
(38). To extend our knowledge
about the secretome of IR-induced senescent cancer cells in this
study, we profiled the top 20 cytokines from IR-induced senescent
cancer cells (Table I) and selected
OPG, MDK and ApoE3 for further investigation by examining their
effects on the tumor microenvironment during radiotherapy.
Furthermore, we verified that the secretions of OPG, MDK, and ApoE3
are commonly increased in MCF7 breast, H460 lung and HCT116 colon
carcinoma cell lines following premature senescence induced by
exposure to 6 Gy IR.
OPG, an essential secreted protein in bone, was
originally identified for the role it plays as a decoy receptor for
the receptor activator of nuclear factor-κB ligand (RANKL)
(39). The RANKL-RANK-OPG system is
important for bone homeostasis, as it regulates osteoclasts
(40). However, OPG acts in various
biological functions due to its ability to bind to additional
ligands in vascular, immune, and tumor tissues. OPG is also known
to be involved in cell survival, cell proliferation and cell
migration (41,42). Specifically, OPG increases
endothelial cell survival and migration and has
metastasis-promoting effects (17,42).
However, the role of OPG in cancer remains unclear. There is
conflicting evidence of indirect antitumoral effects and
pro-tumoral effects mediated by TRAIL inhibition (43). Coppé et al (34) reported that OPG secretion increased
following replicative senescence induced by 10 Gy of IR or various
oncogenes; however, they did not validate the effects of secreted
OPG on the tumor microenvironment. In the present study, we
demonstrated that OPG exhibits pro-tumoral effects evidenced by the
fact that treatment of CM with neutralizing OPG antibodies
successfully diminished the pro-tumoral effects of
senescence-inducing CM. Our results indicate that OPG secreted from
IR-induced senescent cancer cells likely plays a crucial role in
promoting the migration of neighboring cancer cells and that OPG
could be a pivotal therapeutic target that can overcome the
detrimental effects of senescent cancer cells generated by
radiotherapy.
MDK, a heparin-binding growth factor, was originally
identified in embryonal carcinoma cells. MDK expression is
increased during digestion and decreased thereafter (44,45).
Whereas MDK expression is low in normal adult tissues, it is highly
expressed in various cancers, indicating that MDK impacts tumor
development (44,46,47).
Additionally, it has been demonstrated that MDK is involved in many
biological processes, including development, inflammation, tissue
protection, and blood pressure regulation (45).
ApoE has been identified as an essential constituent
of plasma lipoproteins that is responsible for cholesterol
transport and metabolism (48).
ApoE is also involved in several biological processes not directly
related to cholesterol, such as cell proliferation, the
inflammatory response and endocytosis (49,50).
ApoE is produced mainly in the liver, adrenal glands, kidney, and
macrophages, and secreted ApoE serves as an autocrine and paracrine
growth factor (51). ApoE has four
isoforms: ApoE1, E2, E3 and E4, which differ at amino acid residues
112 and 158 (52). The most
prevalent and well-established genetic risk factor associated with
ApoE is Alzheimer's disease. However, differences in the structure
and function between the ApoE isoforms remains an open
question.
To our knowledge, this reports provides the first
evidence that there is increased secretion of MDK and ApoE3 from
senescent tumor cells. We demonstrated that OPG, MDK and ApoE3
impact the tumorigenic, metastatic and angiogenic capabilities of
the cells. Thus, we propose that OPG, MDK and ApoE3 could be
promising therapeutic targets for modulating the pro-tumoral
effects of secreted factors from senescent tumor cells during
radiotherapy. Furthermore, when cancer cells senesce following
treatments with the anticancer drug, doxorubicin, we found that
OPG, MDK and ApoE3 were secreted. These results indicate that OPG,
MDK and ApoE3 could be applied to senescent cancer cells as
therapeutic agents that could diminish the side effects of
radiotherapy and chemotherapy.
The relative concentrations of factors secreted by
senescent tumor cells as well as the concentrations of other
components in the tumor microenvironment likely determine whether
the overall effects of a senescence-inducing secretome is
pro-tumoral or antitumoral. Despite recent advances in our
knowledge of the senescence secretome, many questions still remain.
To minimize the detrimental effects of therapy-induced senescence
or prosenescence therapy, we must validate the most pro-tumoral
secreted factors and develop therapeutics that take advantage of
these validated targets in vitro and in vivo.
Acknowledgments
The present study was supported by the Nuclear
Research and Development Program (grant no. 2012M2B2B1055637) and
the Medical Research Center (MRC) (grant no. 2014009392) of the
National Research Foundation of Korea (NRF) funded by the Korean
government (MSIP) and the Inha University Research Grant (grant no.
INHA-51377).
References
|
1
|
Ben-Porath I and Weinberg RA: The signals
and pathways activating cellular senescence. Int J Biochem Cell
Biol. 37:961–976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu S, Liu S, Wang X, Zhou J, Cao Y, Wang
F and Duan E: The PI3K-Akt pathway inhibits senescence and promotes
self-renewal of human skin-derived precursors in vitro. Aging Cell.
10:661–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Salama R, Sadaie M, Hoare M and Narita M:
Cellular senescence and its effector programs. Genes Dev.
28:99–114. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Campisi J and d'Adda di Fagagna F:
Cellular senescence: When bad things happen to good cells. Nat Rev
Mol Cell Biol. 8:729–740. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Deng Y, Chan SS and Chang S: Telomere
dysfunction and tumour suppression: The senescence connection. Nat
Rev Cancer. 8:450–458. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Michaloglou C, Vredeveld LC, Soengas MS,
Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi
WJ and Peeper DS: BRAFE600-associated senescence-like cell cycle
arrest of human naevi. Nature. 436:720–724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Braig M, Lee S, Loddenkemper C, Rudolph C,
Peters AH, Schlegelberger B, Stein H, Dörken B, Jenuwein T and
Schmitt CA: Oncogene-induced senescence as an initial barrier in
lymphoma development. Nature. 436:660–665. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun P, Yoshizuka N, New L, Moser BA, Li Y,
Liao R, Xie C, Chen J, Deng Q, Yamout M, et al: PRAK is essential
for ras-induced senescence and tumor suppression. Cell.
128:295–308. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Naylor RM, Baker DJ and van Deursen JM:
Senescent cells: A novel therapeutic target for aging and
age-related diseases. Clin Pharmacol Ther. 93:105–116. 2013.
View Article : Google Scholar
|
|
10
|
Coppé JP, Desprez PY, Krtolica A and
Campisi J: The senescence-associated secretory phenotype: The dark
side of tumor suppression. Annu Rev Pathol. 5:99–118. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Campisi J: Cellular senescence: Putting
the paradoxes in perspective. Curr Opin Genet Dev. 21:107–112.
2011. View Article : Google Scholar :
|
|
12
|
Kang TW, Yevsa T, Woller N, Hoenicke L,
Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova
A, et al: Senescence surveillance of pre-malignant hepatocytes
limits liver cancer development. Nature. 479:547–551. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xue W, Zender L, Miething C, Dickins RA,
Hernando E, Krizhanovsky V, Cordon-Cardo C and Lowe SW: Senescence
and tumour clearance is triggered by p53 restoration in murine
liver carcinomas. Nature. 445:656–660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Byun HO, Han NK, Lee HJ, Kim KB, Ko YG,
Yoon G, Lee YS, Hong SI and Lee JS: Cathepsin D and eukaryotic
translation elongation factor 1 as promising markers of cellular
senescence. Cancer Res. 69:4638–4647. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jung SH, Lee HC, Yu DM, Kim BC, Park SM,
Lee YS, Park HJ, Ko YG and Lee JS: Heparan sulfation is essential
for the prevention of cellular senescence. Cell Death Differ. Aug
7–2015.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim BC, Yoo HJ, Lee HC, Kang KA, Jung SH,
Lee HJ, Lee M, Park S, Ji YH, Lee YS, et al: Evaluation of
premature senescence and senescence biomarkers in carcinoma cells
and xenograft mice exposed to single or fractionated irradiation.
Oncol Rep. 31:2229–2235. 2014.PubMed/NCBI
|
|
17
|
Weichhaus M, Segaran P, Renaud A, Geerts D
and Connelly L: Osteoprotegerin expression in triple-negative
breast cancer cells promotes metastasis. Cancer Med. 3:1112–1125.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vakkila J and Lotze MT: Inflammation and
necrosis promote tumour growth. Nat Rev Immunol. 4:641–648. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Joyce JA: Therapeutic targeting of the
tumor microenvironment. Cancer Cell. 7:513–520. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zender L and Rudolph KL: Keeping your
senescent cells under control. Aging (Albany NY). 1:438–441.
2009.
|
|
22
|
Schmitt CA: Cellular senescence and cancer
treatment. Biochim Biophys Acta. 1775:5–20. 2007.
|
|
23
|
de Visser KE, Eichten A and Coussens LM:
Paradoxical roles of the immune system during cancer development.
Nat Rev Cancer. 6:24–37. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gewirtz DA, Holt SE and Elmore LW:
Accelerated senescence: An emerging role in tumor cell response to
chemotherapy and radiation. Biochem Pharmacol. 76:947–957. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ramkumar C, Kong Y, Trabucco SE, Gerstein
RM and Zhang H: Smurf2 regulates hematopoietic stem cell
self-renewal and aging. Aging Cell. 13:478–486. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nardella C, Clohessy JG, Alimonti A and
Pandolfi PP: Pro-senescence therapy for cancer treatment. Nat Rev
Cancer. 11:503–511. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Velarde MC, Demaria M and Campisi J:
Senescent cells and their secretory phenotype as targets for cancer
therapy. Interdiscip Top Gerontol. 38:17–27. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rodier F, Coppé JP, Patil CK, Hoeijmakers
WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR and Campisi
J: Persistent DNA damage signalling triggers senescence-associated
inflammatory cytokine secretion. Nat Cell Biol. 11:973–979. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ren JL, Pan JS, Lu YP, Sun P and Han J:
Inflammatory signaling and cellular senescence. Cell Signal.
21:378–383. 2009. View Article : Google Scholar
|
|
30
|
Novakova Z, Hubackova S, Kosar M,
Janderova-Rossmeislova L, Dobrovolna J, Vasicova P, Vancurova M,
Horejsi Z, Hozak P, Bartek J, et al: Cytokine expression and
signaling in drug-induced cellular senescence. Oncogene.
29:273–284. 2010. View Article : Google Scholar
|
|
31
|
Orjalo AV, Bhaumik D, Gengler BK, Scott GK
and Campisi J: Cell surface-bound IL-1alpha is an upstream
regulator of the senescence-associated IL-6/IL-8 cytokine network.
Proc Natl Acad Sci USA. 106:17031–17036. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cichowski K and Hahn WC: Unexpected pieces
to the senescence puzzle. Cell. 133:958–961. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jiang H, Schiffer E, Song Z, Wang J,
Zürbig P, Thedieck K, Moes S, Bantel H, Saal N, Jantos J, et al:
Proteins induced by telomere dysfunction and DNA damage represent
biomarkers of human aging and disease. Proc Natl Acad Sci USA.
105:11299–11304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz
DP, Goldstein J, Nelson PS, Desprez PY and Campisi J:
Senescence-associated secretory phenotypes reveal
cell-nonautonomous functions of oncogenic RAS and the p53 tumor
suppressor. PLoS Biol. 6:2853–2868. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nickoloff BJ, Lingen MW, Chang BD, Shen M,
Swift M, Curry J, Bacon P, Bodner B and Roninson IB: Tumor
suppressor maspin is up-regulated during keratinocyte senescence,
exerting a paracrine antiangiogenic activity. Cancer Res.
64:2956–2961. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vávrová J and Rezáčová M: The importance
of senescence in ionizing radiation-induced tumour suppression.
Folia Biol (Praha). 57:41–46. 2011.
|
|
37
|
Sabin RJ and Anderson RM: Cellular
Senescence - its role in cancer and the response to ionizing
radiation. Genome Integr. 2:72011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han NK, Kim BC, Lee HC, Lee YJ, Park MJ,
Chi SG, Ko YG and Lee JS: Secretome analysis of ionizing
radiation-induced senescent cancer cells reveals that secreted RKIP
plays a critical role in neighboring cell migration. Proteomics.
12:2822–2832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Baud'huin M, Duplomb L, Teletchea S,
Lamoureux F, Ruiz-Velasco C, Maillasson M, Redini F, Heymann MF and
Heymann D: Osteoprotegerin: Multiple partners for multiple
functions. Cytokine Growth Factor Rev. 24:401–409. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Walsh MC and Choi Y: Biology of the
RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol.
5:5112014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dougall WC: Molecular pathways:
Osteoclast-dependent and osteoclast-independent roles of the
RANKL/RANK/OPG pathway in tumorigenesis and metastasis. Clin Cancer
Res. 18:326–335. 2012. View Article : Google Scholar
|
|
42
|
Reid PE, Brown NJ and Holen I: Breast
cancer cells stimulate osteoprotegerin (OPG) production by
endothelial cells through direct cell contact. Mol Cancer.
8:492009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lamoureux F, Moriceau G, Picarda G,
Rousseau J, Trichet V and Rédini F: Regulation of osteoprotegerin
pro- or anti-tumoral activity by bone tumor microenvironment.
Biochim Biophys Acta. 1805:17–24. 2010.
|
|
44
|
Kishida S, Mu P, Miyakawa S, Fujiwara M,
Abe T, Sakamoto K, Onishi A, Nakamura Y and Kadomatsu K: Midkine
promotes neuroblastoma through Notch2 signaling. Cancer Res.
73:1318–1327. 2013. View Article : Google Scholar
|
|
45
|
Kishida S and Kadomatsu K: Involvement of
midkine in neuroblastoma tumourigenesis. Br J Pharmacol.
171:896–904. 2014. View Article : Google Scholar :
|
|
46
|
Yao J, Li WY, Li SG, Feng XS and Gao SG:
Midkine promotes perineural invasion in human pancreatic cancer.
World J Gastroenterol. 20:3018–3024. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hao H, Maeda Y, Fukazawa T, Yamatsuji T,
Takaoka M, Bao XH, Matsuoka J, Okui T, Shimo T, Takigawa N, et al:
Inhibition of the growth factor MDK/midkine by a novel small
molecule compound to treat non-small cell lung cancer. PLoS One.
8:e710932013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen YC, Pohl G, Wang TL, Morin PJ,
Risberg B, Kristensen GB, Yu A, Davidson B and Shih IeM:
Apolipoprotein E is required for cell proliferation and survival in
ovarian cancer. Cancer Res. 65:331–337. 2005.PubMed/NCBI
|
|
49
|
Li Y, Cam J and Bu G: Low-density
lipoprotein receptor family: Endocytosis and signal transduction.
Mol Neurobiol. 23:53–67. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lynch JR, Morgan D, Mance J, Matthew WD
and Laskowitz DT: Apolipoprotein E modulates glial activation and
the endogenous central nervous system inflammatory response. J
Neuroimmunol. 114:107–113. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Su WP, Chen YT, Lai WW, Lin CC, Yan JJ and
Su WC: Apolipoprotein E expression promotes lung adenocarcinoma
proliferation and migration and as a potential survival marker in
lung cancer. Lung Cancer. 71:28–33. 2011. View Article : Google Scholar
|
|
52
|
Wolf AB, Valla J, Bu G, Kim J, LaDu MJ,
Reiman EM and Caselli RJ: Apolipoprotein E as a
β-amyloid-independent factor in Alzheimer's disease. Alzheimers Res
Ther. 5:382013. View Article : Google Scholar
|