Introduction
Hepatocyte cell adhesion molecule (hepaCAM), also
known as GliaCAM, is located on chromosome 11q24 and encodes a
putative Ig-like cell adhesion molecule with 416 amino acids. It is
a newly identified cell adhesion molecule which belongs to the
immunoglobulin superfamily (1,2). It
was discovered in normal liver tissues, but is decreased during the
development of human hepatocellular carcinoma (3). The loss of hepaCAM in cancer could
help to promote tumorigenesis. Overexpression of hepaCAM in cancer
cells was found to inhibit cell growth and induce cellular
senescence and differentiation (4,5). Our
previous studies showed that exogenous hepaCAM inhibits renal cell
growth by arresting cells at the G0/G1 phase and promotes c-myc
degradation (6,7). We also discovered that overexpression
of hepaCAM inhibited the cell proliferation of human bladder
carcinoma (8). These findings
suggest that hepaCAM acts as a tumor-suppressor gene. Nevertheless,
little is known about the mechanisms of low hepaCAM expression in
bladder cancer.
Abnormal hypermethylation in the promoter plays a
crucial role in cancer by silencing tumor-suppressor genes.
Particularly, downregulation of many tumor-suppressor genes and DNA
repair genes, such as p16, p15, Rb, VHL, E-cadherin, GSTP1, MGMT
and hMLH1, is associated with aberrant methylation in cancer cell
lines and primary tumors (9). In
mammalians, DNA methylation refers to the addition of a methyl
group to the cytosine ring of those cytosines that precede a
guanosine (referred to as CpG dinucleotides) to form
5-methylcytosine (10), which is
catalyzed by three different enzymes, DNMT1, DMNT3A and DNMT3B.
DNMT3A and DNMT3B are most likely responsible for de novo
cytosine methylation at previously unmethylated CpG sites, whereas
the maintenance methyltransferase DNMT1 copies pre-existing
methylation patterns onto the new DNA strand during DNA replication
(11,12).
Abundant evidence shows that DNA methyltransferases
(DNMTs) are overexpressed in various types of cancers (13–15).
DNMTs have become valuable therapeutical targets of cancers by the
use of DNMT inhibitors (DNMTi) (16). 5-Azacytidine (AZAC) and
5-aza-2-deoxycytidine (decitabine), characterized as DNMT
inhibitors, were initially used as antimetabolites and cytotoxic
agents in phase I/II studies of malignancies (17,18).
Meanwhile, aberrant hypermethylation of DNA can be reverted by
DNMTi. We hypothesized that overexpression of DNMT3A/3B may also
contribute to hepaCAM silencing in bladder cancer.
In the present study, we detected the expression of
DNMT3A/3B and hepaCAM in bladder cancer tissues. There was a
negative linear correlation between DNMT3A/3B and hepaCAM
expression in the same bladder carcinoma patients. High expression
of DNMT3A/3B and aberrant hypermethylation in promoter CpG islands
of the hepaCAM gene were found in bladder cancer cell lines.
Furthermore, we found that downregulation of DNMT3A/3B expression,
after treatment with AZAC, reversed the hypermethylation and
expression of hepaCAM. Furthermore, AZAC inhibited the growth of
bladder cancer in vitro and in vivo, providing a new
insight into the therapeutic strategy of bladder cancer.
Materials and methods
Tissue specimens
Thirty-one urothelial carcinoma samples and 22
corresponding adjacent tissues were collected from patients who
underwent total cystectomy. Nine patients were treated with
transurethral resection of bladder carcinoma but no adjacent tissue
was obtained. Patients were enrolled at the Department of Urology
at the First Affiliated Hospital of Chongqing Medical University.
All tissue specimens were confirmed to be bladder cancer or normal
histologically, and histological grade and stage were determined
according to UICC guidelines. Written informed consent was received
from all participants. This study was approved by the Ethics
Committee of Chongqing Medical University. All tissue specimens
were stored at −80°C before the experiment.
Immunohistochemistry
The assay was performed as described previously
(19). In brief, paraffin
wax-embedded tissue sections were dewaxed, rehydrated and
microwaved for 30 min in sodium citrate buffer to retrieve antigen
epitopes. Endogenous peroxidase activity was suppressed by 3%
H2O2 and blocked by goat serum 5% BSA.
Diluted primary polyclonal rabbit antibodies against hepaCAM
(ProteinTech, China), DNMT3A and DNMT3B (Immunoway Biotechnology,
China) were added and left at 4°C overnight. As secondary reagents,
we used biotin-labeled anti-IgG and avidin-biotin horseradish
peroxidase complex, followed by staining with the chromogen
diaminobenzidine (Zhongshan, China) until a brown color developed.
Slides were counterstained with Mayer's hematoxylin and
differentiated in a solution containing 1% hydrochloric acid and
99% ethanol. Cell nuclei were stained blue using lithium carbonate.
Sections were dehydrated and a transparent coverslip was added to
enable observation by microscopy.
Cells and culture
Human bladder cancer cell lines (T24, EJ and BIU-87)
were obtained from the American Type Culture Collection (ATCC;
USA). All the cells were cultured in RPMI-1640 medium supplemented
with 10% fetal bovine serum (both from Gibco, USA) under standard
culture conditions (5% CO2 at 37°C).
MTT assay
To determine the optimal drug doses, bladder cancer
cells (T24, EJ and BIU-87) were seeded in 96-well plates at a
density of 1×104 cells/well for 12 h and treated with
5-azacytidine (Sigma, USA) at different concentrations (0.5, 1, 2,
3, 4, 5, 6 and 7 µg/ml). Three plates were seeded, and the
cells were cultured for 24, 48, 72 and 96 h, respectively. Before
removal from the incubator, the cells were incubated with 20
µl 0.5 mg/ml MTT (Sigma) for an additional 4 h. Culture
medium was discarded, and 150 µl dimethylsulfoxide (DMSO;
Sigma) was added to dissolve the formazan crystals. The absorbance
was measured in a microplate reader at an optical density (OD) of
492 nm. Inhibition rate was calculated using the formula: IR = (1 −
experiment group/control group) × 100%.
CCK-8 assay
To evaluate the cell viability, the CCK-8 Kit
(Beyotime, China) was applied according to the manufacturer's
instructions. The results were repeated in triplicate. The OD of
the untreated controls was measured as 100% survival.
Colony formation assay
Bladder cancer cells were seeded into 6-well plates
at a density of 300 cells/well. After a 48-h incubation, cells were
treated with AZAC or DMSO. After 13 days, the cells were fixed with
methanol for 20 min and stained with crystal violet. Colonies were
observed and images were captured under a microscope.
Flow cytometric assay
Bladder cancer cells (1×106) treated with
AZAC or DMSO were cultured in 6-well plates for 48 h. The cells
were collected in cold phosphate-buffered solution (PBS), fixed in
70% ethanol, and stored at 4°C for subsequent cell cycle analysis.
Procedures for testing the cell cycle have been previously
described (7).
Methylation-specific polymerase chain
reaction assay
Genomic DNA was extracted from bladder cancer cells
according to the instructions provided in the TIANamp Genomic DNA
kit (Tiangen Biotech, China). The bisulfite modification procedures
were carried out using EZ DNA Methylation-Gold™ kit (Zymo Research,
USA). Methylation-specific polymerase chain reaction (MSP) was
performed to detect the methylation status of the hepaCAM gene (M,
methylation status; U, unmethylation status). The PCR conditions
were as follows: pre-denaturation at 98°C for 10 min, denaturation
at 95°C for 10 sec; annealing at 58.4°C (U)/61.8°C (M) for 60 sec;
extension at 72°C for 30 sec, final extension at 72°C for 5 min.
Primer sequences were: hepaCAM(M) forward, AGAATTCGGTTTCGGAGTTTC
and reverse, CTAAACGACGAC GAATATATCCG; and hepaCAM(U) forward,
AGAATTTGGTTTTGGAGTTTTGA and reverse, CCTAAACAACAACAAATATATCCAAC.
PCR products were separated on 3% agarose gel.
Reverse transcription and RT-PCR
Total RNA was isolated from bladder cancer cells
using RNAiso Plus (Takara, Japan) according to the manufacturer's
instructions. Complementary DNA was synthesized using a reverse
transcription kit (Takara) according to the manufacturer's
protocol. The PCR conditions consisted of predenaturing at 95°C for
5 min, denaturing at 95°C for 10 sec, annealing for 50 sec,
extension at 72°C for 1 min, a total of 35 cycles from denaturing,
and a final extension at 72°C for 5 min. Primer sequences were as
follows: hepaCAM forward, TACTGTAGATGTGCCCATTTCG and reverse,
CTTCTGGTTTCAGGCGGTC; DNMT3A forward, GGCGTTAGTGACAAGAGGG and
reverse, TGGACTGGG AAACCAAATA; DNMT3B forward, CAAAGAGTTGGGC
ATAAAGG and reverse, GCGTGAGTAATTCAGCAGGT; and β-actin forward,
CACCACACCTTCTACAATGAGC and reverse, GTGATCTCCTTCTGCATCCTGT. The
β-actin gene was used as an internal standard. All RT-PCR reactions
were executed in triplicate. Each PCR product was
electrophoretically tested on 1.5% agarose gel.
Protein extraction and western blot
analysis
Total protein extraction from cells and tissues was
performed using RIPA buffer supplemented with protease inhibitor
(PMSF) and phosphatase inhibitors (NaF and
Na3VO4) (Roche, Switzerland). The BCA Protein
Assay kit (Beyotime) was used to detect the quantity of each sample
according to the manufacturer's instructions. Proteins (100
µg) were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (Invitrogen, USA). Then
the proteins were electrotransferred onto polyvinylidene difluoride
membranes (Millipore, USA). The membranes were blocked with 5% skim
milk at room temperature for 1 h and then incubated with the
primary antibody against hepaCAM, DNMT3A, DNMT3B and β-actin
(Zhongshan, China) overnight at 4°C. Membranes were washed twice
with TBST and once with TBS, and then incubated with the
HRP-conjugated secondary antibody for 1 h at room temperature.
Proteins were visualized using a chemiluminescence (ECL) reagent
(Millipore, Billerica, MA, USA), and the densities of the bands
were quantified and normalized to that of β-actin by Quantity One
software. The experiments were performed as described previously
(20).
Tumor model
Nude mice (4–6 weeks of age) were purchased from the
Animal Institute of the Chinese Medical Academy (Beijing, China).
Viable EJ cells (3×106) resuspended in PBS were injected
into the right flanks of 8 male nude mice. The mice were randomized
into two groups: group A (DMSO group) and group B (AZAC group)
after 2 weeks. Subsequently, AZAC was injected into mice in group B
and DMSO in group A every three days for 6 times. The weight of
each nude mice was determined daily for 3 weeks. After 3 weeks, all
mice were sacrificed. The xenograft tumor weight was measured at
the terminal time, and tumor tissues were resected for hematoxylin
and eosin (H&E) staining and immunohistochemical examination of
hepaCAM, DNMT3A and DNMT3B. All animal experiments were approved by
the Institutional Animal Care and Use Committee (IACUC) of
Chongqing Medical University.
Statistical analysis
All statistical analyses were performed with SPSS
19.0 software using paired-samples t-test. Data are shown as mean ±
SD, and P<0.05 served as the criterion for statistical
significance.
Results
High expression of DNMT3A/3B and low
expression of hepaCAM are noted in the bladder cancer tissues
To investigate whether there was any correlation
between DNMT3A/3B and hepaCAM expression, we used anti-DNMT3A/3B
and anti-hepaCAM antibodies to detect the expression of DNMT3A/3B
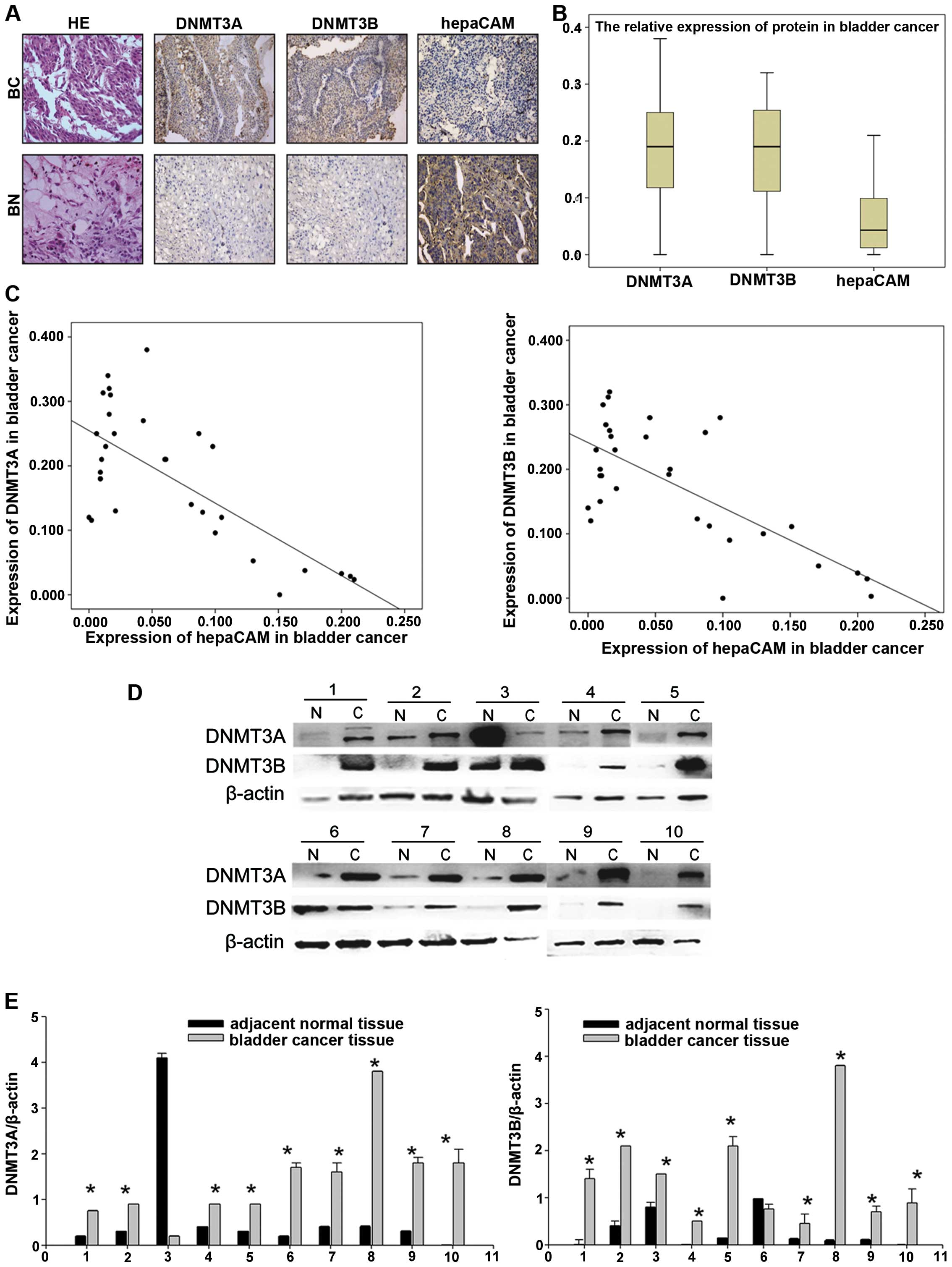
and hepaCAM in the bladder cancer and adjacent tissues. The results
showed that DNMT3A/3B was strongly expressed in the bladder cancer
tissues, but weakly expressed in the adjacent tissues. However, in
regards to hepaCAM, the expression levels in the adjacent tissues
were higher than levels in the cancer tissues (Fig. 1A and B). We made use of the mean
density for estimating the protein expression levels of DNMT3A/3B
and hepaCAM. The results revealed a negative linear correlation
between DNMT3A/3B and hepaCAM expression in the same patients
according to Pearson's analysis (r=−0.7176/−0.7127, P<0.05,
Fig. 1C). However, there was no
significant difference in expression levels in regards to age,
gender, and disease recurrence (Tables
I and II).
 | Table IComparison of the protein expression
of DNMT3A and hepaCAM in the bladder cancer cases according to
clinicopatho-logical parameters. |
Table I
Comparison of the protein expression
of DNMT3A and hepaCAM in the bladder cancer cases according to
clinicopatho-logical parameters.
| Variable | n (%) | DNMT3A
| hepaCAM
| Mean density (mean
± SD)
| P-value
|
|---|
| − | + | − | + | DNMT3A | hepaCAM | DNMT3A | hepaCAM |
|---|
| Tissue | | | | | | | | 0.008a | 0.006a |
| Cancer | 31 | 6 | 25 | 27 | 4 | 0.182±0.104 | 0.065±0.066 | | |
| Adjacent | 22 | 20 | 2 | 3 | 19 | 0.076±0.071 | 0.193±0.119 | | |
| Gender | | | | | | | | 0.067 | 0.116 |
| Male | 26 (84) | 5 | 21 | 24 | 2 | 0.183±0.112 | 0.066±0.045 | | |
| Female | 5 (16) | 1 | 4 | 3 | 2 | 0.181±0.143 | 0.064±0.093 | | |
| Age (years) | | | | | | | | 0.079 | 0.117 |
| ≥60 | 21 (68) | 5 | 16 | 17 | 4 | 0.183±0.161 | 0.065±0.091 | | |
| <60 | 10 (32) | 1 | 9 | 10 | 0 | 0.182±0.132 | 0.064±0.063 | | |
| Histological
stage | | | | | | | | 0.346 | 0.211 |
| Ta-T1 | 9 (29) | 5 | 4 | 8 | 1 | 0.181±0.152 | 0.064±0.054 | | |
| T2-T4 | 22 (71) | 1 | 21 | 19 | 3 | 0.182±0.141 | 0.065±0.028 | | |
| Histological
grade | | | | | | | | 0.098 | 0.113 |
| G1-G2 | 21 (68) | 5 | 16 | 21 | 0 | 0.182±0.133 | 0.064±0.019 | | |
| G3-G4 | 10 (32) | 1 | 9 | 6 | 4 | 0.183±0.167 | 0.066±0.021 | | |
| Occurrence | | | | | | | | 0.198 | 0.214 |
| Primary | 20 (65) | 5 | 15 | 19 | 1 | 0.182±0.107 | 0.064±0.091 | | |
| Recurrent | 11 (35) | 1 | 10 | 8 | 3 | 0.184±0.171 | 0.065±0.089 | | |
| Table IIComparison of the protein expression
of DNMT3B and hepaCAM in the bladder cancer cases according to
clinicopathological parameters. |
Table II
Comparison of the protein expression
of DNMT3B and hepaCAM in the bladder cancer cases according to
clinicopathological parameters.
| Variable | n (%) | DNMT3B
| hepaCAM
| Mean density (mean
± SD)
| P-value
|
|---|
| − | + | − | + | DNMT3B | hepaCAM | DNMT3B | hepaCAM |
|---|
| Tissue | | | | | | | | 0.011a | 0.006a |
| Cancer | 31 | 5 | 26 | 27 | 4 | 0.176±0.094 | 0.065±0.066 | | |
| Adjacent | 22 | 20 | 2 | 3 | 19 | 0.073±0.061 | 0.193±0.119 | | |
| Gender | | | | | | | | 0.087 | 0.116 |
| Male | 26 (84) | 4 | 22 | 24 | 2 | 0.175±0.131 | 0.066±0.045 | | |
| Female | 5 (16) | 1 | 4 | 3 | 2 | 0.177±0.123 | 0.064±0.093 | | |
| Age (years) | | | | | | | | 0.093 | 0.117 |
| ≥60 | 21 (68) | 4 | 17 | 17 | 4 | 0.178±0.114 | 0.065±0.091 | | |
| <60 | 10 (32) | 1 | 9 | 10 | 0 | 0.175±0.123 | 0.064±0.063 | | |
| Histological
stage | | | | | | | | 0.199 | 0.211 |
| Ta-T1 | 9 (29) | 4 | 5 | 8 | 1 | 0.175±0.127 | 0.064±0.054 | | |
| T2-T4 | 22 (71) | 1 | 21 | 19 | 3 | 0.177±0.161 | 0.065±0.028 | | |
| Histological
grade | | | | | | | | 0.099 | 0.113 |
| G1-G2 | 21 (68) | 3 | 18 | 21 | 0 | 0.175±0.132 | 0.064±0.019 | | |
| G3-G4 | 10 (32) | 2 | 8 | 6 | 4 | 0.178±0.176 | 0.066±0.021 | | |
| Occurrence | | | | | | | | 0.215 | 0.214 |
| Primary | 20 (65) | 4 | 16 | 19 | 1 | 0.175±0.161 | 0.064±0.091 | | |
| Recurrent | 11 (35) | 1 | 10 | 8 | 3 | 0.177±0.175 | 0.065±0.089 | | |
In contrast, protein lysates were extracted from 10
bladder cancer patient samples. Importantly, western blotting with
antibodies against DNMT3A/3B showed that 9 of the 10 bladder cancer
tissues had elevated levels of DNMT3A/3B, respectively, whereas
only 1 of the 10 adjacent normal tissues had detectable expression
of DNMT3A/3B (P<0.05, Fig. 1D and
E).
Optimal concentration and treatment time
for AZAC
To confirm the mode of action of AZAC in bladder
cancer cells, we determined the absorption at a variety of
concentrations and times. We calculated the IR with the
above-documented equation. The results indicated that bladder
cancer cell proliferation was markedly inhibited by AZAC in a
concentration- and dose-dependent manner (P<0.05, Table III). According to Table III, 4 µg/ml (BIU-87) and 5
µg/ml (T24 and EJ) was confirmed as the most appropriate
concentrations. The dose-effect curve indicated that the bladder
cancer cell IR gradually increased with prolongation of treatment
time at the same concentration compared with the controls
(P<0.05, Fig. 2A). Based on
these results, concentrations of 4 µg/ml (BIU-87 cells) and
5 µg/ml (T24 and EJ cells) AZAC were selected as the optimal
treatment conditions for the following experiments.
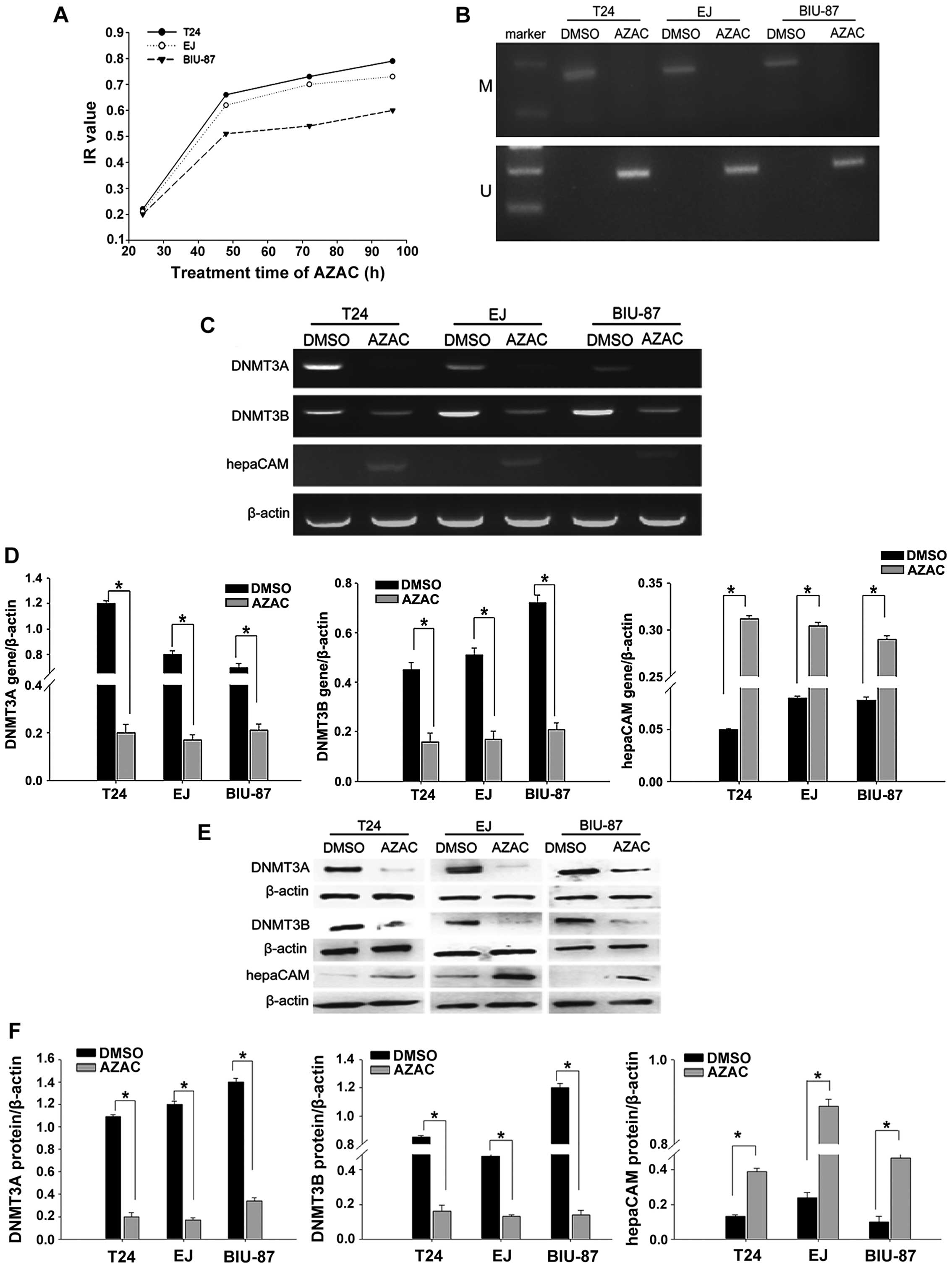 | Figure 2AZAC reverses hepaCAM methylation and
AZAC reduces levels of DNMT3A/3B and induces expression of hepaCAM.
(A) MTT assay results revealed that the DNMT inhibitor, AZAC,
increased the bladder cancer cell inhibition rate in a
time-dependent manner. Time-points included 24, 48, 72 and 96 h.
AZAC at 4 µg/ml (BIU-87) and 5 µg/ml (T24 and EJ) for
48 h were the optimal concentrations and times for use in the
experiment. (B) MSP analysis of the methylation of the hepaCAM gene
in bladder cancer cells. We found that in the blank group, hepaCAM
was hypermethylated which was reversed after treatment of AZAC
compared with the DMSO group (M, methylation; U, unmethylation).
(C) RT-PCR was used to detect mRNA expression of DNMT3A, DNMT3B and
hepaCAM. HepaCAM expression was upregulated and DNMT3A/3B
expression was decreased significantly compared with the DMSO
group. (D) Relative intensities of target bands of PCR were scanned
by Quantity One software; *P<0.05, compared with the
DMSO group. (E) Western blot analysis showed that hepaCAM was
re-activated but DNMT3A and DNMT3B were downregulated in three
bladder cancer cell lines after AZAC treatment compared with the
DMSO group. β-actin was used as control. (F) Intensities were
quantitated by Quantity One software; *P<0.05,
compared with the DMSO group. |
| Table IIIInhibition rates for different
concentrations of AZAC in the bladder cancer cells. |
Table III
Inhibition rates for different
concentrations of AZAC in the bladder cancer cells.
| Cancer cells | AZAC concentration
|
|---|
| 0.5 | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|
| T24 | | | | 25.05 | 27.34 | 32.25 | 39.17 | 44.56 |
| EJ | | | | 3.63 | 6.96 | 33.00 | 34.86 | 35.87 |
| BIU-87 | 8.77 | 12.75 | 12.93 | 28.33 | 34.70 | 48.15 | | |
AZAC reverses the hypermethylation of
hepaCAM
Hypermethylation of the hepaCAM promoter was
apparently observed in the T24, EJ and BIU-87 cell lines by use of
MSP. In addition, the hepaCAM promoter was obviously demethylated
by 5 µg/ml of AZAC in the T24 and EJ cell lines, and 4
µg/ml of AZAC in the BIU-87 cell line at 48 h (Fig. 2B).
Suppression of DNMT3A/3B induces
re-expression of hepaCAM
Based on our findings, we analyzed the mRNA and
protein expression of DNMT3A/3B and hepaCAM by RT-PCR and
immunoblotting. The results revealed that DNMT inhibitor AZAC
inhibited both the mRNA and protein levels of DNMT3A/3B in the
bladder cancer cell lines. AZAC not only reversed the expression of
hepaCAM mRNA (Fig. 2C and D) but
also its protein level (Fig. 2E and
F). These results suggest that the inactivation of DNMT3A/3B by
AZAC treatment may participate in restoring the expression of the
tumor-suppressor gene hepaCAM.
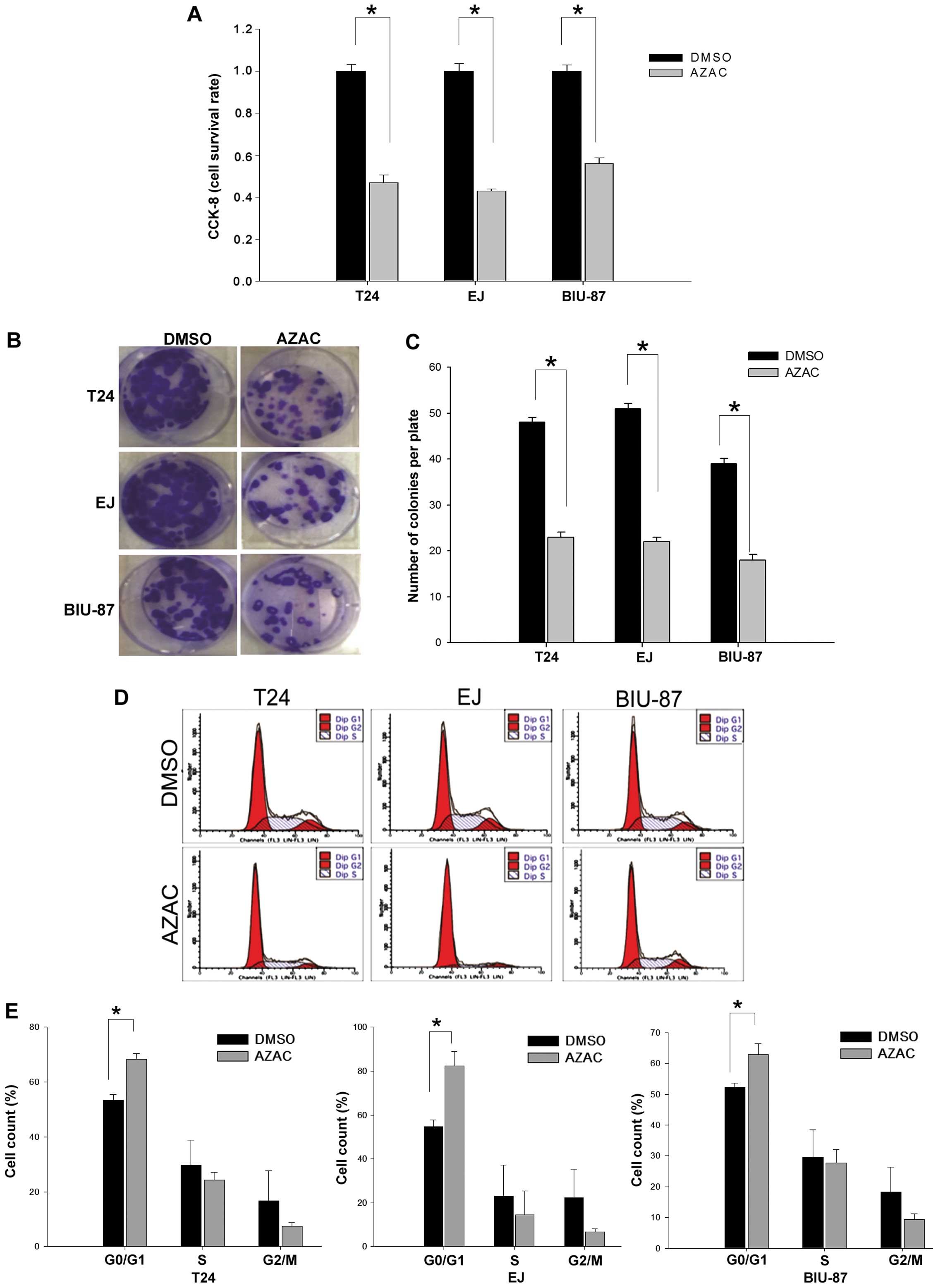
AZAC inhibits cell proliferation
To investigate the effect of AZAC against bladder
cancer cells, cell proliferation was detected by CCK-8 assay,
colony formation assay and flow cytometry. The results showed that
the growth of bladder cancer cells was inhibited after exposure to
AZAC for 48 h compared with the control groups (P<0.05, Fig. 3A). This result was further supported
by a colony formation assay. As shown in Fig. 3B and C, there was a significantly
lower colony formation potential in the AZAC-treated cells compared
to that in the DMSO treatment groups (P<0.05).
It has been reported that the growth inhibition of
AZAC is always associated with cell cycle arrest (21). To evaluate the exact cell cycle
phase, bladder cancer cells were inspected at 48 h, under
stimulation by 4 µg/ml (BIU-87) and 5 µg/ml (T24 and
EJ) AZAC. The results revealed that bladder cancer cells treated
with AZAC had a significant accumulation in the G0/G1 phase
compared to the controls (P<0.05, Fig. 3D and E). These data indicate that
the growth of bladder cancer cells was markedly inhibited by
AZAC.
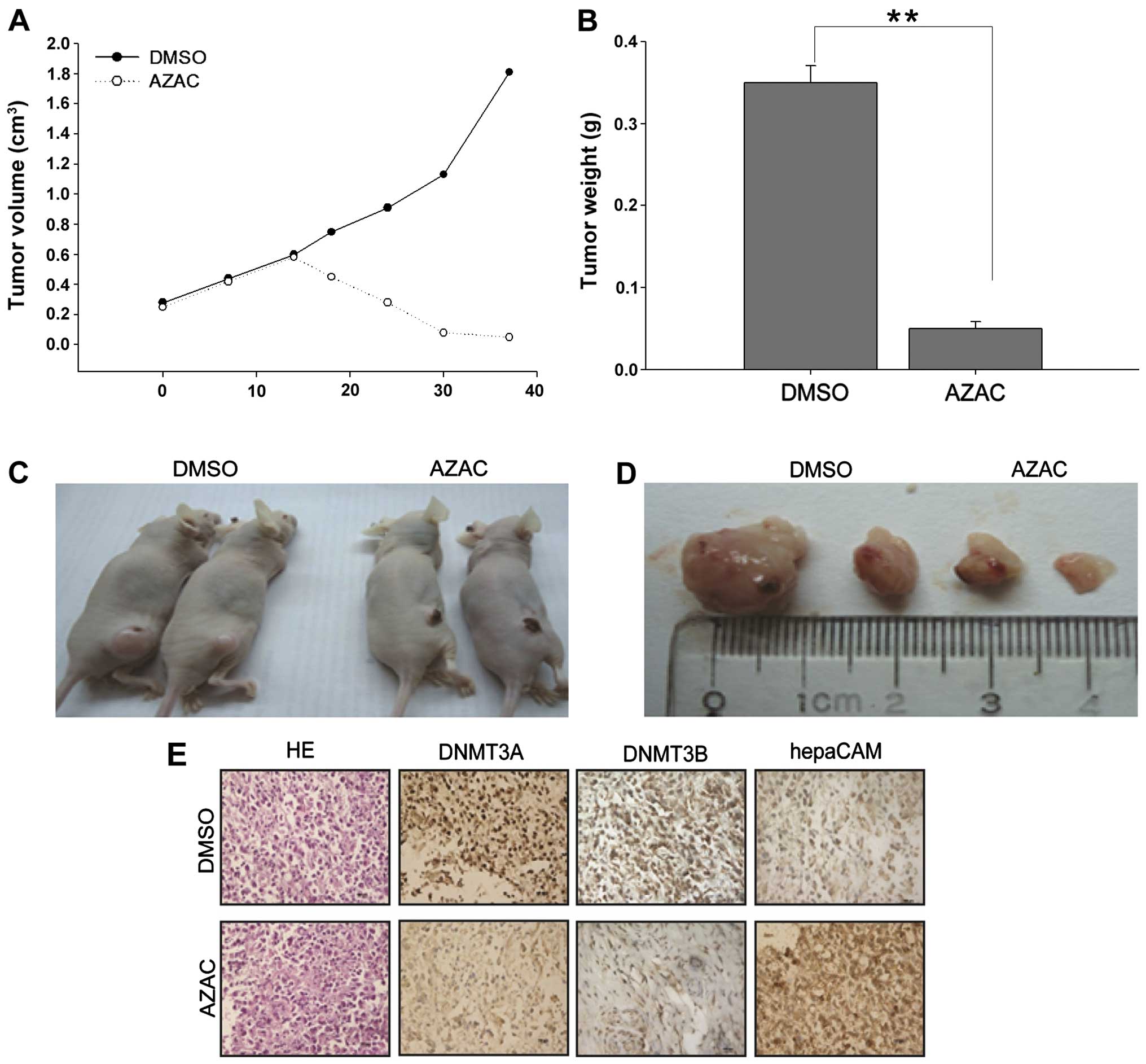
Effects of AZAC on nude mice
To further investigate the tumor-suppressive
function of AZAC and its mechanisms in vivo, the
antitumorigenicity of AZAC was tested in nude mice. Thirty days
after injection, tumors were excised from the tested mice for
further analysis. The volume and weight of the tumors in the AZAC
treatment groups were significantly decreased, compared with the
control groups (P<0.05, Fig.
4A–D). Immunohistochemistry was further performed to analyze
the expression of DNMT3A/3B and hepaCAM in the xenograft tumors.
Numerous tumor cells with high nuclear fragmentation were found in
H&E-stained sections from the DMSO treatment groups compared
with the AZAC groups. Furthermore, low expression of DNMT3A/3B but
high levels of hepaCAM immunostaining were observed in the AZAC
treatment-derived tumor tissues (Fig.
4E). Taken together, these data indicate that AZAC acts as an
antitumor agent, suppressing the proliferation of bladder cancer
and enhancing the expression of hepaCAM via downregulation of the
levels of DNMT3A/3B.
Discussion
Bladder cancer ranks fourth in regards to the
worldwide cancer incidence and is the seventh leading cause of
mortality from cancer (22). The
main features of bladder cancer are disease recurrence and
progression (23). Research has
confirmed that bladder cancer results from interactions among
exogenous, genetic and epigenetic factors. Previous studies have
demonstrated that the most common risk factors of bladder cancer
are smoking and occupational exposure (23–25).
However, more and more studies have aimed to investigate the
molecular mechenisms of bladder carcinogenesis in recent years.
Various studies have discovered that inactivation of one or more
tumor-suppressor genes, such as p16INK4a, FHIT, LAMC2, APC, hMLH1
and E-cadherin, may be an early event in the tumorigenic pathway
leading to bladder cancer (26–29).
HepaCAM localized on the cell surface has a role in
cell-cell adhesion, and acts as a tumor suppressor. We have been
researching the reason for the absence of hepaCAM in bladder
cancer. In a previous study, we found that exon 2 methylation of
hepaCAM may be one of the reasons for its low expression in bladder
cancer tissues and cell lines (30). However, the precise mechanism of low
hepaCAM expression in bladder cancer still needs to be
elucidated.
In cancer, hypermethylation of CpG islands, regarded
as regions of DNA 0.5–4 kb with C+G content >0.5 (31–33),
contributes to transcription silencing of tumor-suppressor genes.
There is mounting evidence that abnormal hypermethylation of the
promoter is the mechanism involved in the silencing of
tumor-suppressor genes in tumorigenesis. For example, RASSF1A is
methylated in lung and breast cancers, and hypermethylation of the
CDH1 promoter is noted in gastric cancer (34). In the past few years, more and more
candidate tumor-suppressor genes have been found to be silenced by
promoter hypemethylation in cancers. In the present study, we
predicted a CpG island in the hepaCAM promoter by an online
database (http://rulai.cshl.org/tools/FirstEF/) and designed
meth-primers (http://www.urogene.org/methprimer/index1.html) to
detect the methylation status of the CpG island in the hepaCAM
promoter. Our results demonstrated that aberrant hypermethylation
occurred in the CpG island of the hepaCAM gene promoter, which
contributed to the absence of hepaCAM in the bladder cancer cells
(T24, EJ and BIU-87). Methylation was catalyzed by DNMTs. Many
previous studies have shown that DNMT3A/3B are overexpressed in
cancers. For example, the level of DNMT3A/3B was significantly
higher in gastric cancer than in normal tissues (35), and its expression in prostate
carcinoma tissues was higher than that in adjacent normal tissues
(36). In the present study, we
detected the expression of DNMT3A/3B and hepaCAM in bladder cancer.
Correlations between DNMT3A/3B and hepaCAM in bladder cancer
patients suggested that a low level of hepaCAM was closely related
with high expression of DNMT3A/3B in bladder carcinomas. Then, we
found that AZAC reversed the high methylation of hepaCAM and
restored its level via downregulation of the expression of
DNMT3A/3B, at not only the mRNA but also the protein level, in the
bladder cancer cell lines. Previous studies also showed similar
findings in prostate cancer cells under mahanine treatment
(37). These studies indicate that
the precise mechanism of low hepaCAM expression in bladder cancer
may be abnormal hypermethylation of hepaCAM induced by high levels
of DNMT3A/3B.
In the clinic, AZAC has been used as a drug for
hematopoietic disorders and as an anticancer treatment strategy for
solid tumors. Fenaux et al showed that AZAC significantly
extended the overall survival of patients with myelodysplastic
syndrome (MDS, a bone marrow stem cell disorder). Likewise, AZAC
was found to suppress the cell growth of three different
neuroendocrine tumors in vitro (38). In the present study, CCK-8 and
colony-formation assays revealed that AZAC inhibited the
proliferation of bladder cancer cells. For the first time, we also
provided evidence for the accumulation of G0/G1 cell cycle markers
following AZAC treatment, suggesting that AZAC inhibited cell
proliferation by inducing G0/G1 cell growth arrest. These data were
in concordance with a previously published report that
adenovirus-hepaCAM treatments induce G0/G1 growth arrest in bladder
cancer (19). Thus, our data
suggest that AZAC may represent an effective therapeutic
intervention for bladder cancer.
To address the regulation mechanism and apparently
central role of AZAC in vitro, we performed in vivo
experiments to validate these results. First, in vivo tests
revealed that AZAC inhibited tumor growth, which was in line with a
previous study (39). Next,
immunohistochemistry showed low protein levels of DNMT3A/3B and
high protein expression of hepaCAM in the AZAC-treated tumors,
which were in accordance with our in vitro tests. However,
the sites of methylation of hepaCAM in bladder cancer cells need to
be further explored.
In summary, our study showed that DNMT3A/3B
expression was increased while hepaCAM protein expression was
decreased in bladder cancer tissues, and there was a negative
linear correlation between them. HepaCAM was silenced by its
promoter hypermethylation and was re-activated by methylation
inhibitor AZAC via downregulation of the expression of DNMT3A/3B.
Furthermore, AZAC inhibits the growth of bladder cancer in
vitro and in vivo, providing a new insight into the
therapeutic strategy of bladder cancer.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant no. 81072086).
Abbreviations:
|
hepaCAM
|
hepatocyte cell adhesion molecule
|
|
AZAC
|
5-azacytidine
|
|
DNMT3A/3B
|
DNA methyltransferase 3A/3B
|
References
|
1
|
Favre-Kontula L, Rolland A, Bernasconi L,
Karmirantzou M, Power C, Antonsson B and Boschert U: GlialCAM, an
immunoglobulin-like cell adhesion molecule is expressed in glial
cells of the central nervous system. Glia. 56:633–645. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chung Moh M, Hoon Lee L and Shen S:
Cloning and charac-terization of hepaCAM, a novel Ig-like cell
adhesion molecule suppressed in human hepatocellular carcinoma. J
Hepatol. 42:833–841. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moh MC, Zhang C, Luo C, Lee LH and Shen S:
Structural and functional analyses of a novel Ig-like cell adhesion
molecule, hepaCAM, in the human breast carcinoma MCF7 cells. J Biol
Chem. 280:27366–27374. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moh MC, Zhang T, Lee LH and Shen S:
Expression of hepaCAM is downregulated in cancers and induces
senescence-like growth arrest via a p53/p21-dependent pathway in
human breast cancer cells. Carcinogenesis. 29:2298–2305. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee LH, Moh MC, Zhang T and Shen S: The
immunoglobulin-like cell adhesion molecule hepaCAM induces
differentiation of human glioblastoma U373-MG cells. J Cell
Biochem. 107:1129–1138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xun C, Luo C, Wu X, Zhang Q, Yan L and
Shen S: Expression of hepaCAM and its effect on proliferation of
tumor cells in renal cell carcinoma. Urology. 75:828–834. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang QL, Luo CL, Wu XH, Wang CY, Xu X,
Zhang YY, Liu Q and Shen SL: HepaCAM induces G1 phase arrest and
promotes c-Myc degradation in human renal cell carcinoma. J Cell
Biochem. 112:2910–2919. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He Y, Wu X, Luo C, Wang L and Lin J:
Functional significance of the hepaCAM gene in bladder cancer. BMC
Cancer. 10:832010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu X, Shan L, Wang F, Wang J, Wang F,
Shen G, Liu X, Wang B, Yuan Y, Ying J, et al: Hypermethylation of
BRCA1 gene: Implication for prognostic biomarker and therapeutic
target in sporadic primary triple-negative breast cancer. Breast
Cancer Res Treat. 150:479–486. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ahmed IA, Pusch CM, Hamed T, Rashad H,
Idris A, El-Fadle AA and Blin N: Epigenetic alterations by
methylation of RASSF1A and DAPK1 promoter sequences in mammary
carcinoma detected in extracellular tumor DNA. Cancer Genet
Cytogenet. 199:96–100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liang G, Chan MF, Tomigahara Y, Tsai YC,
Gonzales FA, Li E, Laird PW and Jones PA: Cooperativity between DNA
methyltransferases in the maintenance methylation of repetitive
elements. Mol Cell Biol. 22:480–491. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ramos MP, Wijetunga NA, McLellan AS,
Suzuki M and Greally JM: DNA demethylation by
5-aza-2′-deoxycytidine is imprinted, targeted to euchromatin, and
has limited transcriptional consequences. Epigenetics Chromatin.
8:112015. View Article : Google Scholar
|
|
13
|
Amara K, Ziadi S, Hachana M, Soltani N,
Korbi S and Trimeche M: DNA methyltransferase DNMT3b protein
over-expression as a prognostic factor in patients with diffuse
large B-cell lymphomas. Cancer Sci. 101:1722–1730. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen MF, Chen WC, Chang YJ, Wu CF and Wu
CT: Role of DNA methyltransferase 1 in hormone-resistant prostate
cancer. J Mol Med Berl. 88:953–962. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Devanand P, Kim SI, Choi YW, Sheen SS, Yim
H, Ryu MS, Kim SJ, Kim WJ and Lim IK: Inhibition of bladder cancer
invasion by Sp1-mediated BTG2 expression via inhibition of DNA
methyltransferase 1. FEBS J. 281:5581–5601. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brueckner B and Lyko F: DNA
methyltransferase inhibitors: Old and new drugs for an epigenetic
cancer therapy. Trends Pharmacol Sci. 25:551–554. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wijermans P, Lübbert M, Verhoef G, Bosly
A, Ravoet C, Andre M and Ferrant A: Low-dose
5-aza-2′-deoxycytidine, a DNA hypo-methylating agent, for the
treatment of high-risk myelodysplastic syndrome: A multicenter
phase II study in elderly patients. J Clin Oncol. 18:956–962.
2000.PubMed/NCBI
|
|
18
|
Issa JP, Garcia-Manero G, Giles FJ,
Mannari R, Thomas D, Faderl S, Bayar E, Lyons J, Rosenfeld CS,
Cortes J, et al: Phase 1 study of low-dose prolonged exposure
schedules of the hypomethylating agent 5-aza-2′-deoxycytidine
(decitabine) in hematopoietic malignancies. Blood. 103:1635–1640.
2004. View Article : Google Scholar
|
|
19
|
Xu B, He Y, Wu X, Luo C, Liu A and Zhang
J: Exploration of the correlations between interferon-γ in patient
serum and HEPACAM in bladder transitional cell carcinoma, and the
interferon-γ mechanism inhibiting BIU-87 proliferation. J Urol.
188:1346–1353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Q, Luo C, Wu X, Du H, Song X and Fan
Y: hepaCAM and p-mTOR closely correlate in bladder transitional
cell carcinoma and hepaCAM expression inhibits proliferation via an
AMPK/mTOR dependent pathway in human bladder cancer cells. J Urol.
190:1912–1918. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alexander VM, Roy M, Steffens KA,
Kunnimalaiyaan M and Chen H: Azacytidine induces cell cycle arrest
and suppression of neuroendocrine markers in carcinoids. Int J Clin
Exp Med. 3:95–102. 2010.PubMed/NCBI
|
|
22
|
Pasin E, Josephson DY, Mitra AP, Cote RJ
and Stein JP: Superficial bladder cancer: An update on etiology,
molecular development, classification, and natural history. Rev
Urol. 10:31–43. 2008.PubMed/NCBI
|
|
23
|
Zeegers MP, Kellen E, Buntinx F and van
den Brandt PA: The association between smoking, beverage
consumption, diet and bladder cancer: A systematic literature
review. World J Urol. 21:392–401. 2004. View Article : Google Scholar
|
|
24
|
Scrima M, De Marco C, De Vita F, Fabiani
F, Franco R, Pirozzi G, Rocco G, Malanga D and Viglietto G: The
nonreceptor-type tyrosine phosphatase PTPN13 is a tumor suppressor
gene in non-small cell lung cancer. Am J Pathol. 180:1202–1214.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yoon JI, Kim SI, Tommasi S and Besaratinia
A: Organ specificity of the bladder carcinogen 4-aminobiphenyl in
inducing DNA damage and mutation in mice. Cancer Prev Res (Phila).
5:299–308. 2012. View Article : Google Scholar
|
|
26
|
Kandimalla R, van Tilborg AA and Zwarthoff
EC: DNA methylation-based biomarkers in bladder cancer. Nat Rev
Urol. 10:327–335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bilgrami SM, Qureshi SA, Pervez S and
Abbas F: Promoter hypermethylation of tumor suppressor genes
correlates with tumor grade and invasiveness in patients with
urothelial bladder cancer. Springerplus. 3:1782014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li G, Liu Y, Yin H, Zhang X, Mo X, Tang J
and Chen W: E-cadherin gene promoter hypermethylation may
contribute to the risk of bladder cancer among Asian populations.
Gene. 534:48–53. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Piaton E, Casalegno JS, Advenier AS,
Decaussin-Petrucci M, Mege-Lechevallier F, Ruffion A and Mekki Y:
p16(INK4a) over-expression is not linked to oncogenic human
papillomaviruses in patients with high-grade urothelial cancer
cells. Cancer Cytopathol. 122:760–769. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pan C, Wu X, Luo C, Yang S, Pu J, Wang C
and Shen S: Exon 2 methylation inhibits hepaCAM expression in
transitional cell carcinoma of the bladder. Urol Int. 85:347–354.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
32
|
Stutterheim J, Ichou FA, den Ouden E,
Versteeg R, Caron HN, Tytgat GA and van der Schoot CE: Methylated
RASSF1a is the first specific DNA marker for minimal residual
disease testing in neuroblastoma. Clin Cancer Res. 18:808–814.
2012. View Article : Google Scholar
|
|
33
|
Tian X, Chen D, Zhang R, Zhou J, Peng X,
Yang X, Zhang X and Zheng Z: Quantitative survey of multiple CpGs
from 5 genes identifies CpG methylation panel discriminating
between high- and low-grade cervical intraepithelial neoplasia.
Clin Epigenetics. 7:42015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jing H, Dai F, Zhao C and Yang J, Li L,
Kota P, Mao L, Xiang K, Zheng C and Yang J: Association of genetic
variants in and promoter hypermethylation of CDH1 with gastric
cancer: A meta-analysis. Medicine (Baltimore). 93:e1072014.
View Article : Google Scholar
|
|
35
|
Ding WJ, Fang JY, Chen XY and Peng YS: The
expression and clinical significance of DNA methyltransferase
proteins in human gastric cancer. Dig Dis Sci. 53:2083–2089. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
He S, Wang F, Yang L, Guo C, Wan R, Ke A,
Xu L, Hu G, Xu X, Shen J, et al: Expression of DNMT1 and DNMT3a are
regulated by GLI1 in human pancreatic cancer. PLoS One.
6:e276842011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Agarwal S, Amin KS, Jagadeesh S, Baishay
G, Rao PG, Barua NC, Bhattacharya S and Banerjee PP: Mahanine
restores RASSF1A expression by down-regulating DNMT1 and DNMT3B in
prostate cancer cells. Mol Cancer. 12:99–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fenaux P, Mufti GJ, Hellstrom-Lindberg E,
Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz
G, List A, et al International Vidaza High-Risk MDS Survival Study
Group: Efficacy of azacitidine compared with that of conventional
care regimens in the treatment of higher-risk myelodysplastic
syndromes: A randomised, open-label, phase III study. Lancet Oncol.
10:223–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Borodovsky A, Salmasi V, Turcan S, Fabius
AW, Baia GS, Eberhart CG, Weingart JD, Gallia GL, Baylin SB, Chan
TA, et al: 5-Azacytidine reduces methylation, promotes
differentiation and induces tumor regression in a patient-derived
IDH1 mutant glioma xenograft. Oncotarget. 4:1737–1747. 2013.
View Article : Google Scholar : PubMed/NCBI
|