Introduction
Hypoxia, a reduction in tissue oxygen levels below
physiological levels, is a nearly universal hallmark of solid
tumors, and commonly develops due to heterogeneous blood flow from
structurally and functionally abnormal blood vessels within the
tumor (1,2). Intratumoral hypoxia is significantly
associated with aggressive tumor progression, resistance to
chemotherapy and radiation, and poor prognosis (3,4).
Tumor cells and tissues adapt to hypoxic micro
environment via the activation of numerous hypoxia-related
molecules, among which hypoxia-inducible factor 1 (HIF-1) is the
predominant one (5). HIF-1 is a
heterodimeric transcription factor composed of an
O2-regulated HIF-1α subunit and a constitutively
expressed HIF-1β subunit, which are basic helix-loop-helix-PAS
domain proteins, only HIF-1α is regulated by the oxygen tension
(6). In normoxic conditions, the
hydroxylation of proline residue 402 and/or 564 by prolyl
hydroxylase domain protein 2 (PHD2) promotes the interaction of
HIF-1α with the von-Hippel-Lindau (VHL) tumor suppressor protein,
which recruits an E3 ubiquitin-protein ligase and thus targets
HIF-1α for degradation by the ubiquitin proteasome system (7). In response to physiological hypoxia,
HIF-1α becomes rapidly stabilized and is localized to then nucleus,
where it binds to HIF-1β to form the HIF-1 complex. HIF-1
specifically binds to a short DNA sequence, 5′-A/GCGTG-3′, known as
the hypoxia-responsive element (HRE) within target genes (8). In the last decade, significant
evidence has accumulated that indicates that HIF-1α overexpression
increases the probability of patient mortality (9). HIF-1 plays a key role in tumor
progression and angiogenesis because activating the transcription
of human VEGF genes allows the encoding of the vascular endothelial
growth factor, a critical regulator for vascularization (10). Because of its importance in cancer,
HIF-1α is viewed as a novel anticancer target for the development
of new anticancer therapeutics (11).
Kamebakaurin, a compound of kaurane diterpenes was
isolated from traditional Chinese medicinal plant Isodon excia
(Maxin.) Hara. Previous studies have shown its
anti-neuroinflammatory actions targeting microglia-mediated
neurodegenerative diseases and inhibited the production of nitric
oxide (NO) and prostaglandin E2 (PGE2) through the inhibition of
nuclear factor-κB (NF-κB) signaling in lipopoly-saccharide
(LPS)-treated RAW264.7 macrophages (12,13).
In the present study, we found that kamebakaurin also inhibited
hypoxia-induced HIF-1 activation. This compound rapidly
downregulates not only HIF-1α by decreasing its protein synthesis
without affecting mRNA levels or protein degradation, but also the
expression of HIF target genes such as vascular endothelial growth
factor (VEGF) and erythropoietin (EPO), which are essential for
tumor growth. Based on these research findings, we further
confirmed our in vitro observations by showing profound
antitumor activity of kamebakaurin in a murine xenograft model with
no apparent toxicity to the animals.
Materials and methods
Cell culture and reagents
HeLa and KM12C cells were grown in DMEM with
penicillin (100 U/ml)-streptomycin (100 U/ml) (Invitrogen,
Carlsbad, CA, USA) and 10% heat-inactivated fetal bovine serum
(Hyclone, Logan, UT, USA). HCT116 and SNU638 cells were maintained
in RPMI-1640 medium supplemented as above. The cells were purchased
from American Type Culture Collection (ATCC, Manassas, VA, USA).
Cobalt chloride (CoCl2), desferrioxamine (DFO), MG-132,
and cycloheximide (CHX) from Sigma Chemical Co. (St. Louis, MO,
USA). Antibody for HIF-1α was obtained from BD Biosciences (San
Diego, CA, USA). CoCl2 was reported as a widely used
mimetic of hypoxia in a large range of cells, the molecule is known
to inhibit prolyl hydroxylases leading to HIF-1α stabilization
(14). Thus, in this study,
CoCl2 was used to induce hypoxia mimicking condition.
Then, the cell culture was kept in a gas-controlled chamber (Thermo
Electron Corp., Marietta, OH, USA) maintained at 5% CO2
and 37°C. Kamebakaurin was isolated from Isodon excia
(Maxin.) Hara and the structure is shown in Fig. 1A. The purity of kamebakaurin was
>98% in HPLC analysis.
Transfection and luciferase reporter
assay
The ability of the compound to inhibit hypoxia
inducible factor was determined by HRE-dependent reporter assay as
previously described (15). In
brief, at 50-80% confluence, HCT116 cells were cotransfected with
the vectors for pGL3-HRE-Luciferase plasmid containing six copies
of HREs derived from the human VEGF gene and with pRL-CMV (Promega,
Madison, WI, USA) using Lipofectamine 2000 reagent (Invitrogen).
Following 24 h of incubation, the cells were treated with various
concentrations of kamebakaurin and incubated for 16 h in hypoxia.
Luciferase assay was performed using Dual-luciferase reporter assay
system according to the instructions of the manufacturer (Promega).
Luciferase activity was determined in Microlumat plus luminometer
(EG&G Berthold, Bad Wildbad, Germany) by injecting 100
µl of assay buffer containing luciferin and measuring light
emission for 10 sec. The results were normalized to the activity of
renilla expressed by cotransfected Rluc gene under the control of a
constitutive promoter. Data were analyzed using ANOVA (analysis of
variance).
Measurement of cell viability by MTT
assay
HCT116 cells were seeded at 1×105
cells/ml in 96-well plates containing 100 µl of RPMI-1640
with 10% FBS and incubated overnight. Kamebakaurin was dissolved in
DMSO and DMSO was added to all plates to compensate the same volume
of DMSO. After 24 h, the cells were pretreated with different
concentrations of kamebakaurin for 24 h. Subsequently, cells were
cultured with MTT solution (5 mg/ml)
[3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide]
(Sigma, St. Louis, MO, USA) for 4 h. The viable cells converted MTT
to formazan, which generated a blue-purple color after dissolving
in 100 µl of DMSO. The absorbance at 570 nm was measured by
micro-plate reader (Molecular Devices, Sunnyvale, CA, USA).
Western blot analysis
Whole-cell extracts were obtained by lysing cells in
ice-cold lysis buffer (50 mM Tris-HCl, pH 7.5, 1% Nonidet P-40, 1
mM EDTA, 1 mM phenylmeth-ylsulfonylfluoride) supplemented with the
protease inhibitor cocktail (Sigma-Aldrich). HIF-1α protein was
analyzed in nuclear extracts prepared from cells using NE-PER
reagent (Pierce, Rockford, IL, USA), according to the instructions
of manufacturer. An aliquot of protein extracts were used to
determine protein concentration by the Bradford method. Fifty
microgram of whole-cell extracts or 30 µg of nuclear extract
protein per lane was separated by SDS-polyacrylamide gels and
followed by transferring to a polyvinylidenedifluoride membrane
(Millipore, Bedford, MA, USA). The membrane was blocked with 5%
skim milk, and then incubated with the corresponding antibody.
Antibody for HIF-1α was obtained from BD Biosciences. The primary
antibodies for VEGF, Topo-I, GLUT1, c-Myc and α-tubulin were
purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The
primary antibody for cyclin D1 was purchased from Cell Signaling
Technology (Beverly, MA, USA). After binding of an appropriate
secondary antibody coupled to horseradish peroxidase, proteins were
visualized by enhanced chemiluminescence according to the
instructions of the manufacturer (Amersham Pharmacia Biotech,
Buckinghamshire, UK).
VEGF ELISA
HCT116 cells were plated in a 96-well plate at a
density of 1×105 cells per well and treated with various
concentrations of kamebakaurin for 12 h under hypoxia conditions.
The VEGF levels in the culture supernatant and the serum were
determined by ELISA using the Duo-Set ELISA development kit
(R&D Systems, Inc., Minneapolis, MN, USA) according to the
manufacturer's instructions.
RT-PCR analysis
Total RNA from HCT116 cells was obtained using RNA
Mini kit (Qiagen, Valencia, CA, USA). Total RNA (2 µg) was
used to perform reverse transcription-PCR (RT-PCR) using RT-PCR kit
(Invitrogen) according to the manufacturer's protocol. The PCR
primers for VEGF were 5′-GCTCTACCTCCACCATGCCAA-3′ (sense) and
5′-TGGA AGATGTCCACCAGGGTC-3′ (antisense); for EPO were
5′-CACTTTCCGCAAACTCTTCCG-3′ (sense) and 5′-GTC ACAGCTTGCCACCTAAG-3′
(antisense); for HIF-1α were 5′-CTCAAAGTCCGACAGCCTCA-3′ (sense) and
5′-CCCT GCAGTAGGTTTCTGCT-3′ (antisense); for GAPDH were
5′-ACCACAGTCCATGCCATCAC-3′ (sense) and 5′-TCCA CCACCCTGTTGCTGTA-3′
(antisense). The oligonucleotide sequences of the reaction products
were confirmed by sequencing.
Measurement of cell cycle
Distribution of cells in different stages of cell
cycle was analyzed by BD Accuri™ C6 flow cytometry
(Becton-Dickinson, Franklin Lakes, NJ, USA). The kit utilizes
propidium iodide (PI) staining to allow quantitative measurements
of percentage of cells in the G0/G1, S and G2/M phases on the Cell
Quest software (Becton-Dickinson). For all assays, 10,000 events
were counted. The ModFit LT V4.0 software package (Verity Software,
Topsham, ME, USA) was used to analyze the data. The cell cycle
analysis was performed according to the manufacturer's protocol,
and as previously described (16).
Briefly, HCT116 cells (5×105 cells/ml) were treated with
different concentrations of kamebakaurin for 12 h and harvested
from culture dishes. After washing with PBS, HCT116 cells were
fixed with ice-cold 70% ethanol at −4°C for 12 h. The cells were
then washed with PBS containing 0.1% Triton X-100, stained with
PI/RNase reagent for 30 min and analyzed by BD Accuri™ C6 flow
cytometry. The ModFit LT V4.0 software package (Verity Software)
was used to analyze the data.
Tumor xenograft assay
All surgical procedures and care applied to the
animals were in accordance with IACUC guidelines. Six weeks old
specific-pathogen-free Crj:BALB/c nu/nu female athymic nude mice
(Vital River, China) were randomly assigned to three groups, each
of which consists of five mice (n=5 per group), and then were
subcutaneously inoculated with 0.2 ml of HCT116 cells
(5×107 cells/ml) in the left flank region. Kamebakaurin,
dissolved in DMSO, was administered orally every other day for 40
days at a dose of 15 and 50 mg/kg body weight starting from day 10
post cell implantation to mice. Tumor weight was calculated every
five days using the equation: [length × (width)2]/2.
Tumors were harvested 4 h after the last treatment, followed by
homogenising in RIPA for western blot analysis.
Statistical analysis
All values are expressed as mean ± SD. A comparison
of the results was performed with one-way ANOVA and Tukey's
multiple comparison tests (Graphpad Software, Inc, San Diego, CA,
USA). Statistically significant differences between groups were
defined as p-values <0.05.
Results
Kamebakaurin inhibits HIF-1α protein
expression in tumor cells
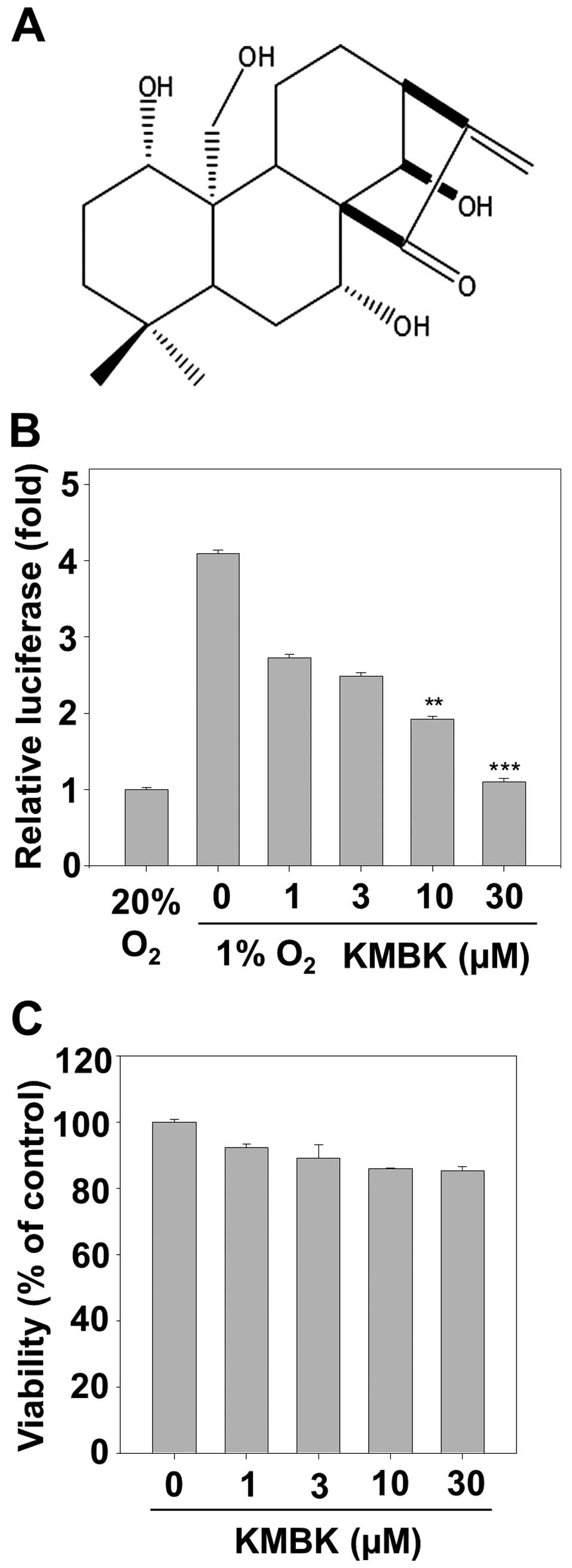
To investigate whether kamebakaurin (Fig. 1A) inhibited HIF-1α transcriptional
activation, we transfected HCT116 cells with a luciferase reporter
gene driven by six specific HRE. A substantial increase of
luciferase activity was observed in cells cultured in hypoxic
conditions, whereas kamebakaurin dose-dependently inhibited
hypoxia-induced luciferase activity (Fig. 1B). Given that the inhibition of
HIF-1α transcriptional activation might be correlated with
kamebakaurin-induced cytotoxicity, parallel studies of cell
viability were performed (Fig. 1C).
After the HCT116 cells were treated with different concentrations
of kamebakaurin for 24 h, no significant alteration of cell
viability was observed relative to the untreated control group.
Kamebakaurin decreases HIF-1a protein
levels in a dose-dependent manner
To explore the underlying mechanism of kamebakaurin
activity, we investigated its effect on HIF-1α protein levels. In
HCT116 cells, HIF-1α protein is undetectable under normoxia,
whereas it is stabilized under hypoxia or in the presence of
CoCl2 and becomes readily detectable by western
blotting. Following 12 h of treatment, kamebakaurin exerted
dose-dependent inhibition of HIF-1α protein levels induced by
hypoxia or CoCl2 in HCT116 cells, with complete
abrogation at 30 µM (Fig.
2A). In contrast to the decrease of HIF-1α levels, kamebakaurin
had almost no effect on the levels of Topo-I protein. Next, in
order to address whether the inhibition of HIF-1α by kamebakaurin
was cell line specific, we extended these studies to a diverse set
of tumor cell lines with tissues of various origins, including the
cervical cancer cell line HeLa, gastric cancer cell line SNU638 and
colorectal-cancer cell line KM12C (Fig.
2A). In addition, induced-accumulation of HIF-1α by
well-characterized hypoxia mimetic reagents, including
CoCl2 and DFO, could also be abrogated by kamebakaurin
(Fig. 2B).
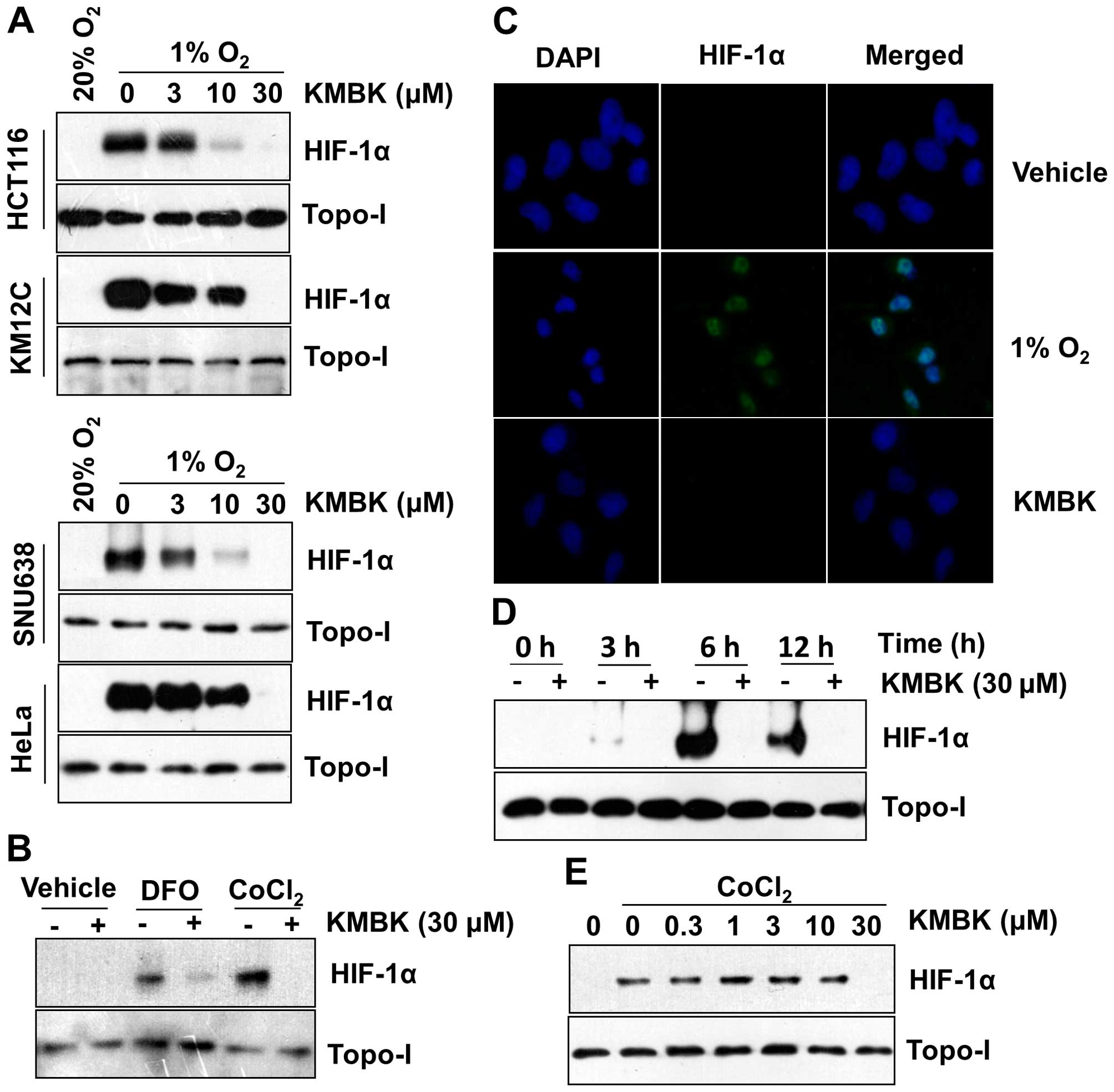 | Figure 2Kamebakaurin inhibits hypoxia-induced
expression of HIF-1α protein. (A) Cancer cells lines (HCT116,
KM12C, SNU638, and HeLa) were pretreated with the indicated
concentrations of kamebakaurin (KMBK) for 30 min and incubated
under normoxia, or hypoxia for 12 h, the nuclear extract for HIF-1α
analyzed by western blotting. The same blot was reprobed with an
anti-Topo-I antibody as a loading control. (B) HCT116 cells were
treated with or without KMBK (30 µM) for 30 min, then
incubated in different hypoxia mimetic reagents, including cobalt
chloride (CoCl2) (200 µM) and desferrioxamine
(DFO) (100 µM). After 12 h of incubation, the nuclear
extract for HIF-1α was analyzed by western blotting. The same blot
was reprobed with an anti-Topo-I antibody as a loading control. (C)
HCT116 cells were cultured in chamber slides under normoxia or
hypoxia conditions and treated with or without KMBK (30 μM)
for 12 h. After fixation, the slides were stained with anti-HIF-1α
(1:100) antibody and Alexa fluor® 488 goat anti-mouse
lgG (H+L) and examined by fluorescence microscopy. DAPI staining
shows the location and size of the nuclei. Left column, DAPI;
Middle column, HIF-1α; Right column, Merge; magnification, ×40. (D)
HCT116 cells were treated with (CoCl2) (200 µM)
for 30 min, then 0, 3, 6 and 12 h in the presence or absence of
KMBK (30 µM). (E) HCT116 cells were treated with
(CoCl2) (200 µM) or not for 30 min, then treated
with dose concentrations of KMBK. |
We next performed an immunofluorescence assay to
evaluate the effect of kamebakaurin on HIF-1α expression in HCT116
cells. Following 12 h of treatment, kamebakaurin (30 µM)
exerted almost complete inhibition of HIF-1α protein levels in cell
nuclei induced by hypoxia in HCT116 cells (Fig. 2C). To further confirm the effects of
kamebakaurin, we conducted both time-course experiments and
dose-response experiments to determine the expression of HIF-1α
protein in the presence of kamebakaurin. The addition of
kamebakaurin to the culture medium remarkably inhibited HIF-1α
accumulation, and this inhibition persisted as long as the drug was
present, at least up to 12 h (Fig.
2D). Dose-response experiments indicated that kamebakaurin
dose-dependently inhibited hypoxia-induced accumulation of HIF-1α
in HCT116 cells, with complete abrogation at 30 µM (Fig. 2E).
Kamebakaurin inhibits the protein
synthesis of HIF-1α but not its degradation
Generally, the accumulation of HIF-1α is dependent
on the balance between its protein synthesis and degradation. To
further address the mechanism by which kamebakaurin inhibits HIF-1α
protein level, we examined whether kamebakaurin modulates HIF-1α
protein synthesis in the presence of a proteasome inhibitor MG-132
to prevent HIF-1α degradation. As expected, addition of proteasome
inhibitor caused the increased accumulation of HIF-1α protein
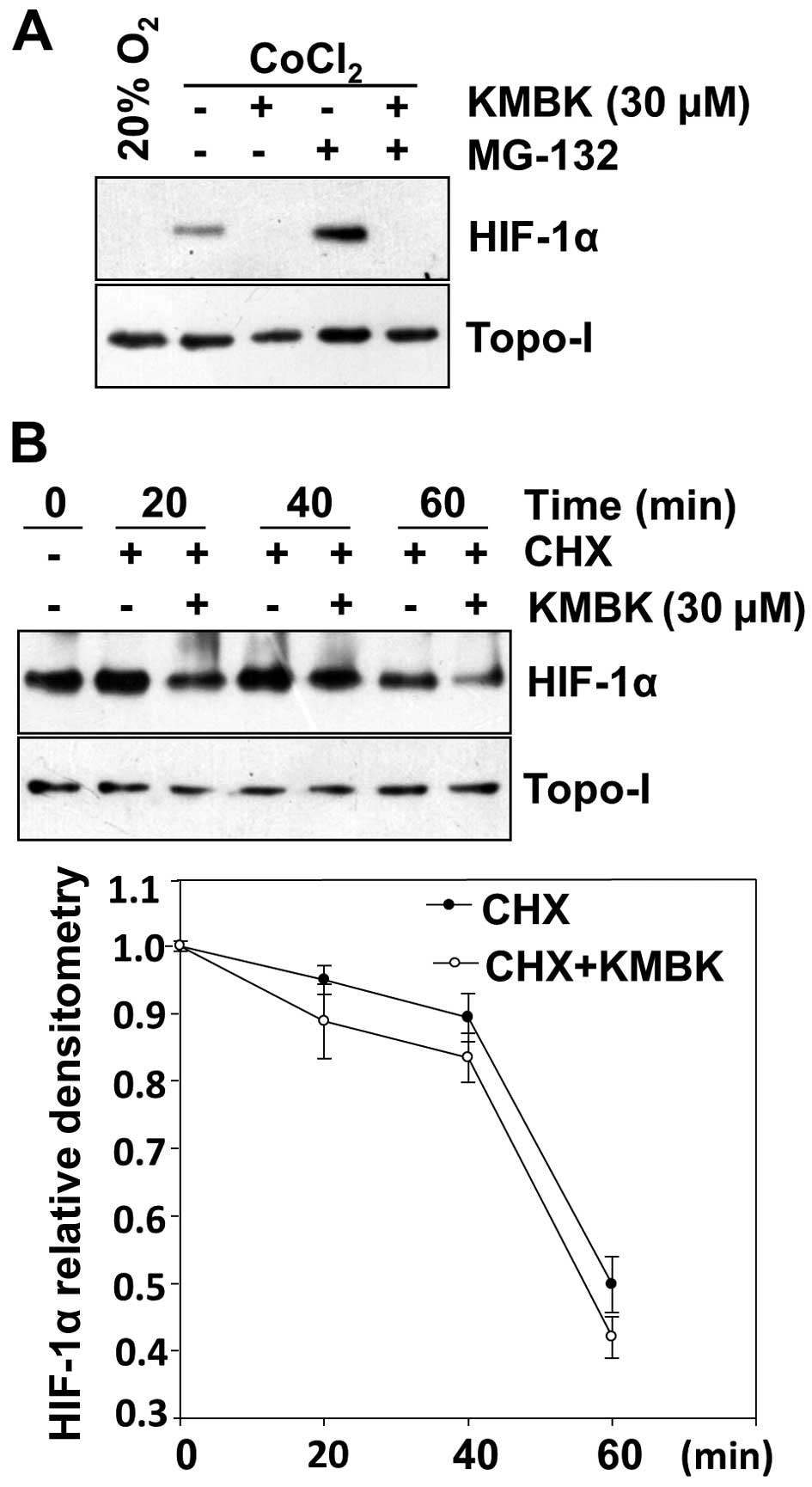
levels in the presence of CoCl2 (Fig. 3A). Kamebakaurin inhibited the
accumulation of HIF-1α protein induced by CoCl2 despite
of the presence of MG-132. No significant effects were observed on
Topo-I levels.
To address the effect of kamebakaurin on HIF-1α
protein stability, the protein translation inhibitor cycloheximide
(CHX) was used to prevent de novo HIF-1α protein synthesis
and then the cells were exposed to normoxia for increasing periods
up to 60 min. The nuclear extracts were prepared to detect HIF-1α
protein by western blotting. Under these conditions, HIF-1α protein
levels mainly reflected the rate of HIF-1α degradation. As shown in
Fig. 3B, the degradation of HIF-1α
was similar in the kamebakaurin-treated and the control cells.
Therefore, it is confirmed in our experiments that kamebakaurin
does not promote the degradation of HIF-1α. To determine whether
HIF-1α synthesis inhibition by kamebakaurin was a downstream effect
from decreased HIF-1α gene transcription or HIF-1α mRNA stability,
we analyzed HIF-1α mRNA levels by RT-PCR. Kamebakaurin did not
reduce HIF-1α mRNA levels significantly (Fig. 4A). This suggests that
kamebakaurin-mediated decrease of HIF-1α synthesis is likely due to
downregulation of HIF-1α mRNA translation.
Kamebakaurin suppresses expression of
HIF-1α target genes
The expression of VEGF and EPO, which are involved
in tumor cells proliferation, angiogenesis, invasion and
metastasis, is known to be regulated by HIF-1α (5,11). We
therefore examined whether kamebakaurin decreases the expression of
these genes. VEGF and EPO mRNA levels were measured by RT-PCR
analysis in HCT116 cells. Treatment of the cells with kamebakaurin
resulted in a dose-dependent inhibition of VEGF and EPO mRNA
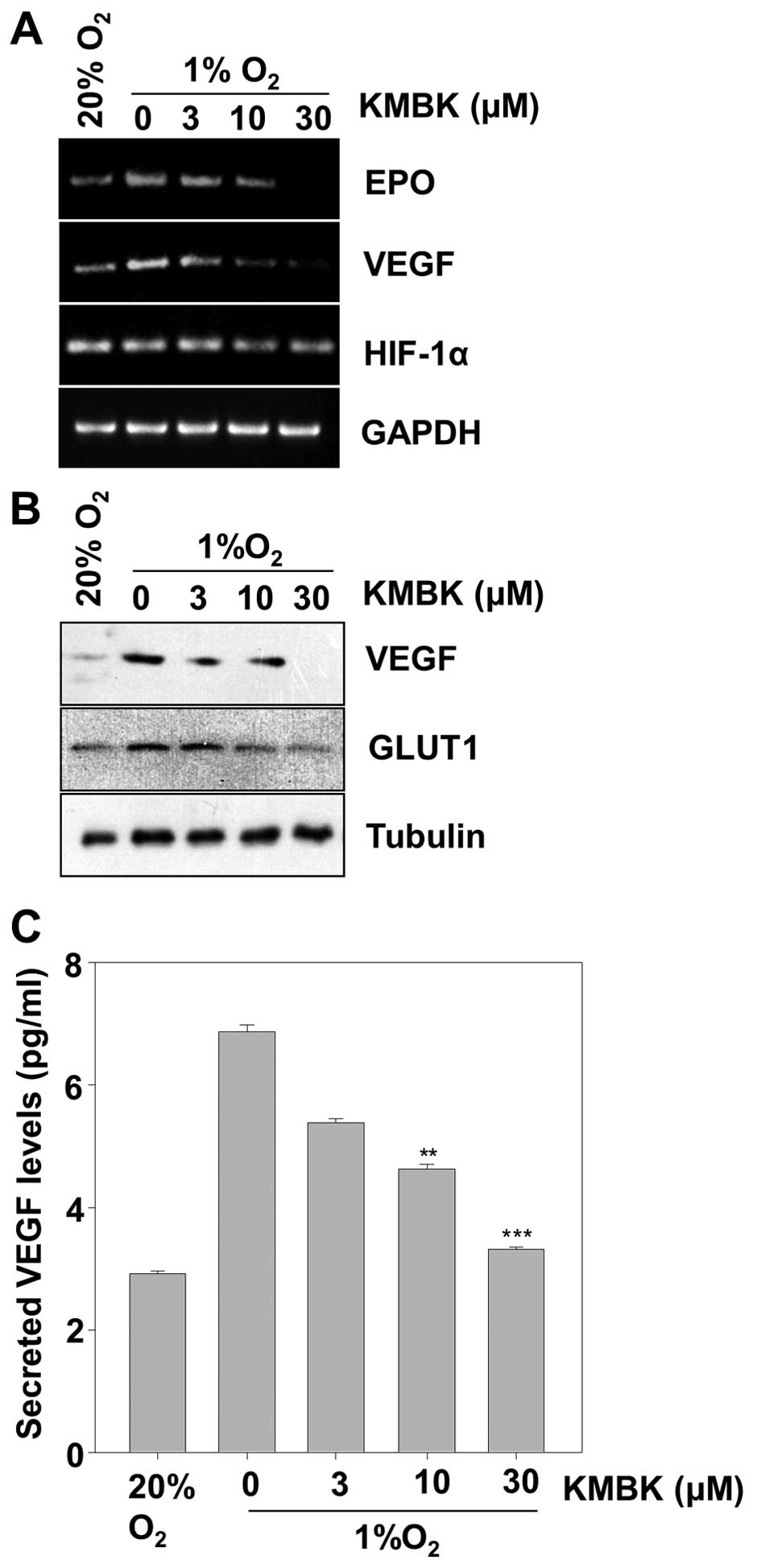
expression (Fig. 4A). Then, we also
examined the hypoxia induction of VEGF or GLUT1 (glucose
transporter 1) protein expression. Consistently, they were
dose-dependently inhibited by kamebakaurin (Fig. 4B). The concentrations to inhibit the
expression of HIF-1α target genes were comparable with those of
HIF-1α protein accumulation. This result led us to measure the VEGF
protein concentration in the culture supernatant by ELISA.
Consistently, the hypoxic induction of secreted VEGF protein was
dose-dependently inhibited by kamebakaurin (Fig. 4C).
Kamebakaurin inhibits the proliferation
of HCT116 cells via blocking cell cycle progression in the G1 phase
and downregulates cyclin D1 and c-Myc
To evaluate the effect of kamebakaurin on cell
proliferation, we tested whether the antiproliferative effect of
kamebakaurin is associated with cell cycle arrest by measuring the
DNA content of nuclei of HCT116 cells in flow cytometric analysis.
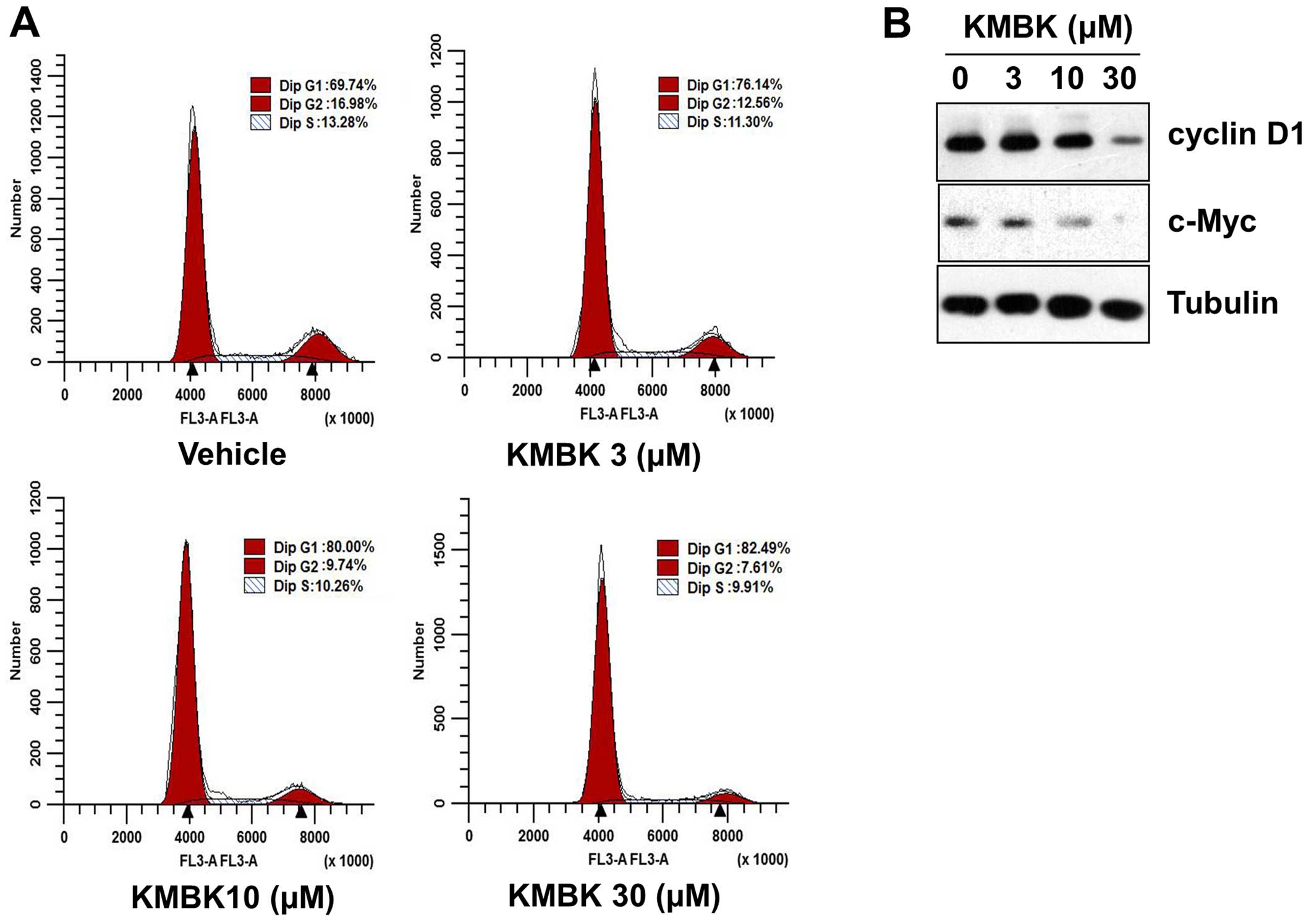
As shown in Fig. 5B, treatment with
30 µM kamebakaurin markedly induced G1/S phase cell cycle
arrest. FACS analysis revealed that 24 h exposure to kamebakaurin
increased the population of G1/S phase cells in a dose-dependent
manner. Cells at the G1/S phase increased from 69.74% in medium
alone to 76.14, 80.00 and 82.49% in the presence of 3, 10 and 30
µM kamebakaurin, respectively (Fig. 5A). While kamebakaurin treatment
retarded the progression of G1 to S/G2 phase.
Next, we determined the specific cell cycle
regulators responsible for the cell cycle arrest induced by
kamebakaurin by western blot analysis using antibodies specific to
cyclin D1 and c-Myc. The result showed that cyclin D1 and c-Myc
were decreased by kamebakaurin dose-dependently (Fig. 5B). Taken together, these results
clearly suggest that antiproliferative effect of kamebakaurin is
associated with its induction of cell cycle arrest at G1 phase, and
further confirmed that kamebakaurin downregulates cyclin D1 and
c-Myc protein levels on the cell cycle.
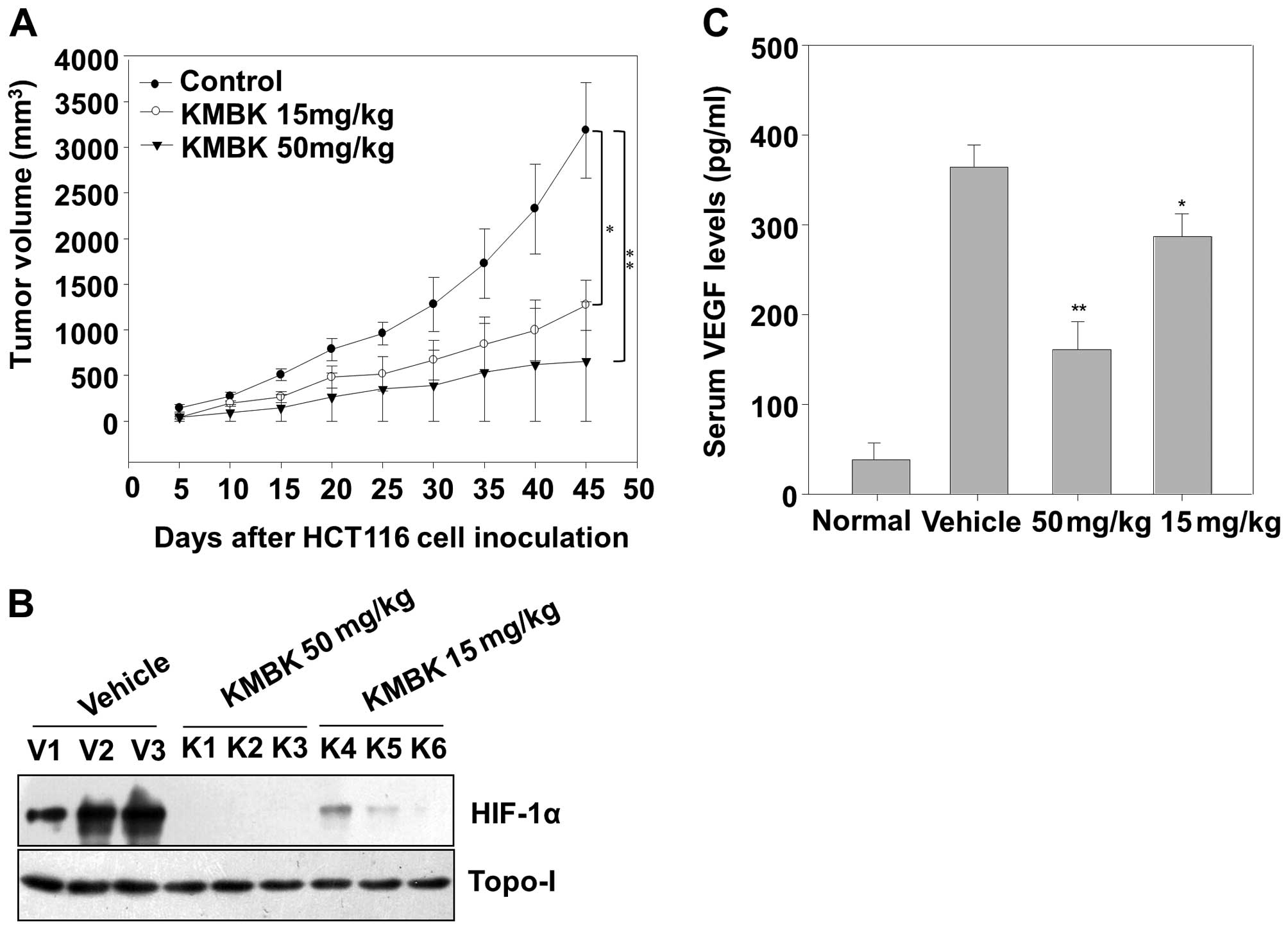
Kamebakaurin inhibits the growth of
HCT116 cells in a xenograft tumor model
To further reveal the effect of kamebakaurin on the
expression of HIF-1α and VEGF in vivo, we next determined
whether these results could be translated into an in vivo
xenograft model. HCT116 cells were subcutaneously implanted in
athymic nude mice, and the experimental mice were treated with
kamebakaurin (15 and 50 mg/kg) every other day until the end of the
study. As expected, kamebakaurin (50 mg/kg) produced significant
growth inhibition of HCT116 cells in a tumor xenograft model,
compared to that of the vehicle-treated control group (Fig. 6A). Due to the key roles of HIF-1α in
tumor angiogenesis, we studied its expression in the tumors by
western blotting. Consistent with the finding in cultured cells,
kamebakaurin significantly decreased the protein levels of HIF-1α
in the tumors, whereas no significant difference was observed in
Topo-I levels (Fig. 6B).
VEGF has key roles in tumor angiogenesis, therefore,
we measured the concentration of secreted VEGF in the serum of
xenograft mouse. Consistent with the findings in cultured cells,
kamebakaurin significantly decreased the serum VEGF levels
dose-dependently (Fig. 6C). Taken
together, our study showed that the downregulation of HIF-1α by
kamebakaurin could contribute to the inhibition of tumor growth and
VEGF secretion in a tumor xenograft model with HCT116 cells.
Discussion
Regions of hypoxia in tumors are associated with a
poor prognosis including treatment failure, metastasis, and
inferior survival (17). Therefore,
proteins that allow tumors to adapt to hypoxic conditions such as
HIF-1 represent critical targets for cancer treatments. HIF-1 plays
a central role in tumor progression and angiogenesis in
vivo. Exposure to a variety of growth factors has also been
shown to increase HIF-1 activity in normoxic and hypoxic
conditions. HIF-1α is overexpressed in many human cancers and has
been associated with tumor angiogenesis and VEGF is one of the most
potent angiogenic factors currently known (18). After VEGF binds to its receptors, it
functions not only as a proliferating factor, but also as an
anti-apoptotic for vascular endothelial cells (19). In addition, tumor growth and
angiogenesis in xenograft tumors also depends on HIF-1 activity and
the expression level of HIF-1α (20). In this study, we identified
kamebakaurin as an inhibitor of HIF-1α activation and VEGF
production.
The level of HIF-1α in cells is dependent on the
balance between its protein degradation and protein synthesis.
HIF-1α is oxygen-sensitive and is degraded mainly by
ubiquitin-proteasome systems (4).
We found that kamebakaurin strongly inhibited HIF-1α protein
accumulation, without affecting the expression level of HIF-1α mRNA
or degradation of HIF-1α protein. These observations may support
the hypothesis that the kamebakaurin-dependent reduction of HIF-1α
accumulation is due to the decrease of de novo HIF-1α
protein synthesis.
Kamebakaurin was also found to inhibit cell
proliferation through cell cycle arrest at G1 phase. This
inhibition was correlated with a reduction in the expression of
cell proliferation proteins such as cyclin D1 and c-Myc. Cyclin D1
is overexpressed in several cancers and is a biomarker of cancer
phenotype and disease progression, indicating that targeting of
cyclin D1 oncogene appears to be an attractive therapeutic strategy
(21). Myc is documented to play a
role in tumor initiation and regulation of cell growth and
proliferation. Inhibiting Myc function has been shown to be a
possible therapeutic strategy (22). c-Myc is one of the myc family of
transcription factors which activates the expression of a myriad of
genes by binding to consensus sequences and recruiting histone
acetyltransferases. A potent proto-oncogene, c-Myc, is often found
to be upregulated in many types of cancers. c-Myc over expression
stimulates gene amplification (23), presumably through DNA
overreplication, which can have a profound effect on the control of
cell growth. In this regard, our study demonstrates that
kamebakaurin downregulates cyclin D1 and c-Myc, leading to cell
growth inhibition through G1-phase arrest.
In conclusion, we have shown that kamebakaurin
decreased HIF-1α protein levels and inhibited hypoxia-induced VEGF
and EPO expression. In addition, our study provides evidence that
kamebakaurin can inhibit the proliferation of cancer cells through
the cell cycle arrest at G1 phase. These results may provide a
rationale for the development of kamebakaurin as an anticancer
drug.
Acknowledgments
This study was supported by National Natural Science
Foundation of China, nos. 81160250 and 81360496. This study also
received assistance from Administration of Traditional Chinese
Medicine of Jilin Province (2014-ZD27) and Jilin Province Science
and Technology Development Plan item (20150101229JC).
Abbreviations:
|
HIF-1
|
hypoxia-inducible factor-1
|
|
PHD2
|
prolyl hydroxylase domain protein
2
|
|
VHL
|
von-Hippel-Lindau
|
|
NO
|
nitric oxide
|
|
PGE2
|
prostaglandin E2
|
|
NF-κB
|
nuclear factor-κB
|
|
LPS
|
lipopolysaccharide
|
|
CoCl2
|
cobalt chloride
|
|
DFO
|
desferrioxamine
|
|
CHX
|
cycloheximide
|
|
VEGF
|
vascular endothelial growth factor
|
|
Topo-I
|
topoisomerase-I
|
|
EPO
|
erythropoietin
|
|
HRE
|
hypoxia response element
|
References
|
1
|
Danquah MK, Zhang XA and Mahato RI:
Extravasation of polymeric nanomedicines across tumor vasculature.
Adv Drug Deliv Rev. 63:623–639. 2011. View Article : Google Scholar
|
|
2
|
Dewhirst MW, Cao Y and Moeller B: Cycling
hypoxia and free radicals regulate angiogenesis and radiotherapy
response. Nat Rev Cancer. 8:425–437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brahimi-Horn MC, Chiche J and Pouysségur
J: Hypoxia and cancer. J Mol Med Berl. 85:1301–1307. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Semenza GL: Defining the role of
hypoxia-inducible factor 1 in cancer biology and therapeutics.
Oncogene. 29:625–634. 2010. View Article : Google Scholar :
|
|
5
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl
Acad Sci USA. 92:5510–5514. 1995. View Article : Google Scholar
|
|
7
|
Jaakkola P, Mole DR, Tian YM, Wilson MI,
Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji
M, Schofield CJ, et al: Targeting of HIF-alpha to the von
Hippel-Lindau ubiquitylation complex by O2-regulated
prolyl hydroxylation. Science. 292:468–472. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang GL and Semenza GL: Purification and
characterization of hypoxia-inducible factor 1. J Biol Chem.
270:1230–1237. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rankin EB and Giaccia AJ: The role of
hypoxia-inducible factors in tumorigenesis. Cell Death Differ.
15:678–685. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsuzuki Y, Fukumura D, Oosthuyse B, Koike
C, Carmeliet P and Jain RK: Vascular endothelial growth factor
(VEGF) modulation by targeting hypoxia-inducible
factor-1alpha--> hypoxia response element--> VEGF cascade
differentially regulates vascular response and growth rate in
tumors. Cancer Res. 60:6248–6252. 2000.PubMed/NCBI
|
|
11
|
Wilson WR and Hay MP: Targeting hypoxia in
cancer therapy. Nat Rev Cancer. 11:393–410. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim BW, Koppula S, Kim IS, Lim HW, Hong
SM, Han SD, Hwang BY and Choi DK: Anti-neuroinflammatory activity
of Kamebakaurin from Isodon japonicus via inhibition of c-Jun
NH(2)-terminal kinase and p38 mitogen-activated protein kinase
pathway in activated microglial cells. J Pharmacol Sci.
116:296–308. 2011. View Article : Google Scholar
|
|
13
|
Lee JH, Choi JK, Noh MS, Hwang BY, Hong YS
and Lee JJ: Anti-inflammatory effect of kamebakaurin in in vivo
animal models. Planta Med. 70:526–530. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hervouet E, Cízková A, Demont J,
Vojtísková A, Pecina P, Franssen-van Hal NL, Keijer J, Simonnet H,
Ivánek R, Kmoch S, et al: HIF and reactive oxygen species regulate
oxidative phosphorylation in cancer. Carcinogenesis. 29:1528–1537.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cai XF, Jin X, Lee D, Yang YT, Lee K, Hong
YS, Lee JH and Lee JJ: Phenanthroquinolizidine alkaloids from the
roots of Boehmeria pannosa potently inhibit hypoxia-inducible
factor-1 in AGS human gastric cancer cells. J Nat Prod.
69:1095–1097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li L, Dai HJ, Ye M, Wang SL, Xiao XJ,
Zheng J, Chen HY, Luo YH and Liu J: Lycorine induces cell-cycle
arrest in the G0/G1 phase in K562 cells via HDAC inhibition. Cancer
Cell Int. 12:492012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vaupel P and Mayer A: Hypoxia in cancer:
Significance and impact on clinical outcome. Cancer Metastasis Rev.
26:225–239. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Obermair A, Bancher-Todesca D, Bilgi S,
Kaider A, Kohlberger P, Müllauer-Ertl S, Leodolter S and Gitsch G:
Correlation of vascular endothelial growth factor expression and
microvessel density in cervical intraepithelial neoplasia. J Natl
Cancer Inst. 89:1212–1217. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tseng PL, Tai MH, Huang CC, Wang CC, Lin
JW, Hung CH, Chen CH, Wang JH, Lu SN, Lee CM, et al: Overexpression
of VEGF is associated with positive p53 immunostaining in
hepato-cellular carcinoma (HCC) and adverse outcome of HCC
patients. J Surg Oncol. 98:349–357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maxwell PH, Dachs GU, Gleadle JM, Nicholls
LG, Harris AL, Stratford IJ, Hankinson O, Pugh CW and Ratcliffe PJ:
Hypoxia-inducible factor-1 modulates gene expression in solid
tumors and influences both angiogenesis and tumor growth. Proc Natl
Acad Sci USA. 94:8104–8109. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Denis N, Kitzis A, Kruh J, Dautry F and
Corcos D: Stimulation of methotrexate resistance and dihydrofolate
reductase gene amplification by c-myc. Oncogene. 6:1453–1457.
1991.PubMed/NCBI
|