Introduction
Although our understanding of cancer has improved
compared with previous years, effective treatment is still hampered
by the highly complex nature of the disease (1). Chemotherapy is usually the primary
treatment for most patients, but multidrug resistance (MDR) often
causes chemotherapy failure (2).
ATP-binding cassette (ABC) transporters have been found to be
involved in MDR. The most important ABC proteins are P-glycoprotein
(Pgp) and breast cancer resistance protein (BCRP) (3,4), both
of which are glycoproteins and can be affected by glycosylation in
MDR (5).
Pgp acts as an ATP-dependent drug efflux pump
(6), and its role in MDR was
described in 1987 by Pastan and Gottesman (7). Human Pgp contains 10 consensus
sequences for N-linked glycosylation, but only three of
these sequences are glycosylated in the first extracellular loop of
Pgp (8). Pgp is synthesized as a
160 kDa protein (8) and is located
at the plasma membrane and in intercellular compartments (9–11).
BCRP, the other drug transporter protein, is also implicated in the
development of the MDR phenotype and has been reported to be
overexpressed in many solid tumors (12). It has a molecular mass of 72 kDa and
is termed a half-transporter (13)
since dimerization is required for substrate transport. The BCRP
N-linked glycosylation sites correspond to Asn596 in the
extracellular loop (14,15).
Glycosylation has been described as the most common
post-translational modification of proteins (16). It is involved in various cellular
processes, including protein folding, protein secretion,
intracellular trafficking, stability and substrate specificity
(15,17). Properly folded proteins are secreted
or targeted to their final intracellular or extracellular
destination, whereas misfolded proteins are recognized as aberrant
products and targeted for endoplasmic reticulum (ER)-associated
degradation (ERAD) (18,19) or delivered to lysosomes for
degradation (16).
Glycosylation can be inhibited by different
inhibitors that are capable of blocking different stages of this
process. The most important inhibitor is tunicamycin, which is used
to block the glycosylation of Pgp and BCRP. Other important
inhibitors of glycosylation are brefeldin A (BFA) and
castanospermine (CAS) (8).
BFA is characterized as an antiviral antibiotic
(20). In primary cultured rat
hepatocytes, BFA inhibits the secretion of plasma proteins at an
early step in the secretory pathway (21,22),
as well as the anterograde movement of the membrane beyond the
mixed ER/Golgi system (20).
Investigators have observed the breakdown of the Golgi apparatus
and redistribution of proteins into the ER in the presence of BFA,
and these effects were rapidly and completely reversed by removal
of the drug (20). BFA treatment of
the Golgi apparatus resulted in the rapid induction of membrane
tubules (20) and the
redistribution of many Golgi enzymes to the ER (23) due to the inhibition of coat protein
(β-COP) assembly on Golgi cisternae (24). In summary, BFA has been shown to
inhibit the exocytotic pathway of proteins from the ER to the
Golgi, irrespective of whether these proteins are secretory or
membrane-bound, but the effect of the BFA blockade is considered to
be only temporary (21).
Furthermore, BFA is a potent inducer of apoptosis in human cancer
cells based on DNA fragmentation (25), and it induces apoptotic cell death
in ovarian carcinoma cell lines by activating the mitochondrial
pathway and the caspase-8 and Bid-dependent pathway (26–28)
but not in a p53-dependent mechanism (25,29).
Additionally, BFA has been shown to reverse the function of the
multidrug resistance-associated protein (MRP) function (30). Although the cytotoxic activity of
BFA has been reported, BFA has not yet been applied as an
anticancer drug.
CAS is an indolizidine alkaloid that was isolated
from the Australian rainforest plant Castanospermum australe
(31). The main function of CAS is
inhibition of glucosidases I and II and obligatory enzyme trimming
during the synthesis of N-linked oligosaccharides on
glycoproteins (32). In various
studies, the ability of CAS to prevent the rejection of organ
transplants has been evaluated (33), and it has been shown to display
antiviral activity against retroviruses including HIV (34,35).
Combination therapy of 6-O-butanoyl castanospermine
(Celgosivir) with peginterferon and/or ribavirin has been
undergoing investigation in phase II clinical trials for the
treatment of patients with chronic HCV (36). CAS significantly inhibits tumor
growth in nude mice, reduces the adhesion of tumor cells to the
vascular endothelium, and decreases antimetastatic activity by
inhibiting platelet aggregation of metastatic cells (32). Furthermore, CAS has been shown to
inhibit cellular transformation by altering oncogene glycosylation,
angiogenesis in drug-treated animals (32) and concentration-dependent inhibition
of C6 glioblastoma cell proliferation (37).
Unfortunately, to date, no studies have been
conducted to examine the correlation between the changes in MDR
protein glycosylation and cancer MDR. Previously, we tested
tunicamycin (tun), one of the most well-known inhibitors of
glycosylation (38). Research was
very encouraging, and therefore we decided to expand our
investigation. In the present study, we analyzed BFA and CAS, which
block different stages of the glycosylation process. We decided to
test the impact of the two N-glycosylation inhibitors on Pgp
and BCRP, two of the most common MDR proteins, the tumor
overexpression of which is the main cause of cancer chemotherapy
failure.
Materials and methods
Chemicals and drugs
Doxorubicin, topotecan, paclitaxel, BFA, CAS, BMA,
MG132, Rho123, H33342 and RIPA lysis buffer were obtained from
Sigma (St. Louis, MO, USA). RPMI-1640 medium and Eagle's Minimum
Essential Medium (EMEM), fetal bovine serum (FBS),
antibiotic-antimycotic solution and L-glutamine were also purchased
from Sigma. PNGase F was obtained from New England Biolabs
(Hitchin, UK). Bradford dye reagent was obtained from Bio-Rad
Laboratories (Hemel Hempstead, UK). Polyvinylidene difluoride
(PVDF) membrane was obtained from GE Healthcare (Buckinghamshire,
UK). Cell Proliferation Kit I (MTT) and protease inhibitor cocktail
were purchased from Roche Diagnostics GmbH (Mannheim, Germany). The
rabbit anti-ABCG2 polyclonal Ab (H-70) used for western blotting
(WB), rabbit anti-GADPH polyclonal antibody (Ab) (FL-335), goat
anti-mouse HRP-conjugated Ab and goat anti-rabbit HRP-conjugated Ab
were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Mouse monoclonal anti-Pgp Ab (C219) was obtained from Alexis
Biochemicals (Lörrach, Germany). The mouse monoclonal anti-p53 Ab
and mouse monoclonal anti-ABCG2 used for the immunofluorescence
experiments were obtained from Novus Biologicals (Germany).
Human/mouse cleaved caspase-3 (Asp175) monoclonal antibody (MAB835)
was obtained from R&D Systems (Abingdon, UK). The MFP488
fluorescent secondary antibodies was obtained from MoBiTec
(Goettingen, Germany). Mounting medium with DAPI was obtained from
Santa Cruz Biotechnology. The 4X Laemmli sample buffer and 4–20%
mini-PROTEAN® TGX™ precast gels were obtained from
Bio-Rad Laboratories.
Cell culture
Two resistant ovarian cancer cell lines were used in
the present study. From the W1 cell line {primary ovarian cancer
cell line, established from ovarian cancer tissues that were
obtained from an untreated patient in December 2009 [these cell
lines were derived in our laboratory as described previously by
januchowski et al (39)]},
we generated the W1TR subline, which is resistant to topotecan
(top) (W1 is topotecan resistant to a top concentration of 24
ng/ml), and the W1PR subline, which is resistant to paclitaxel
(pac) (W1 is paclitaxel resistant to a pac concentration of 1,100
ng/ml). The second ovarian cancer cell line, A2780T1, is a
topotecan-resistant subline that was generated from the
commercially available established human ovarian carcinoma cell
line A2780 (obtained from ATCC, Poland). The drug-resistant
sublines were generated by the exposure of the drug-sensitive cells
to incrementally higher concentrations of each drug. The human
colon adenocarcinoma doxorubicin (dox)-resistant cell line LoVo and
LoVo/Dx were also used (obtained from ATCC). LoVo/Dx was cultured
in the presence of 200 ng/ml dox to maintain its resistant
phenotype.
The LoVo, LoVo/Dx, A2780 and A2780T1 cell lines were
cultured in EMEM; and the W1, W1PR and W1TR cell lines were
cultured in RPMI-1640 medium. The media were supplemented with 10%
FBS, 2 mM L-glutamine and 1% antibiotic-antimycotic solution. The
cells were cultured at 37°C in a humidified atmosphere with 5%
CO2 (v/v).
The W1TR and A2780TR1 cells were shown to
overexpress BCRP, while the W1PR and LoVo/Dx cells overexpress Pgp
(39,40).
Cell viability assay
To determine the cell viability we used the Cell
Proliferation Kit I (MTT) according to the manufacturer's
instructions. The survival assay was performed to estimate the
extent of cell resistance to the chemotherapeutic agents, BFA and
CAS. Briefly, the cells were seeded at 4×103 cells/well
(200 μl) in 96-well culture plates and pre-incubated for 48
h. To examine the effect of BFA, CAS or chemotherapeutic drugs on
cell survival, the cells were treated with increasing
concentrations of BFA, CAS or the different drugs for 72 h.
Subsequently, 10 μl of MTT labeling reagent was added to the
medium (the final concentration of MTT was 0.5 mg/ml) for 4 h and
100 μl of the solubilized solution was then added to each
well. After an overnight incubation, the absorbance was measured in
a microplate reader at 570 nm with a reference wavelength of 720
nm.
In addition, we examined the effect of BFA and CAS
on cellular resistance to chemotherapeutic drugs. The cells were
seeded at 4×103 cells/well in 96-well culture plates.
After 48 h, the cells were pre-treated with BFA or CAS for another
48 h, and the medium was subsequently replaced with fresh medium
supplemented with a cytostatic drug and BFA or CAS. The BFA
concentration used was based on previously established values for
each cell line: 10 ng/ml for LoVo/Dx, 7 ng/ml for W1PR, 6 ng/ml for
W1TR and 8 ng/ml for A2780T1 cells; for CAS: 10 μg/ml for
LoVo/Dx, 50 μg/ml for W1PR and W1TR and 60 μg/ml for
A2780T1 cells. After 72 h of incubation, cell proliferation was
assessed as previously described. As a control, we used cells that
were treated with only the cytostatic drugs for 72 h. All
experiments were repeated three times.
Preparation of cell lysates and
glycosidase treatment
For the immunoblot analysis of protein expression,
the cells were pre-incubated for 48 h (1×106 cells/75
cm2 culture flask). The medium was then replaced with
fresh medium only or fresh medium supplemented with BFA or CAS at
different concentrations. After 72 h of incubation, the cells were
harvested by trypsinization and pelleted by centrifugation. The
cell pellets were washed once with phosphate-buffered saline (PBS),
and the cells were lysed with RIPA lysis buffer supplemented with
protease inhibitor cocktail for 5 min on ice. The cell lysates were
centrifuged at 8,000 × g at 4°C for 10 min, and the total protein
concentration in the supernatant was measured. The protein
concentration was determined using Bradford dye reagent with BSA as
the standard. Glycosidase treatment was performed by incubating 35
μg of the cell lysate with 5 μl of PNGase F at 37°C
for 10 min.
Western blot analysis
Western blot analysis was conducted under reducing
conditions. The cell lysate samples (35 μg) were initially
mixed with 4X Laemmli sample buffer and incubated at room
temperature (RT) for 20 min. The lysates were then
electrophoretically separated on a 4–20% mini-PROTEAN®
TGX™ precast gel and then electroblotted onto a PVDF membrane. The
membrane was blocked with 5% (w/v) dry milk in TBS at RT for 1 h.
The membranes were then incubated with anti-Pgp, anti-BCRP,
anti-p53 primary (1:500 dilution) and anti-CAS 3 (1:1,000 dilution)
antibodies at 4°C overnight. The membranes were also probed with
the anti-GAPDH antibody (1:1,000 dilution) as a loading control.
For the secondary antibody, horseradish peroxidase (HRP)-linked
anti-species antibody was used at a dilution of 1:2,000.
HRP-dependent luminescence was developed with the Femto SuperSignal
reagent and detected using a UVP imaging system.
Immunofluorescence analysis
Cells were fixed and permeabilized in 4%
paraformaldehyde for 15 min, followed by ice-cold methanol for 15
min at −20°C and washed with PBS. The cells were then incubated
with 5% HSA for 1 h. To detect Pgp, the cells were treated with a
mouse monoclonal antibody against Pgp at a dilution of 1:30 for 1 h
at RT. The cells were then washed five times with PBS and incubated
with an MFP488-labeled anti-mouse antibody for 1 h at RT in the
dark. To visualize the cell nuclei, the cells were mounted with a
DAPI-containing mounting medium (blue). To detect BCRP, the cells
were treated with a mouse monoclonal anti-BCRP antibody (Novus
Biologicals) at a dilution of 1:200 for 1 h at RT. Subsequently,
the cells were washed with PBS and incubated with an MFP488-labeled
anti-mouse secondary antibody. Finally, the coverslips were mounted
with DAPI mounting medium. Fluorescent images were collected using
a ZEISS light microscope. The expression of Pgp and BCRP was
analyzed by pseudo-color representations of the fluorescence
intensity (green).
Flow cytometric analysis
To assess the efflux activity of Pgp and BCRP, whole
resistant cell lines were incubated with BFA or CAS and drugs,
using previously described conditions. We used sensitive W1, A2780
and LoVo cell lines as a control for the inability to eliminate the
drugs from cells. In addition, W1PR, W1TR, A2780T1 and LoVo/Dx
cells were used as a positive control for drug efflux. We used the
fluorescent dye Rhodamine 123 (Rho123) as an index of Pgp activity
and Hoechst33342 (H33342) as an index of BCRP activity. The cell
suspensions (1×106/ml) were incubated with 0.5
μg/ml Rho123 or 2.5 μg/ml H33342 for 1 h at 37°C in
medium. Next, the cells were washed twice in cold PBS with 500
μM verapamil (a known MDR inhibitor), and cellular uptake of
Rho123 or H33342 was immediately analyzed using a FACSAria III (BD,
Warsaw, Poland) with the FCS express Plus software program. In each
analysis, 10,000 events were recorded. The fluorescent emission was
collected at 488 nm for Rho123 and at 375 nm for H33342.
Statistical analysis
The statistical analysis was performed using
Microsoft Excel software. The statistical significance of the
differences was determined by applying the Student's t-test. A
p-value ≤0.05 was considered to indicate a statistically
significant result.
Results
Effect of BFA and CAS on cell
viability
To identify the impact of BFA and CAS on cell
viability, the cells were treated with BFA or CAS at increasing
concentrations for 72 h. BFA and CAS induced a dose-dependent
decrease in cell viability in all of the drug-resistant cell lines
(data not shown). In subsequent experiments, we selected an
inhibitor concentration that would only decrease cell viability to
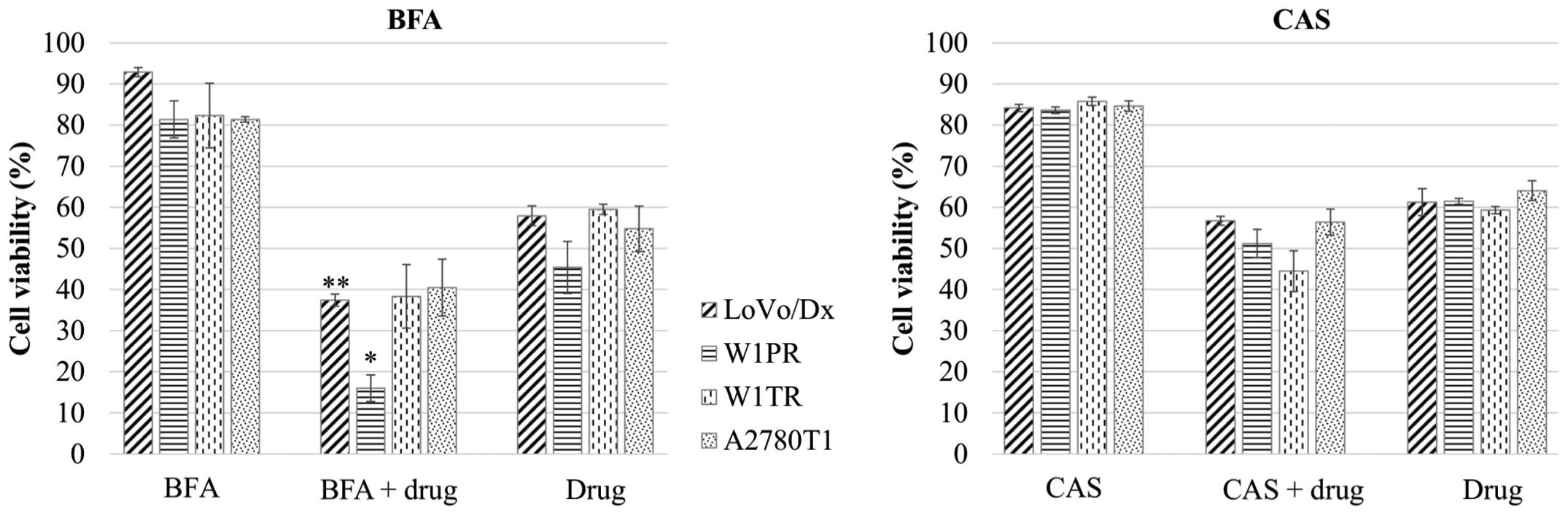
81–93% in each cell line (Fig. 1).
We decided to use an inhibitor concentration that was not too toxic
to the cells, but that provided an observable effect. To achieve
this goal, BFA was applied at a concentration of 10 ng/ml for
LoVo/Dx, 7 ng/ml for W1PR, 6 ng/ml for W1TR and 8 ng/ml for A2780T1
cells, and CAS was used at a concentration of 10 μg/ml for
LoVo/Dx, 50 μg/ml for W1PR and W1TR and 60 μg/ml for
A2780T1 cells.
Next, we tested whether BFA or CAS and the
cytostatic drugs had a synergistic effect on the drug-resistant
cell lines. Cells were treated with BFA or CAS at the indicated
concentrations for 48 h. The medium was then replaced with fresh
medium supplemented with BFA or CAS and one of the cytostatic drugs
at the following concentrations: dox 200 ng/ml, top 24 ng/ml and
pac 1,100 ng/ml. The cells were incubated for another 72 h. We also
treated cells with the cytostatic drugs alone for 72 h. As a
control, cells were assessed in the absence of cytostatic
drugs.
In the LoVo/Dx and W1PR cell lines, we observed a
statistically significant synergistic effect of BFA and the drugs
(Fig. 1). Furthermore, the
viability of the LoVo/Dx cell line in response to the co-treatment
with BFA and dox showed the most significant decrease (p<0.01)
in comparison to the cells that were treated with dox. Co-treatment
of the W1TR cell line with BFA and top resulted in reduced cell
viability in comparison to cells that were treated with top alone,
but the changes were not statistically significant (p<0.1). In
the A2780T1 cells supplemented with BFA and top, there were no
significant changes in viability in comparison to cells that were
treated with top alone (Fig.
1).
CAS treatment had no statistically significant
effect on the sensitivity of tumor cells to cytostatic drugs
(Fig. 1), although in W1PR, W1TR
and A2790T1 cells, CAS reduced the cell viability in comparison to
cells that were treated with the drug alone, but the changes were
not statistically significant (p<0.1).
Effect of BFA and CAS on the expression
of Pgp and BCRP proteins
Western blot analyses were conducted to assess
whether BFA or CAS could affect the expression of Pgp and BCRP
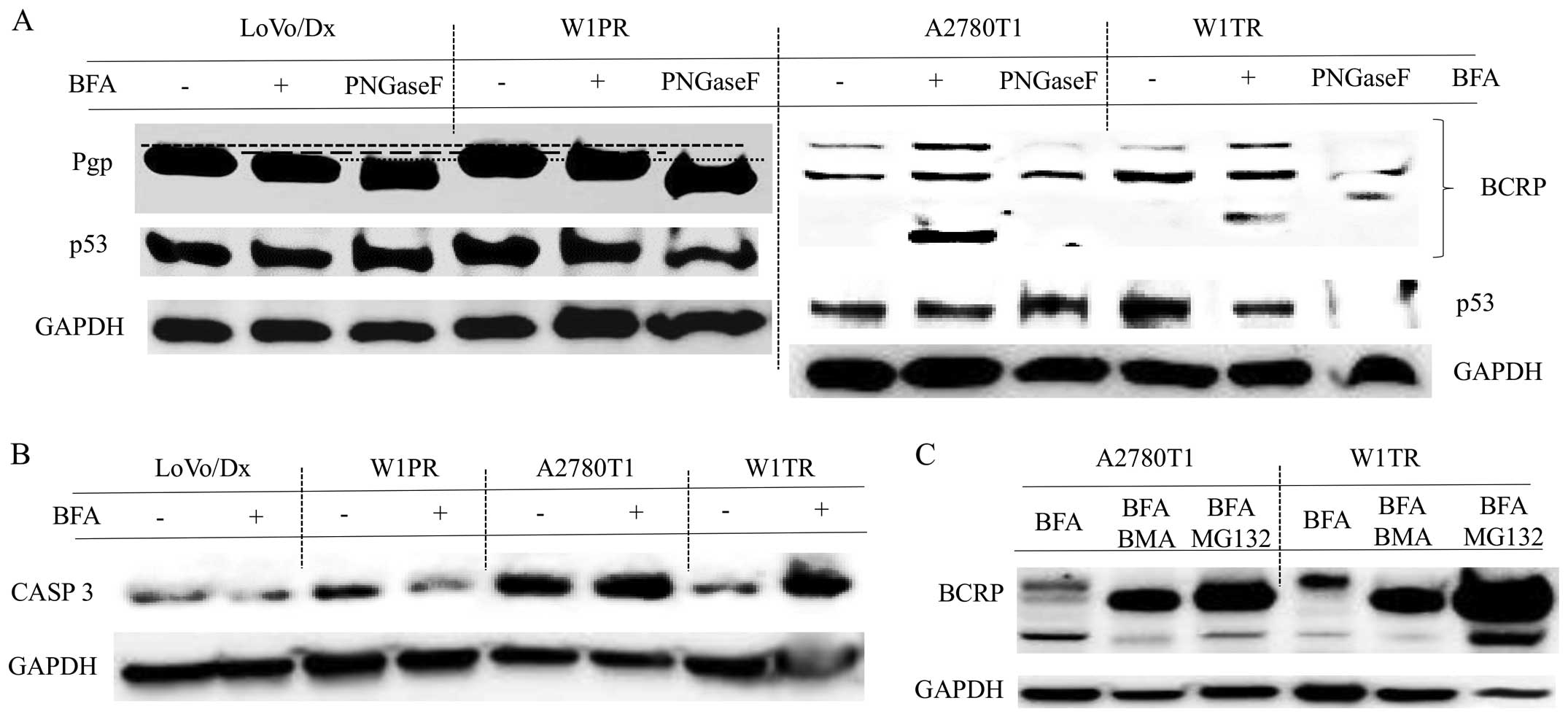
proteins. After BFA treatment, Pgp displayed a molecular mass that
was approximately a few kDa less than that of the fully
glycosylated protein (Fig. 2A). As
a control, the LoVo/Dx and W1PR cell lysates were incubated with
N-glycosidase F (PnGase F) to remove all of the
N-linked glycans. After incubation with PNGase F, we
observed bands at an even lower molecular weight then those
observed for the BFA-treated cells. We did not observe any changes
in the level of p53 protein expression in the LoVo/Dx or W1PR cells
after BFA treatment (Fig. 2A).
For BCRP, it was characteristic to identify two
bands in the control samples, indicating two forms of the BCRP
protein (Figs. 2A and 3). After incubation with PNGase F, only
the band with the lower molecular weight was observed (Fig. 2A). The lower band was consistent
with the size of the unglycosylated protein. In the BFA-treated
cells, we observed a third, additional band (Fig. 2A), and a more fully glycosylated
form of BCRP. We did not notice any changes in p53 expression in
the A2780T1 cells following treatment with BFA, in contrast to W1TR
cells in which the level of p53 protein was reduced after BFA
supplementation (Fig. 2A). BFA
treatment increased and decreased the expression of CASP 3 in the
W1TR and W1PR cell lines, respectively (Fig. 2B).
Due to the observation of a third band of BCRP in
BFA-treated A2780TR1 and W1TR cells, we decided to further
investigate the mechanism underlying this phenomenon. We suspect
that BFA could affect BCRP degradation. Protein degradation occurs
at two major sites: the lysosome and the proteasome. To examine
whether the lysosomal or the ubiquitin-proteasomal pathway was
involved in BCRP degradation, we pre-cultured A2780TR1 and W1TR
cells in the presence or absence of 2 μM MG132 (a
proteasomal degradation inhibitor) or 10 nM BMA (a lysosomal
degradation inhibitor) for 3 h and subsequently replaced the medium
with fresh medium supplemented with BFA and MG132 or BMA for
another 72 h. We verified that the concentrations of the protein
degradation inhibitors were non-toxic to the cells. We found that
pre-treatment with MG132 or BMA led to an increase in the
expression only of unglycosylated BCRP, but the lowest band was
still observed (Fig. 2C). Moreover,
BCRP protein expression was increased in the W1TR cells that were
co-treated with BFA and MG132, despite the lower total protein
concentration represented by GAPDH (Fig. 2C).
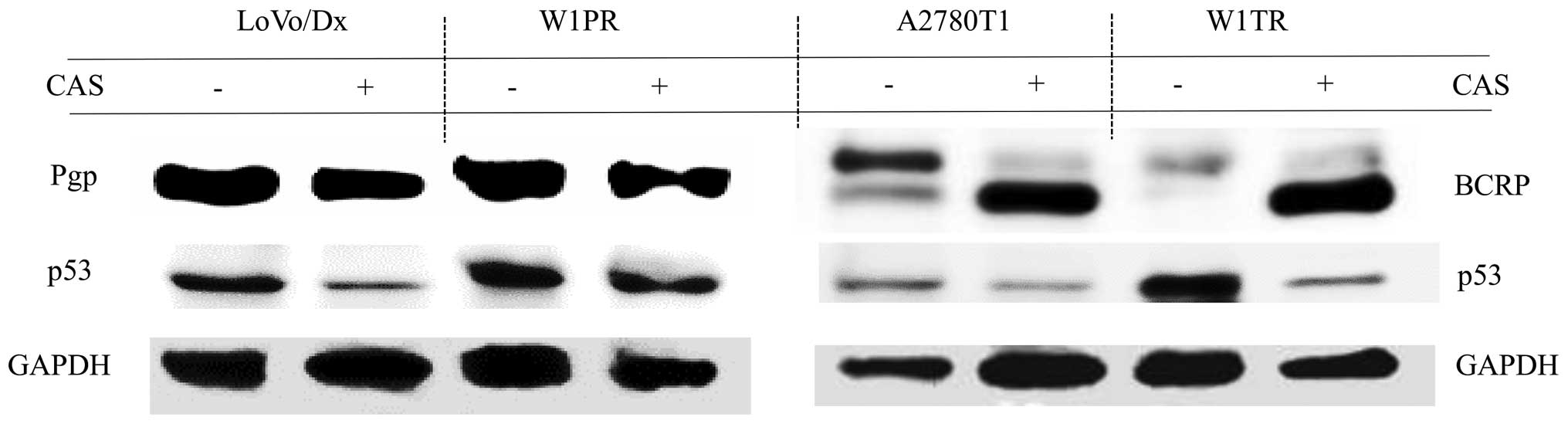
Pgp bands of the same size were obtained from
LoVo/Dx and W1PR cells treated with CAS compared with the controls
without drug (Fig. 3). BCRP was
detected in the form of two bands in the A2780T1 and W1TR cell
lines. The upper bands corresponding to the fully glycosylated form
of BCRP were more apparent. In cells treated with CAS, BCRP was
mainly detected as a lower-molecular-weight protein (Fig. 3) corresponding to the size of
unglycosylated BCRP. Reduced expression of p53 was detected in all
of the cells treated with CAS (Fig.
3).
Effect of BFA and CAS on the cellular
localization of Pgp and BCRP
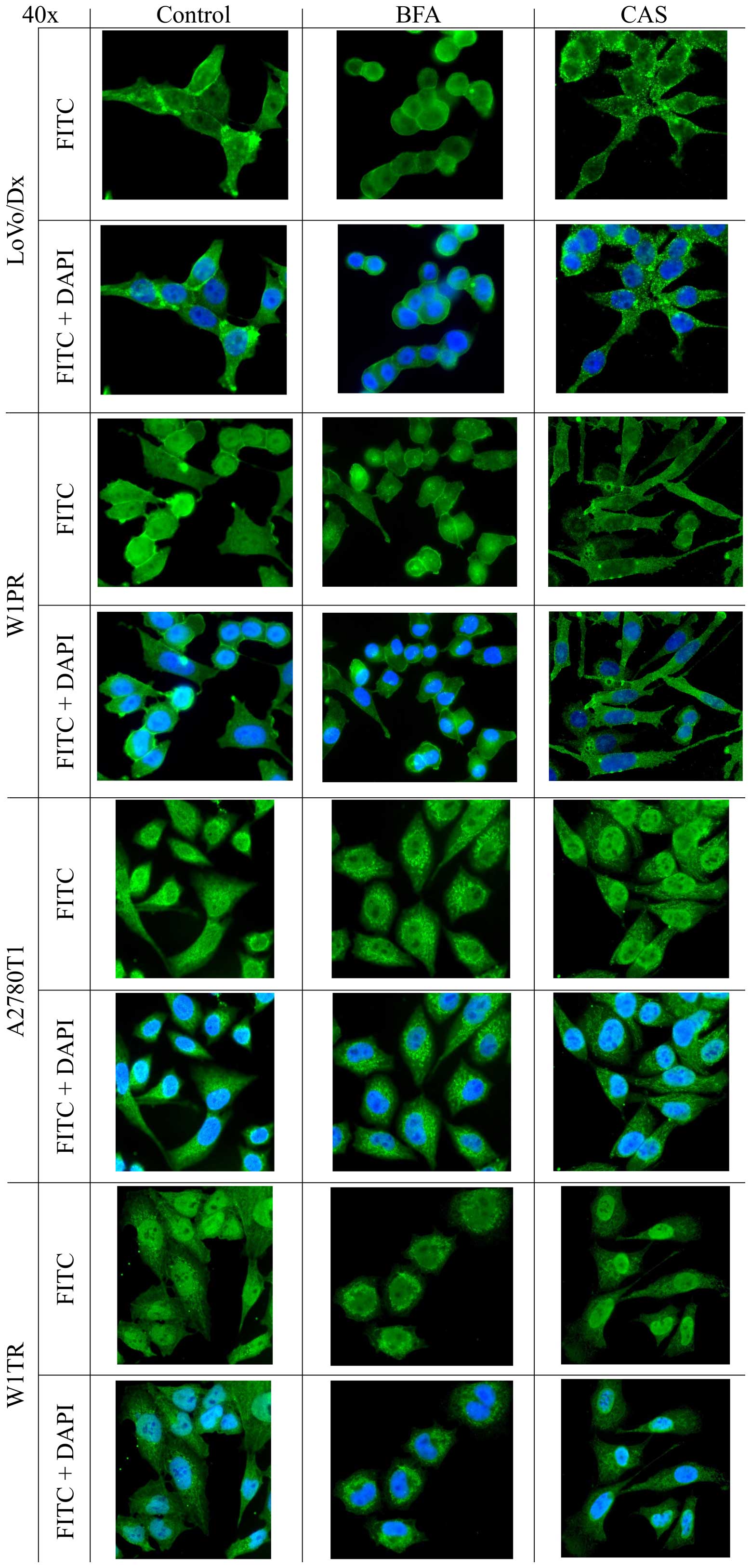
We were interested in whether BFA or CAS affected
the cellular localization of Pgp in W1PR and LoVo/Dx cells and the
localization of BCRP in W1TR and A2780T1 cells. In LoVo/Dx cells,
Pgp was mostly detected at the plasma membrane, in the cytoplasm
and around the nuclear envelope in the absence of BFA or CAS
treatment (Fig. 4). After BFA
treatment, we noticed changes in the morphology of the cells; they
became rounder, while the Pgp signal remained the same as in the
control. In contrast, in CAS-treated cells, Pgp was mainly observed
within the cytoplasm in the form of granules (Fig. 4). In W1PR cells, the localization of
Pgp was similar to that in the LoVo/Dx cells; it was observed at
the plasma membrane, in the cytoplasm and close to the nuclear
envelope. BFA treatment caused a Pgp signal in the cytoplasm to
move to the nuclear periphery. In the CAS W1PR cells, the
fluorescence signal was mostly cytoplasmic, which was a little
different compared with the other cells (Fig. 4).
In the untreated A2780T1 and W1TR cells, BCRP was
detected in the cytoplasm (Fig. 4).
After BFA treatment, the localization of BCRP in the A2780T1 cell
line changed to a granular signal. The morphology of W1TR cells
changed after BFA treatment, exhibiting a rounder appearance
compared with the control (Fig. 4).
CAS treatment affected BCRP localization only in A2780T1 cells, in
which BCRP protein aggregates were observed in the cytoplasm
(Fig. 4).
Effect of BFA and CAS on the cellular
accumulation of Rho123 or H33342
To verify whether BFA or CAS could have a functional
role in drug uptake and efflux, fluorescence accumulation and
efflux were investigated in the absence and presence of BFA or CAS.
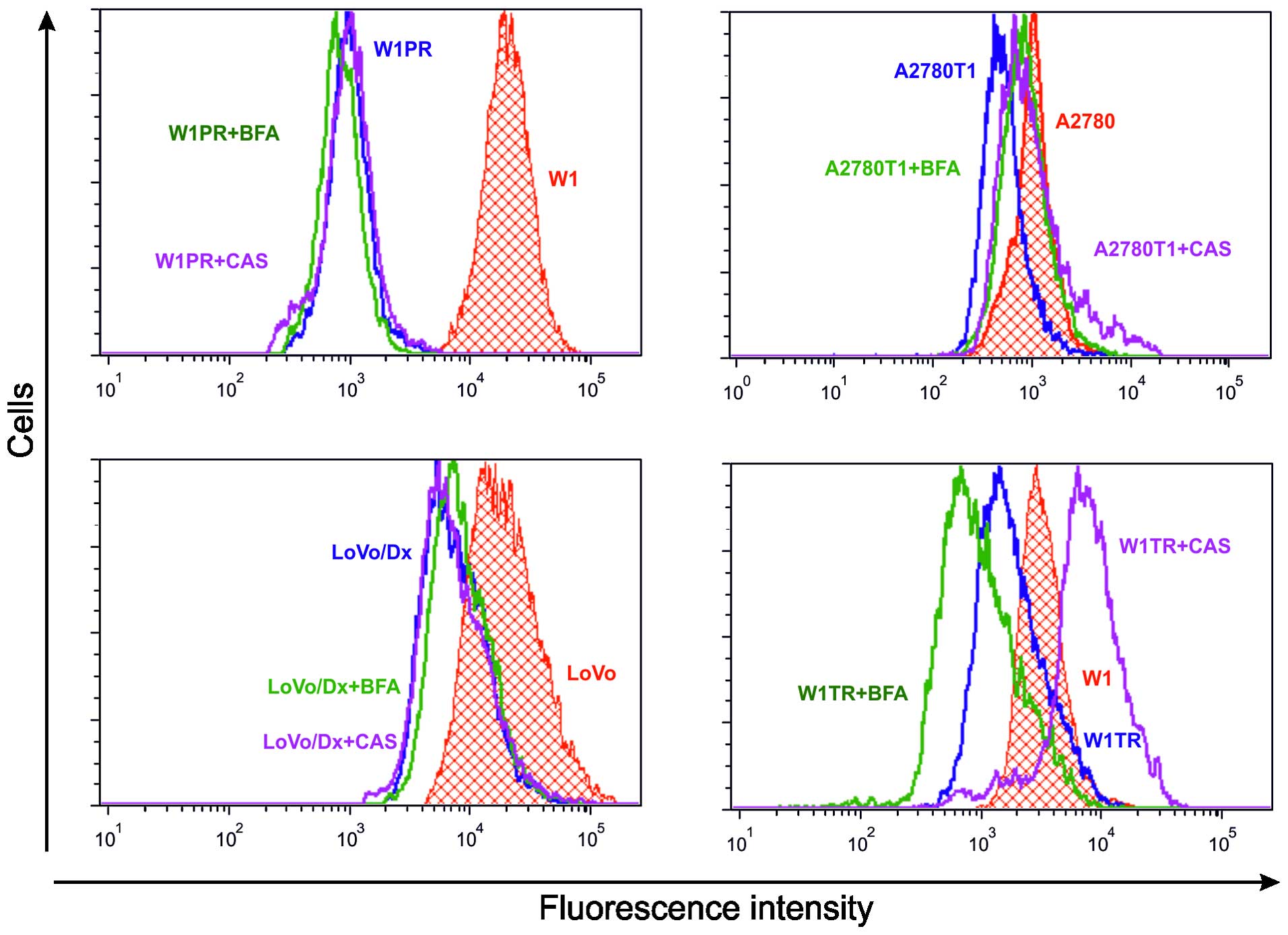
The intracellular uptake of the fluorescent dye Rho123 or H33342
was evaluated in sensitive and MDR cell lines, and in MDR cells
treated with BFA or CAS. A precise analysis was conducted based on
the mean fluorescence intensity (MFI). The results indicated that
the amount of Rho123 or H33342 accumulation in sensitive cells was
increased compared with that in resistant cells in all cell lines
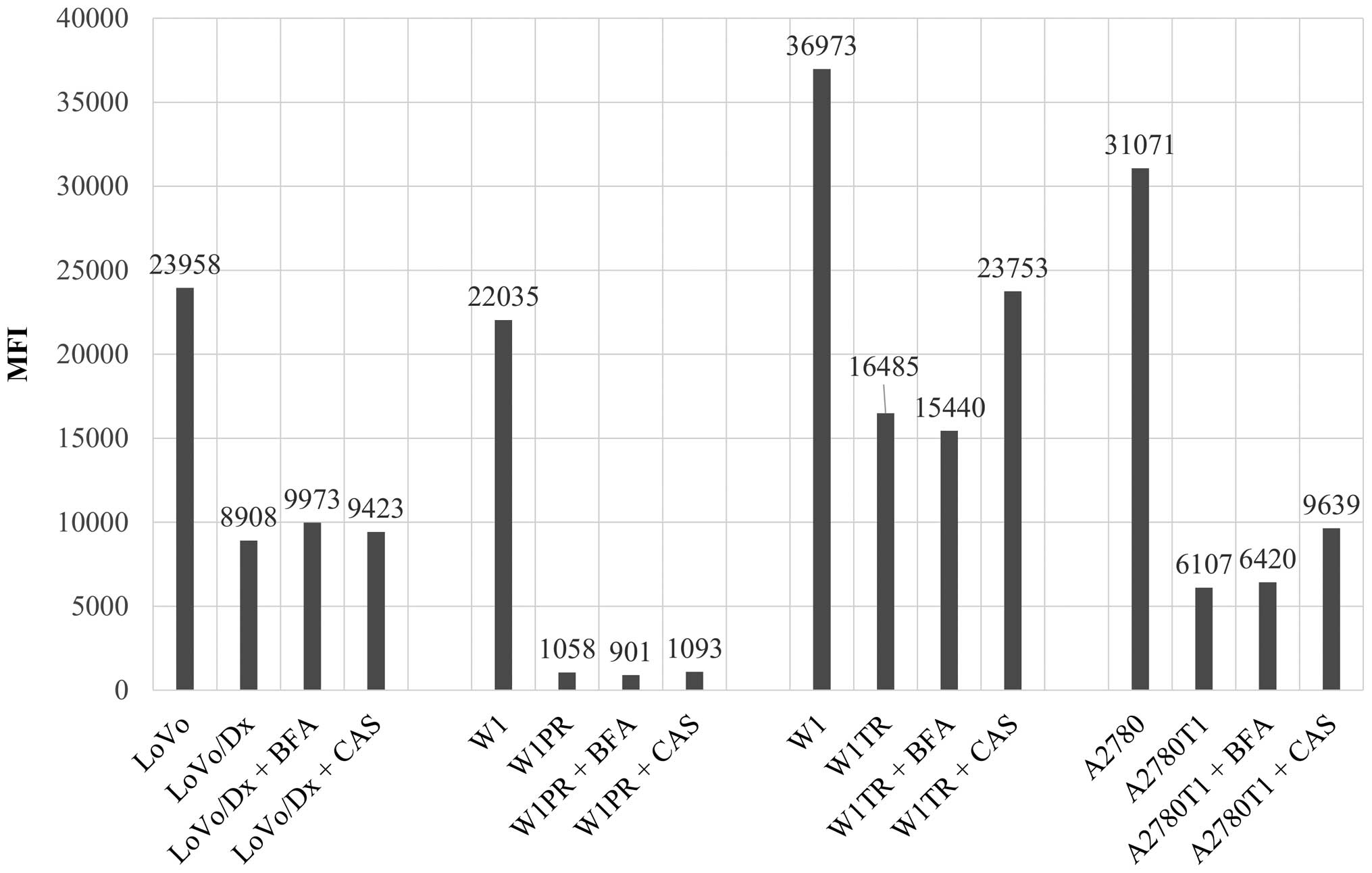
(Fig. 5). The level of
intracellular Rho123 was ~2.7 times higher in LoVo than that in the
LoVo/Dx cells and 20 times higher in W1 than that in the W1PR cells
(Fig. 6). Similarly, the
intracellular level of H33342 was ~2.2 times higher in W1 than that
in the W1TR cells and 5 times higher in A2780 than that in the
A2780T1 cells (Fig. 6). We observed
an increase in the MFI of Rho123 in the LoVo/Dx cells treated with
BFA and CAS in comparison with the LoVo/Dx control sample, but
these changes were not significant (Figs. 5 and 6). No differences in the MFI of Rho123
were detected in the W1PR cells after BFA and CAS stimulation. The
addition of BFA to W1TR cells had no effect on MFI, but CAS led to
a significant increase in the level of intracellular H33342 in the
resistant W1TR cells (Figs. 5 and
6). The analogous effect was
observed in A2780T1 cells, in which CAS increased the MFI of H33342
in comparison to BFA (Figs. 5 and
6).
Discussion
The expression of MDR proteins, including Pgp and
BCRP, is responsible for chemotherapy failure in numerous cancers.
Consequently, the application of drugs that block the activity and
function of MDR proteins represents an opportunity in current
research in oncology. New strategies are needed to circumvent the
effects of MDR in cancer chemotherapy. Since both proteins are
glycosylated, it can be anticipated that the use of glycosylation
inhibitors will change their expression and increase the
sensitivity of cells to cytostatic drugs.
In the present study, we tested two inhibitors of
N-linked glycosylation, BFA and CAS, as potential factors
facilitating an increase in the efficiency of chemotherapy. BFA
inhibits protein transport from the ER to the Golgi apparatus
(20). In the present study, BFA
increased sensitivity to cytostatic drugs in LoVo/Dx and W1PR cell
lines characterized by Pgp overexpression. This finding is
consistent with the results of Fu et al, who also showed
that BFA increased the intracellular accumulation of the Pgp
substrate daunorubicin, resulting in a higher toxicity in HeLa
cells (41). Fu et al stated
that this phenomenon resulted from an increase in the localization
of Pgp in the ER (41). We
suspected that BFA had a similar effect on Pgp in the present
analyses. BFA also significantly increased the intercellular
accumulation of ZDV, another Pgp substrate, in a pre-adipocyte cell
line (42). We suggest that BFA
induces an increase in the accumulation of chemotherapeutic agents
in the cytoplasm of cells that are characterized by Pgp
overexpression. Furthermore, Rhodes et al demonstrated that
BFA blocks non-Pgp-mediated MDR activity in the COR-L23/R cell line
(43). In contrast, other
researchers reported that BFA has no effect on the cellular drug
level (44–46). Therefore, we agree with the opinion
that BFA may have a sensitizing effect, depending on the drug
resistance level, mainly by reversing Pgp-mediated resistance
(30).
The expression of BCRP in W1TR and A2780TR1 cell
lines is responsible for top resistance. We did not observe any
significant changes in top resistance following BFA co-treatment in
these cell lines. It has been reported that BFA has both a
sensitizing and a protective effect, depending on the drug
resistance level (30). This
finding could explain why we did not notice any significant changes
in W1TR and A2780T1 cell viability during co-treatment with BFA and
drugs. The BFA concentrations used in this experiments may not have
been sufficient for reversal of the BCRP resistant effect.
Wlodkowic et al showed that BFA had a different effect on
cell proliferation in various human follicular lymphoma cell lines
(28). We suspect that ovarian
cancer cell lines may not be susceptible to BFA.
The results of the immunoblot analysis demonstrated
that BFA partially blocked the N-linked glycosylation of Pgp
in the W1PR and LoVo/Dx cell lines. We observed bands that were
approximately a few kDa smaller than the control bands, but they
were not characteristic of Pgp lacking sugar as determined by
treatment with PNGase F. It is possible that the concentration of
BFA used in the present study was insufficient to completely
inhibit Pgp glycosylation. BFA has not been used as an anticancer
drug to date, but it has been shown to induce p53-independent
apoptosis in human cancer cells (25,29,47),
and internucleosomal DNA fragmentation associated with apoptosis
(25). Thus, we assessed the level
of p53 expression in all of the tested cell lines. Our results were
inconsistent with some of the literature since we did not observe
any differences in p53 expression in cells that were treated with
BFA but rather in the W1TR cell line, which displayed lower p53
expression. An explanation for this phenomenon is provided by Lee
et al, who showed that BFA caused concentration-dependent
changes in apoptosis-related protein levels and that these effects
were also linked to an increase in p53 levels (26). The discrepancy in p53 expression may
be a consequence of the association of apoptosis induced by BFA and
mitochondrial breaching and caspases activation (27,28).
Thus, we evaluated the expression of CASP 3 in BFA-treated cells.
All of the control cells without BFA possessed the protein, which
is consistent with the previous supplementation of the cells with
cytostatic drugs that could initiate the apoptotic cascade. In
BFA-treated W1TR cells, we observed elevated CASP 3 expression,
demonstrating that in this example, BFA potently induced apoptosis
associated with caspase activation. This finding is in agreement
with a previous report and confirms the involvement of caspase in
cell death in this model (28). In
contrast, in W1PR cells treated with BFA, a lower level of CASP 3
expression was observed, revealing that in various cell lines BFA
can affect other signaling pathways. Western blot analysis was also
used to detect BCRP protein in the A2780T1 and W1TR cell lines
before and after BFA supplementation. In the control samples, two
bands were identified that were consistent with antibody
specifications. These two bands represented the unglycosylated and
fully glycosylated forms of BCRP. In the BFA-treated cells, in
addition to these two bands, we observed another band corresponding
to a protein with a smaller molecular weight. Nakagawa et al
also identified additional bands in Flip-In-293/ABCG2 cells that
had been incubated with BFA and suggested that they were
unglycosylated or immature forms of BCRP (15). We suggest that BFA caused the
partial degradation of BCRP, probably due to ERAD in which
misfolded proteins are retrotranslocated from the ER to the cytosol
and subsequently degraded by the ubiquitin-proteasome system
(16,22). We demonstrated that N-linked
glycosylation was essential for BCRP expression and that the
disruption of this process resulted in protein expression,
confirming a previous study (14).
To examine the potential impact of BFA on BCRP protein degradation
in both cell lines, we used two inhibitors of protein degradation,
BMA and MG132. However, in the presence of these inhibitors, the
degraded BCRP was barely visible, which may indicate that BFA did
not result in proteasomal or lysosomal degradation alone but that a
more complicated pathway could be involved, as claimed by Ferris
et al (16).
In LoVo/Dx and W1PR cells treated with BFA, we did
not observe any changes in the localization of the
immunofluorescent Pgp signal in comparison to the control without
drugs. However, Labroille et al showed a 35% decrease in the
level of surface Pgp when resistant cells were treated for 24 h
with BFA (48). Similarly, Fu et
al showed that 5 μg/ml BFA completely blocked
intracellular trafficking of Pgp to the cell surface in HeLa cells
(41). In the present study, a
similar phenomenon was not detected through 72 h of incubation with
BFA. We suspect that the inhibition of BFA was not completely
efficient, and therefore Pgp was still observed in the cell
membrane in the W1PR cell line. The time needed to transport Pgp
from the ER to the Golgi and finally to the plasma membrane is
12–48 h (41), and therefore our
72-h incubation should have been sufficient. However, another study
showed that the total level of Pgp was stable in BFA-treated K562
MDR cells (48), indicating that
the surface Pgp pool is continuously supplied by the cytoplasmic
pool via the migration of Golgi vesicles containing Pgp towards the
cell surface (48).
Immunofluorescent analysis of Pgp revealed granules in W1PR cells
treated with BFA and similar granules with positive staining for
BCRP in the A2780T1 cell line. These findings could be explained by
a higher condensation of Pgp/BCRP in the cytoplasm induced by BFA
disorders in the intercellular protein trafficking of newly
secreted proteins. Furthermore, in the BFA-treated W1PR cells, we
observed an accumulation of Pgp close to the nucleus. Our
explanation is consistent with that of other researchers. The
entrapment of insoluble forms of a protein in the ER is commonly
observed (16). Granules can be
describe as a trapped, newly synthesized Pgp in the fused Golgi-ER
compartment (41) since BFA could
cause the collapse of Golgi membranes into the ER (47). The accumulation of protein may be
associated with an inefficient detection of misfolded protein
(19) induced by BFA treatment, or
it could indicate that the protein was degraded through ERAD
(16). BFA did not have a
significant effect on Pgp expression in LoVo/Dx cells, probably due
to the large intracellular pool of Pgp. We observed a clear change
only in the morphology of the cells, which become rounder. A
similar effect was observed for BCRP in the W1TR cell line after
BFA incubation. This finding may indicate that the concentration of
BFA was slightly toxic to the cells. However, we did not observe
nuclear condensation or fragmentation, which is characteristic of
apoptotic cells (26,28). The results may have been caused by
the ability of BFA to block cell adhesion, as demonstrated in the
GR and oVCAR-3 cell lines (24,26).
BFA started to induce apoptosis associated with mitochondrial
breaching and caspase activation, as demonstrated in human
follicular lymphoma cell lines (28). We confirmed that the morphological
changes induced in BFA-exposed cells could be dependent on cell
cycle arrest (24).
CAS is an α-glucosidase inhibitor. During the
simultaneous treatment with CAS and drugs, we observed a decrease
in cell viability, but this change was not statistically
significant. This result could indicate that CAS did not
significantly affect the activity of glycoproteins. Nakajima et
al also showed that CAS treatment had an insignificant effect
on UGTA9 activity in HEK293 cells (17). In contrast to our results, a smaller
reversal effect of CAS on cellular resistance has been shown in
Flip-In-293/ABCG2 cells (15). It
has also been demonstrated that CAS reduced the proliferative
properties of C6 glioblastoma cells (37). Its analogues SO-OCS and CO-OCS
reduced cell proliferation by inducing cell cycle arrest in breast
cancer cell lines and induced cell death in cancer cells via
Bcl2 family proteins (49).
The discrepancies between the literature and our results may be
explained by differential effects of CAS on different cell
types.
The results of the western blot analysis showed that
CAS had no effect on Pgp expression. In control samples of BCRP
using the A2780T1 cell line, two bands were observed, which was
similar to the results obtained with BFA. These two bands
represented the unglycosylated and fully glycosylated forms of
BCRP. In the W1TR cells, we noted only one, the heavier band,
suggesting that this sample contained mainly one form of BCRP, the
fully glycosylated form. In both cases, in the A2780T1 and W1TR
cell lines, CAS treatment resulted principally in the appearance of
the lower bands. In our opinion, this result demonstrated that CAS
blocked N-glycosylation of the BCRP protein. In contrast,
Nakagawa et al showed that incubation with CAS only reduced
the level of BCRP protein expression in Flip-In-293/ABCG2 cells,
but no additional bands with a lower molecular weight were observed
(15). Notably, the level of p53
expression was reduced in all of the cells that were supplemented
with CAS. A few possible explanations may explain the reduced
expression of p53. One explanation, which has been described in
HL-60 cells, is that the gene was deleted or mutations occurred in
the p53 promoter or within regulatory regions (50). It has been well established that the
loss of p53 prevents cellular senescence and apoptosis (51), which likely explains why we were
unable to detect a decrease in cell viability in response to CAS
treatment.
We wanted to determine whether CAS had any influence
on the cellular localization of MDR proteins. The
immunofluorescence results revealed that the Pgp signal around the
nucleus was diminished in cells treated with CAS. Furthermore, in
the LoVo/Dx cells, we observed a granular signal in the cytoplasm.
Similarly, a granular and tubular signal was observed for BCRP in
the A2780T1 cells. These results indicated that CAS did not
significantly modify the export of surface glycoproteins and that
it could abrogate protein transport in the cytoplasm. Lin et
al showed that CAS may lead to an accumulation of unfolded
proteins in the ER due to improper glycosylation (52). We propose a similar explanation for
the effects of CAS treatment in the present study. We also suggest,
in agreement with a previous report, that the disruption of BCRP
N-linked glycosylation may enhance ubiquitin-mediated
proteasomal degradation (15). The
N-linked glycan of BCRP could be a checkpoint, and it has
been considered to be important for the stabilization of nascent
BCRP protein (15). We did not
observe any changes in W1TR cells supplemented with CAS.
Schraen-Maschke and Zanetta also showed that C6 glioblastoma cells
treated with CAS displayed reduced re-aggregation (37).
We assessed whether BFA or CAS increases the
intercellular accumulation of drugs in resistant cell lines. We
showed that whole, sensitive versions of cancer cell lines retained
a greater amount of fluorescent dye in comparison with the
resistant cell lines. Similarly, Meschini et al observed
high levels of dox in LoVo cells and only very low amounts of drug
in LoVo/Dx cells (6). Furthermore,
Verovski et al showed significantly reduced Rho123 uptake in
Pgp-positive H134AD cells relative to H134 cells (30). These findings confirmed the
hypothesis that resistant cell lines express MDR proteins that
protect the cells against the cytotoxic effects of drugs. For this
reason, the MFI values for Rho123 or H33342 were reduced in
resistant cells. We noted that BFA increased the accumulation of
fluorescent dye only in the LoVo/Dx cells. This result confirmed
our findings using the MTT test, in which BFA caused the most
statistically significant decrease in cell viability in LoVo/Dx
cells. Janneh et al also showed that BFA increased the
accumulation of zidovudine in 3T3-F442A pre-adipocytes (42). We agree with the finding that BFA
had a toxic effect mainly toward LoVo/Dx cells that was similar to
the effect reported for oVCAR-3 cells (26). In W1TR cells, BFA had a small
protective effect (decrease in MFI). Similar results have been
described by Verovski et al, who showed a decrease in
cellular dox content in PSN1/ADR cells that were treated with BFA
(30). It is possible that BFA
plays a role in drug resistance associated with the Pgp
transporter, but only in selected types of cancers. Flow cytometric
analyses showed that CAS had no effect on the accumulation of
Rho123 in W1PR cells, but in the remaining cell lines, it was very
important for pump activity. We detected the greatest increase in
H33342 accumulation in W1TR cells, a somewhat smaller increase in
A2780T1 cells and the smallest increase in LoVo/Dx cells. These
results differ from those obtained for the MTT assay. Although the
cell viability did not decrease in response to CAS administration,
an increased accumulation of drug was observed by flow cytometry.
In our opinion, this finding indicates that the pumps work in the
presence of CAS but function at a somewhat slower rate. As a
result, using flow cytometry, we observed an increased
concentration of drug shortly after exposure of the cells to the
dyes. These results are difficult to discuss since similar
experiments with CAS have not been reported.
These results suggest that BFA could be a candidate
anticancer drug supplement, mainly in cells that are characterized
by Pgp overexpression. CAS has therapeutic potential, but it must
be evaluated in other cell lines. These compounds are likely to be
useful as supplements in anticancer therapy.
Acknowledgments
The present was supported by the Poznan University
of Medical Sciences 502-14-02229373-10079 to K.W.
Abbreviations:
|
BFA
|
brefeldin A
|
|
CAS
|
castanospermine
|
|
ABC
|
ATP-binding cassette superfamily
|
|
BCRP/ABCG2
|
breast cancer resistance protein
|
|
BSA
|
bovine serum albumin
|
|
CASP 3
|
caspase 3
|
|
FBS
|
fetal bovine serum
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
MDR
|
multidrug resistance
|
|
EMEM
|
Eagle's Minimum Essential Medium
|
|
MFI
|
mean fluorescence intensity
|
|
PBS
|
phosphate-buffered saline
|
|
Pgp
|
glycoprotein P
|
|
PNGase F
|
N-glycosidase F
|
|
TBS
|
Tris-buffered saline
|
References
|
1
|
Christiansen MN, Chik J, Lee L, Anugraham
M, Abrahams JL and Packer NH: Cell surface protein glycosylation in
cancer. Proteomics. 14:525–546. 2014. View Article : Google Scholar
|
|
2
|
Persidis A: Cancer multidrug resistance.
Nat Biotechnol. 18(Suppl): IT18–IT20. 2000.
|
|
3
|
Sharom FJ: ABC multidrug transporters:
Structure, function and role in chemoresistance. Pharmacogenomics.
9:105–127. 2008. View Article : Google Scholar
|
|
4
|
Ma H, Miao X, Ma Q, Zheng W, Zhou H and
Jia L: Functional roles of glycogene and n-glycan in multidrug
resistance of human breast cancer cells. IUBMB Life. 65:409–422.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li K, Sun Z, Zheng J, Lu Y, Bian Y, Ye M,
Wang X, Nie Y, Zou H and Fan D: In-depth research of multidrug
resistance related cell surface glycoproteome in gastric cancer. J
Proteomics. 82:130–140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meschini S, Calcabrini A, Monti E, Del
Bufalo D, Stringaro A, Dolfini E and Arancia G: Intracellular
P-glycoprotein expression is associated with the intrinsic
multidrug resistance phenotype in human colon adenocarcinoma cells.
Int J Cancer. 87:615–628. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pastan I and Gottesman M: Multiple-drug
resistance in human cancer. N Engl J Med. 316:1388–1393. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wojtowicz K, Szaflarski W, Januchowski R,
Zawierucha P, Nowicki M and Zabel M: Inhibitors of N-glycosylation
as a potential tool for analysis of the mechanism of action and
cellular localisation of glycoprotein P. Acta Biochim Pol.
59:445–450. 2012.PubMed/NCBI
|
|
9
|
Shapiro AB, Fox K, Lee P, Yang YD and Ling
V: Functional intracellular P-glycoprotein. Int J Cancer.
76:857–864. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baldini N, Scotlandi K, Serra M, Shikita
T, Zini N, Ognibene A, Santi S, Ferracini R and Maraldi NM: Nuclear
immunolocalization of P-glycoprotein in multidrug-resistant cell
lines showing similar mechanisms of doxorubicin distribution. Eur J
Cell Biol. 68:226–239. 1995.PubMed/NCBI
|
|
11
|
Calcabrini A, Meschini S, Stringaro A,
Cianfriglia M, Arancia G and Molinari A: Detection of
P-glycoprotein in the nuclear envelope of multidrug resistant
cells. Histochem J. 32:599–606. 2000. View Article : Google Scholar
|
|
12
|
Noguchi K, Katayama K and Sugimoto Y:
Human ABC transporter ABCG2/BCRP expression in chemoresistance:
Basic and clinical perspectives for molecular cancer therapeutics.
Pharm Genomics Pers Med. 7:53–64. 2014.
|
|
13
|
Nakanishi T and Ross DD: Breast cancer
resistance protein (BCRP/ABCG2): Its role in multidrug resistance
and regulation of its gene expression. Chin J Cancer. 31:73–99.
2012. View Article : Google Scholar
|
|
14
|
Wakabayashi-Nakao K, Tamura A, Furukawa T,
Nakagawa H and Ishikawa T: Quality control of human ABCG2 protein
in the endoplasmic reticulum: Ubiquitination and proteasomal
degradation. Adv Drug Deliv Rev. 61:66–72. 2009. View Article : Google Scholar
|
|
15
|
Nakagawa H, Wakabayashi-Nakao K, Tamura A,
Toyoda Y, Koshiba S and Ishikawa T: Disruption of N-linked
glycosylation enhances ubiquitin-mediated proteasomal degradation
of the human ATP-binding cassette transporter ABCG2. FEBS J.
276:7237–7252. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ferris SP, Kodali VK and Kaufman RJ:
Glycoprotein folding and quality-control mechanisms in
protein-folding diseases. Dis Model Mech. 7:331–341. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakajima M, Koga T, Sakai H, Yamanaka H,
Fujiwara R and Yokoi T: N-Glycosylation plays a role in protein
folding of human UGT1A9. Biochem Pharmacol. 79:1165–1172. 2010.
View Article : Google Scholar
|
|
18
|
Tannous A, Pisoni GB, Hebert DN and
Molinari M: N-linked sugar-regulated protein folding and quality
control in the ER. Semin Cell Dev Biol. 41:79–89. 2015. View Article : Google Scholar :
|
|
19
|
Ruggiano A, Foresti O and Carvalho P:
Quality control: ER-associated degradation: protein quality control
and beyond. J Cell Biol. 204:869–879. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Klausner RD, Donaldson JG and
Lippincott-Schwartz J: Brefeldin A: Insights into the control of
membrane traffic and organelle structure. J Cell Biol.
116:1071–1080. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oda K, Fujiwara T and Ikehara Y: Brefeldin
A arrests the intracellular transport of viral envelope proteins in
primary cultured rat hepatocytes and HepG2 cells. Biochem J.
265:161–167. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Misumi Y, Misumi Y, Miki K, Takatsuki A,
Tamura G and Ikehara Y: Novel blockade by brefeldin A of
intracellular transport of secretory proteins in cultured rat
hepatocytes. J Biol Chem. 261:11398–11403. 1986.PubMed/NCBI
|
|
23
|
Yardin C, Terro F, Esclaire F, Rigaud M
and Hugon J: Brefeldin A-induced apoptosis is expressed in rat
neurons with dephosphorylated tau protein. Neurosci Lett. 250:1–4.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pommepuy I, Terro F, Petit B, Trimoreau F,
Bellet V, Robert S, Hugon J, Labrousse F and Yardin C: Brefeldin A
induces apoptosis and cell cycle blockade in glioblastoma cell
lines. Oncology. 64:459–467. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shao RG, Shimizu T and Pommier Y:
Brefeldin A is a potent inducer of apoptosis in human cancer cells
independently of p53. Exp Cell Res. 227:190–196. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee SA, Kim YJ and Lee CS: Brefeldin A
induces apoptosis by activating the mitochondrial and death
receptor pathways and inhibits focal adhesion kinase-mediated cell
invasion. Basic Clin Pharmacol Toxicol. 113:329–338.
2013.PubMed/NCBI
|
|
27
|
Salles FT, Hespanhol AM, Jaeger RG and
Marques MM: Brefeldin-A induces apoptosis in human adenoid cystic
carcinoma cultured cells. Oral Oncol. 40:585–590. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wlodkowic D, Skommer J and Pelkonen J:
Brefeldin A triggers apoptosis associated with mitochondrial breach
and enhances HA14-1- and anti-Fas-mediated cell killing in
follicular lymphoma cells. Leuk Res. 31:1687–1700. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wallen E, Sellers RG and Peehl DM:
Brefeldin A induces p53-independent apoptosis in primary cultures
of human prostatic cancer cells. J Urol. 164:836–841. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Verovski VN, Van den Berge DL, Delvaeye
MM, Scheper RJ, De Neve WJ and Storme GA: Low-level doxorubicin
resistance in P-glycoprotein-negative human pancreatic tumour
PSN1/ADR cells implicates a brefeldin A-sensitive mechanism of drug
extrusion. Br J Cancer. 73:596–602. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hohenschutz LD, Bell EA, Jewess PJ,
Leworthy DP, Pryce RJ, Arnold E and Clardy J: Castanospermine, a
1,6,7,8-tetrahydroxyoctahydroindolizine alkaloid from seeds of
Castanospermum austarale. Phytochemistry. 20:811–814. 1981.
View Article : Google Scholar
|
|
32
|
Pili R, Chang J, Partis RA, Mueller RA,
Chrest FJ and Passaniti A: The α-glucosidase I inhibitor
castanospermine alters endothelial cell glycosylation, prevents
angiogenesis, and inhibits tumor growth. Cancer Res. 55:2920–2926.
1995.PubMed/NCBI
|
|
33
|
Grochowicz PM, Hibberd AD, Bowen KM, Clark
DA, Pang G, Grochowicz LK, Willenborg DO and Cowden WB: Synergism
of castanospermine and FK 506. Transplant Proc. 27:355–356.
1995.PubMed/NCBI
|
|
34
|
Vlietinck AJ, De Bruyne T, Apers S and
Pieters LA: Plant-derived leading compounds for chemotherapy of
human immunodeficiency virus (HIV) infection. Planta Med.
64:97–109. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chang J, Block TM and Guo JT: Antiviral
therapies targeting host ER alpha-glucosidases: Current status and
future directions. Antiviral Res. 99:251–260. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kato A, Hirokami Y, Kinami K, Tsuji Y,
Miyawaki S, Adachi I, Hollinshead J, Nash RJ, Kiappes JL, Zitzmann
N, et al: Isolation and SAR studies of bicyclic iminosugars from
Castanospermum australe as glycosidase inhibitors. Phytochemistry.
111:124–131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schraen-Maschke S and Zanetta JP: Role of
oligomannosidic N-glycans in the proliferation, adhesion and
signalling of C6 glioblastoma cells. Biochimie. 85:219–229. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wojtowicz K, Januchowski R, Nowicki M and
Zabel M: Inhibition of protein glycosylation reverses the MDR
phenotype of cancer cell lines. Biomed Pharmacother. 74:49–56.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Januchowski R, Wojtowicz K,
Sujka-Kordowska P, Andrzejewska M and Zabel M: MDR gene expression
analysis of six drug-resistant ovarian cancer cell lines. BioMed
Res Int. 2013:2417632013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Szaflarski W, Sujka-Kordowska P,
Januchowski R, Wojtowicz K, Andrzejewska M, Nowicki M and Zabel M:
Nuclear localization of P-glycoprotein is responsible for
protection of the nucleus from doxorubicin in the resistant LoVo
cell line. Biomed Pharmacother. 67:497–502. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fu D, Bebawy M, Kable EP and Roufogalis
BD: Dynamic and intracellular trafficking of P-glycoprotein-EGFP
fusion protein: Implications in multidrug resistance in cancer. Int
J Cancer. 109:174–181. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Janneh O, Owen A, Bray PG, Back DJ and
Pirmohamed M: The accumulation and metabolism of zidovudine in
3T3-F442A pre-adipocytes. Br J Pharmacol. 159:484–493. 2010.
View Article : Google Scholar :
|
|
43
|
Rhodes T, Barrand MA and Twentyman PR:
Modification by brefeldin A, bafilomycin A1 and
7-chloro-4-nitrobenz-2-oxa-1,3-diazole (nBD) of cellular
accumulation and intracellular distribution of anthracyclines in
the non-P-glycoprotein-mediated multidrug-resistant cell line
COR-L23/R. Br J Cancer. 70:60–66. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wood DJT, Rumsby MG and Warr JR: Monensin
and verapamil do not alter intracellular localisation of
daunorubicin in multidrug resistant human KB cells. Cancer Lett.
108:41–47. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Marquardt D and Center MS: Drug transport
mechanisms in HL60 cells isolated for resistance to adriamycin:
Evidence for nuclear drug accumulation and redistribution in
resistant cells. Cancer Res. 52:3157–3163. 1992.PubMed/NCBI
|
|
46
|
Gong Y, Wang Y, Chen F, Han J, Miao J,
Shao N, Fang Z and Ou Yang R: Identification of the subcellular
localization of daunorubicin in multidrug-resistant K562 cell line.
Leuk Res. 24:769–774. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Togawa A, Ito H, Kimura F, Shimizu H,
Ohtsuka M, Shimamura F, Yoshidome H, Katoh A and Miyazaki M:
Establishment of gemcitabine-resistant human pancreatic cancer
cells and effect of brefeldin-A on the resistant cell line.
Pancreas. 27:220–224. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Labroille G, Belloc F, Bilhou-Nabera C,
Bonnefille S, Bascans E, Boisseau MR, Bernard P and Lacombe F:
Cytometric study of intracellular P-gp expression and reversal of
drug resistance. Cytometry. 32:86–94. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Allan G, Ouadid-Ahidouch H,
Sanchez-Fernandez EM, Risquez-Cuadro RÃ, Fernandez JÃMG,
Ortiz-Mellet C and Ahidouch A: New castanospermine glycoside
analogues inhibit breast cancer cell proliferation and induce
apoptosis without affecting normal cells. PLoS One. 8:e764112013.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Durland-Busbice S and Reisman D: Lack of
p53 expression in human myeloid leukemias is not due to mutations
in transcriptional regulatory regions of the gene. Leukemia.
16:2165–2167. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Muller PAJ, Vousden KH and Norman JC: p53
and its mutants in tumor cell migration and invasion. J Cell Biol.
192:209–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lin ZP, Boller YC, Amer SM, Russell RL,
Pacelli KA, Patierno SR and Kennedy KA: Prevention of brefeldin
A-induced resistance to teniposide by the proteasome inhibitor
MG-132: Involvement of NF-kappaB activation in drug resistance.
Cancer Res. 58:3059–3065. 1998.PubMed/NCBI
|