Introduction
IER3 (immediate early response gene 3) was
first identified in fibroblasts from mouse in 1993 (1), and human IER3, formerly
referred to as the immediate early gene X-1 (IEX-1), was
first identified in human squamous cell carcinomas. The human
IER3 gene is located on the short arm of chromosome 6
(6p21.3) and has 2 exons. The gene encodes 156 amino acids
(2). IER3 protein is rapidly and
transiently induced by a variety of stimuli, including ionizing
radiation, inflammatory cytokines, viral infection, anti-cancer
drugs and growth factors (2,3), and
is associated with apoptosis and cell proliferation (3,4). As it
is involved in cell growth, IER3 expression has been examined in
several human tumors, including pancreatic carcinoma, ovarian
carcinoma, breast cancer, and myelodysplastic syndrome (5–8).
However, there has been no study of IER3 expression in human lung
carcinoma.
Protein phosphatase 2A (PP2A), which consists of
structural subunit A, regulatory subunits B, and catalytic subunit
C, has a broad spectrum of functions due to the variety of
regulatory subunits, of which there are more than 20 kinds
(9). PP2A can act as a tumor
suppressor (10,11). B56 regulatory subunits are involved
in cell proliferation signaling (11,12).
Among them, PP2A-B56γ1 dephosphorylates Thr202 of
phosphorylated extracellular signal-regulated kinase (pERK),
leading to inactivation of ERK (13). Furthermore, Garcia et al
(14) found that the regulation of
cell growth by IER3 is mediated by ERK, which also plays a central
role in regulation of a variety of cellular events, including cell
proliferation, differentiation, migration, and apoptosis (15,16).
IER3 regulates ERK activity through its inhibitory activity on
B56-containing PP2A, which dephosphorylates ERK (13). Recently it has been clarified that
activated ERK induces IER3 protein synthesis, and then IER3
inhibits B56γ1-PP2A, forming the ERK/IER3/B56γ1-PP2A positive
feedback loop, which eventually leads to sustained activation of
ERK (17). The duration of ERK
activation is considered to be one of the factors that determine
cellular events (18,19). Thus, IER3 and PP2A should contribute
to multiple cellular processes, including tumorigenesis, via
regulation of ERK.
In this context, mutation of the B56γ gene
(PPP2R5C), which is located at 14q32.31, in lung carcinomas
as well as other carcinomas has been reported (20). Deletion of 14q has also been
reported in lung carcinoma (21).
PPP2R5C is frequently involved in loss of heterozygosity
(LOH) in 14q32 in a variety of carcinomas (22,23),
which suggests an important role of B56γ in carcinogenesis.
Furthermore, BL6 mouse melanoma cells, in which B56γ is truncated,
are highly metastatic (24). In a
patient with Sezary syndrome, translocation of t(14;14) (q11;q32)
caused rearrangement of PPP2R5C (25). All of these findings indicate an
important role of B56γ in carcinogenesis. We recently showed that
the ERK/IER3/B56γ1 loop is activated by the growth factor-induced
RAS/RAF/MEK/ERK cascade (17).
After growth factor binds to epidermal growth factor receptor
(EGFR), RAS, RAF, MEK and ERK are activated sequentially by
phosphorylation or binding (26).
The cascade is frequently constitutively activated by mutation or
gene amplification in lung adenocarcinomas, including mutation of
EGFR, amplification of HER2, mutation of RAS,
and mutation of RAF. Among these changes, mutations of
EGFR and RAS are most frequent. Activating
EGFR mutations are found in 5–30% (as high as 50–60% in
Asian populations) of non-small cell lung carcinomas, while
K-RAS mutations are found in 20–12%, and RAF
mutations in <1% (27,28). Furthermore, it was reported that
phosphorylation of ERK was closely associated with mutation of
RAS or EGFR (29).
The aim of this study was to examine whether the
IER3/B56γ1-PP2A/ERK-positive feedback loop is involved in
carcinogenesis of non-small cell lung carcinoma. We also report the
high prevalence in these carcinomas of allelic deletion of B56γ
gene, which is a major cause of secondary overexpression of
IER3/pERK.
Materials and methods
Tumor samples
Tumor samples, including normal tissues, were
obtained from 16 lung cancer patients (12 adenocarcinomas and 4
squamous cell carcinomas) who were operated at Kanazawa University
Hospital from September 2013 to December 2014. Tissue specimens
were frozen at −80°C.
This work was approved by the ethics committee of
Kanazawa University. We obtained informed consent from all
patients, and all studies were performed on the basis of the
declaration of Helsinki and the ethical guidelines for human genome
and gene analysis of the Ministry of Health, Labour and Welfare,
Japan.
Immunohistochemistry
Sections (5 µm) were cut from formalin-fixed,
paraffin-embedded (FFPE) tissues. To unmask epitopes, the sections
were treated with Target Retrieval Solution (pH 9.0, Dako, Uppsala,
Sweden). Endogenous peroxidase activity was blocked with 3%
H2O2. Sections were reacted with specific
rabbit antibodies against human IER3 antibody (Abnova, Taipei,
Taiwan) at 1:500 dilution, or specific antibodies against
Thr202/Tyr204-phosphorylated p42/44 ERK (Cell
Signaling Technology, Danvers, MA, USA) at 1:100 dilution, and then
reacted with peroxidase-labeled dextran goat anti-rabbit IgG
(Dako). Diaminobenzidine 4HCl (Histofine SAB-PO (M) kit, Nichirei,
Tokyo, Japan) was used for visualization. Negative controls were
done on serial tissue sections using 0.5% BSA or with
antigen-absorbed antibody.
Glutathione-S-transferase (GST) fusion IER3 protein,
which was used to absorb antibodies against IER3, was purified with
GST beads. Fusion IER3 protein (1 mg) was added to 20 µl of
antibody solution, and rocked for 60 min at room temperature. Then,
the reaction mixture was centrifuged at 14,000 rpm for 10 min. To
evaluate possible differences of positivity between non-tumor
epithelia and carcinoma cells, the ratio of positive cells were
counted in 5 randomly selected areas.
Laser capture microdissection
Frozen sections of lung tumor tissues were fixed
with 100% ethanol for 2 min and stained with 0.1% toluidine blue
(Merck, Darmstadt, Germany). Approximately 1,000 carcinoma cell
sections were cut out using a Laser Microdissection system (LMD
7000, Leica Microsystems, Inc., Bensheim, Germany), and collected
in a 0.5 ml RNase-free PCR tube. Extraction of genomic DNA was
performed from the frozen sections using a ReliaPrep™ gDNA Tissue
Miniprep System (Promega, Madison, WI, USA). Extraction of RNA was
performed using an SV Total Isolation system (Promega), and reverse
transcription to cDNA was performed using an RT-PCR system.
Direct sequencing
Extraction of genomic DNA from frozen sections was
performed using a ReliaPrep gDNA Tissue Miniprep System (Promega).
Specific primer sets were used for amplification of human EGFR,
K-RAS, PPP2R5C (B56γ1 gene) and IER3. Exons 18, 19, 20
and 21 in EGFR and exon 2 in K-RAS, which are hot
spots of mutation (30), were
evaluated (Table I). All of the
exons of PPP2R5C (13 exons) and IER3 (2 exons) were
amplified and sequenced. PCR amplification was performed with KOD
Fx Neo (Toyobo Co., Ltd., Tokyo, Japan). Cycle sequencing reaction
was performed using a Big Dye Terminator v1.1 Cycle Sequencing kit
(Applied Biosystems, Foster City, USA). The sequencing reactions
were conducted with a capillary sequencer (ABI PRISM 3130x1 Genetic
Analyzer, Applied Biosystems).
 | Table IThe primer sets for direct
sequencing. |
Table I
The primer sets for direct
sequencing.
| Genes | 1st pair of PCR
primers | Nested primers |
|---|
| EGFR |
GCTCTGTAGAGAAGGCGAC |
GCTCTGTAGAGAAGGCGTAC |
| Exon 18 |
TCCCAAACACTCAGTGAAACAAA |
TTGGTCTCACAGGACCACTG |
| EGFR |
CCCCAGCAATATCAGCCTTA |
GTGCATCGCTGGTAACATCC |
| Exon 19 |
GTGGATACCAGCATGGGAGA |
TGTGGAGATGAGCAGGGTCT |
| EGFR |
TGAAACTCAAGATCGCATTCA |
ATCGCATTCATGCGTCTTCA |
| Exon 20 |
TGGGACAGGCACTGATTTGT |
ATCCCCATGGCAAACTCTTG |
| EGFR |
AGCCATAAGTCCTCGACGTG |
GCTCAGAGCCTGGCATGAA |
| Exon 21 |
TGGCTCACACTACCAGGAGA |
CATCCTCCCCTGCATGTGT |
| KRAS |
ACGTCTGCAGTCAACTGGAA |
GGAGTATTTGATAGTGTATTAACCT |
| Exon 2 |
ACCCACTGTATGAGGGTTCC |
AGAATGGTCCTGCACC |
| IER3 |
CATAAATTACCTCTGCCGGC |
CTCACTTGGCCTTACACTCC |
| Exon 1 |
TCAAGTTGCCTCGGAAGTCC |
AAACAGGAGACAGGTCAGGT |
| IER3 |
CATAAATTACCTCTGCCGGC |
ACCTGACCTGTCTCCTGTTT |
| Exon 2 |
TCAAGTTGCCTCGGAAGTCC |
CGCCTGGTGTTTCTTTGTGG |
| PPP2R5C |
TGCTGACATCACGAACCAGC |
TTCCCGCTGAAGTCTAG |
| Exon 1 |
CACCCAACCTCCTCTGGTTA |
CCGATTCAGTGAACACAC |
| PPP2R5C |
CTAACAGTGCTCTCAACATGG |
ATATGACCAGCGACTAGCTG |
| Exon 2 |
TAGGCTCAGAGATCTTGGCC |
AAATCAGAACTGGGACAC |
| PPP2R5C |
TACGGAGGAGCTAAGTTACC |
GCGGCTACTGTTAGAATTACC |
| Exon 3 |
ACATCTTCACTGGCTGTTG |
CATGCTACTGAGGAGGAGAG |
| PPP2R5C |
TGTAGTCTGTGGGTTTCACC |
GTGGCTTTGAGAGGCTAATA |
| Exon 4–5 |
TCCTGTCAGAGGAGACGATG |
AATGAGCATCACCACACAC |
| PPP2R5C |
AGATGCGCTTCTTGTTGCCTG |
ATGGCTCCTCCTAGAGCATTG |
| Exon 6 |
TCTAAGAGCTCACCAACTCC |
CAGCTGGACTCAATGAAATG |
| PPP2R5C |
ACAGGTGCATGGCAACATGC |
GCTGGGTTTTGATGGTGA |
| Exon 7 |
AGGCCGGATTCACTATCTCG |
CTGTGTTCCTAGAGTCCTG |
| PPP2R5C |
GTTAATGCCCGTTAATCACAC |
GGGAAGGTGTTTAACGATG |
| Exon 8 |
CCATGAATGAAATGAGCCTG |
GCAATTTGGTGAAGCAGATG |
| PPP2R5C |
CACAGACTTCTTACCATGCAG |
CATCAGTCACTCCACGTGTC |
| Exon 9 |
TGCATGAGAGGCAAAGCATG |
GCTACGCATGGAACTTTTCC |
| PPP2R5C |
GGTTACAGGTTACAGTCTAG |
TCTGGTCCAAGGTAGTTCAT |
| Exon 10 |
GAGTCCACAAAGCAACTGAC |
ATGGCAGCCAGTTCTTTC |
| PPP2R5C |
CAAGTAACGCGGAATGAGCAG |
ACTGTTGGAATATGGAGCAG |
| Exon 11 |
GCTTTCGAGTGAAGAATACCG |
GTGGAAGAGGTTTACTTAGG |
| PPP2R5C |
CTCAACATGCCTGTGCCTT |
TGTGTTCTTGTGTCTGATGC |
| Exon 12 |
CACAGTCTCAGGCTGTATTC |
ACGTTAGTGAAATCGAGACC |
| PPP2R5C |
TGTTGCAGGTGTAGGCGAGT |
ATGAGAAGCTTGGAGTTCAG |
| Exon 13 |
CAGCTTAAACAGAAGGCACTG |
TGCAAATCACATCGCCTAC |
| PPP2R5C |
TGGATGCGGCCAACTCCAAT |
AACTCCAATGGGCCTTTCC |
| Exon 1–2 |
GAGGAAGGTGGTAATGTTCG |
ACGTTGGTTCATCTTCCTCC |
| PPP2R5C |
CGCAACACTGACGTAACTTC |
CAGATGTTCCTCCTGCTGAT |
| Intron 1 |
ATATGACCAGCGACTAGCTG |
GGAATAGCCTTTGTTTACCC |
TA cloning
Fresh PCR products amplified using taq DNA
polymerase (Takara Ex Taq®, Takara, Kusatsu, Japan) were
ligated into linearized pCR™ 2.1-TOPO® vectors in a TOPO
TA cloning kit (Invitrogen, Carlsbad, CA, USA) using T4 DNA ligase.
The ligated vectors were transformed into competent TOP10 cells and
grown overnight on LB agar plates containing ampicillin and X-gal.
Blue-white screening was performed for inserts. The ligated clones
were purified and the inserts were sequenced using the M13 (-20)
forward primer and a Big Dye Terminator v1.1 Cycle Sequencing
kit.
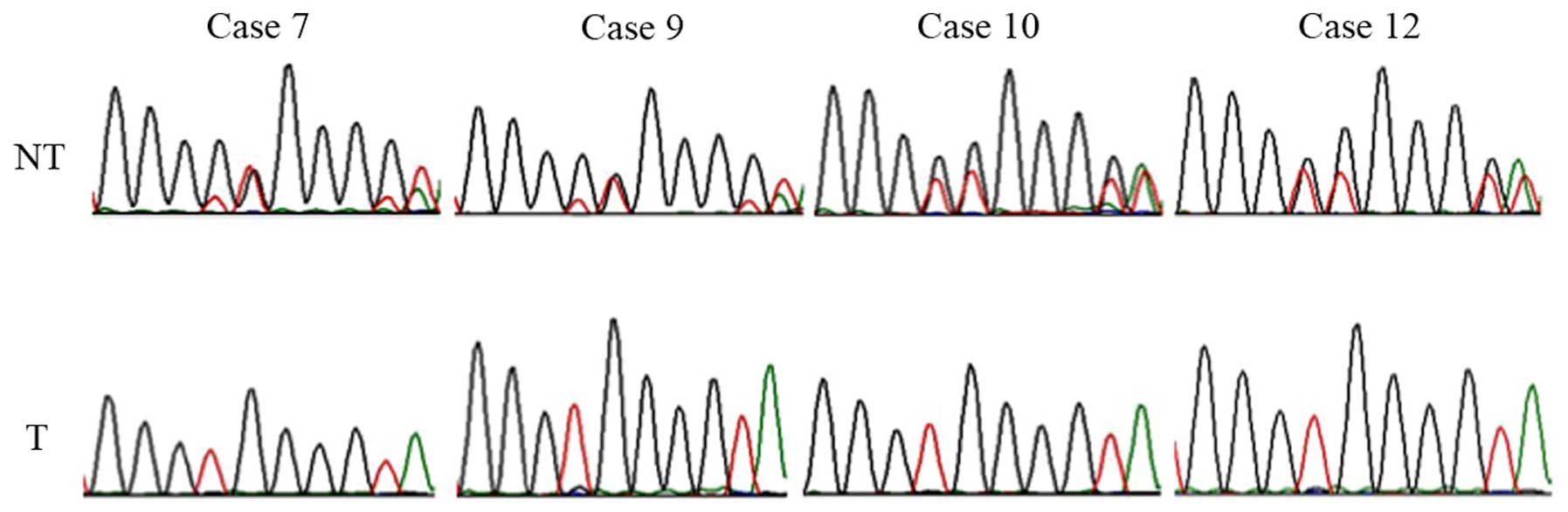
Analysis of LOH in PPP2R5C
A single nucleotide insertion in intron 1 in
PPP2R5C, found in the present study, was used for analyzing
LOH. The 3′ portions of intron 1 and exon 2 were amplified with the
primer sets used to sequence exon 2 (Table I). When the single nucleotide
insertion/deletion was found in one allele and not found in the
other allele (i.e., duplicated peaks were seen in the
electropherogram of the normal lung tissue), the status was
considered informative. When the duplicated peaks were not found in
the electropherogram of the corresponding tumor, the status was
considered LOH.
Statistics
One-way analysis of variance was used for comparing
the numbers of immunohistochemically positive carcinoma cells,
normal bronchiolar epithelia or alveolar epithelia. When a
significant difference was found, multivariate analysis using
Dunnett's multiple comparison test was applied.
Results
Immunohistochemical detection of
IER3
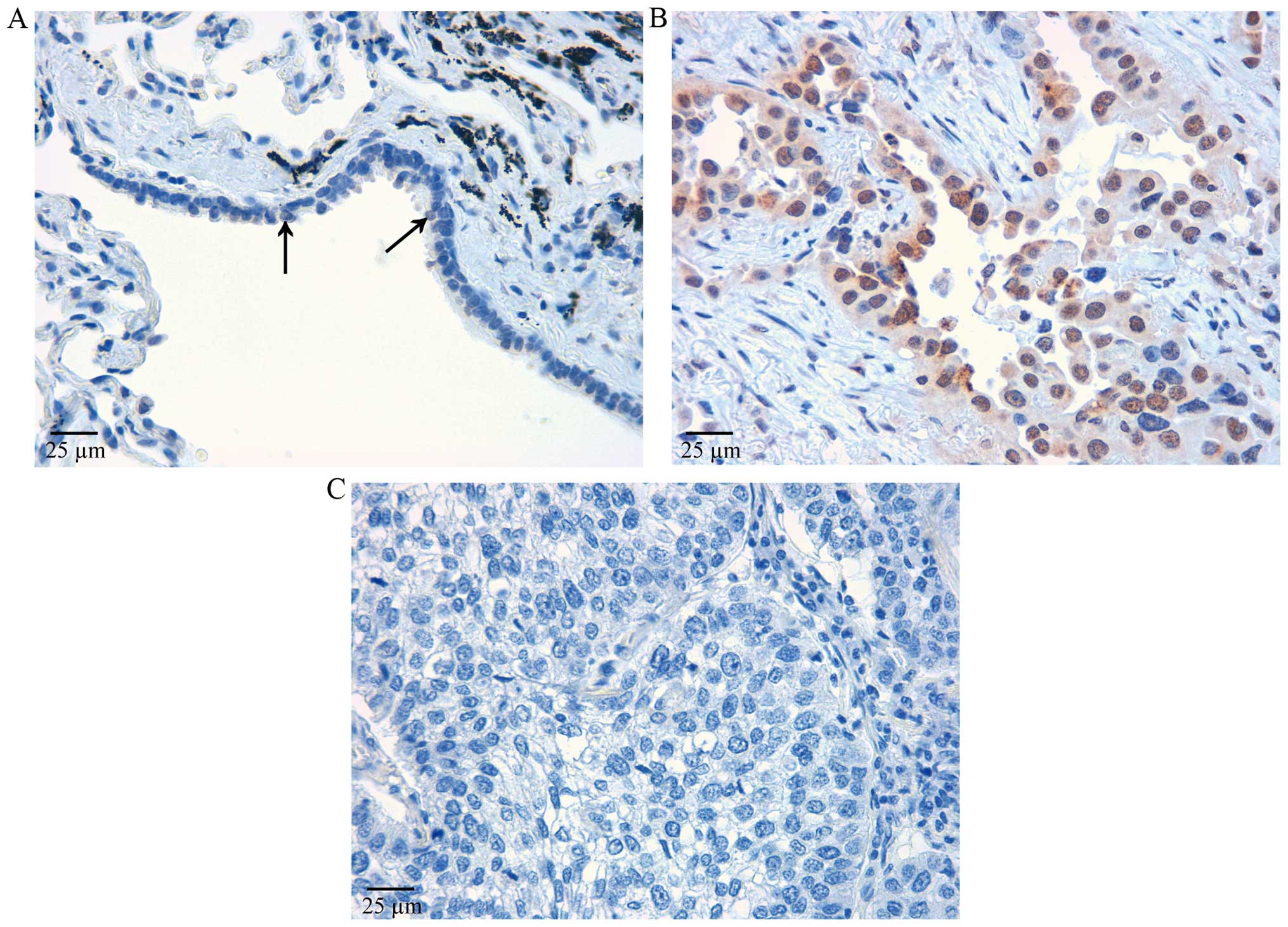
A few bronchiolar epithelial cells were positive to
anti-IER3 antibodies (Fig. 1A).
IER3 was immunolocalized in the nuclei. In adenocarcinoma, IER3 was
strongly immunostained in nuclei and faintly in cytoplasm (Fig. 1B). Granular staining remained
positive in cytoplasm when antigen-absorbed antibody was used,
suggesting that it was non-specific. Therefore, we considered that
only the nuclear staining was positive in the present study. All
the adenocarcinomas showed large numbers of positive cells, whereas
squamous cell carcinomas showed far fewer positive cells (Fig. 1C).
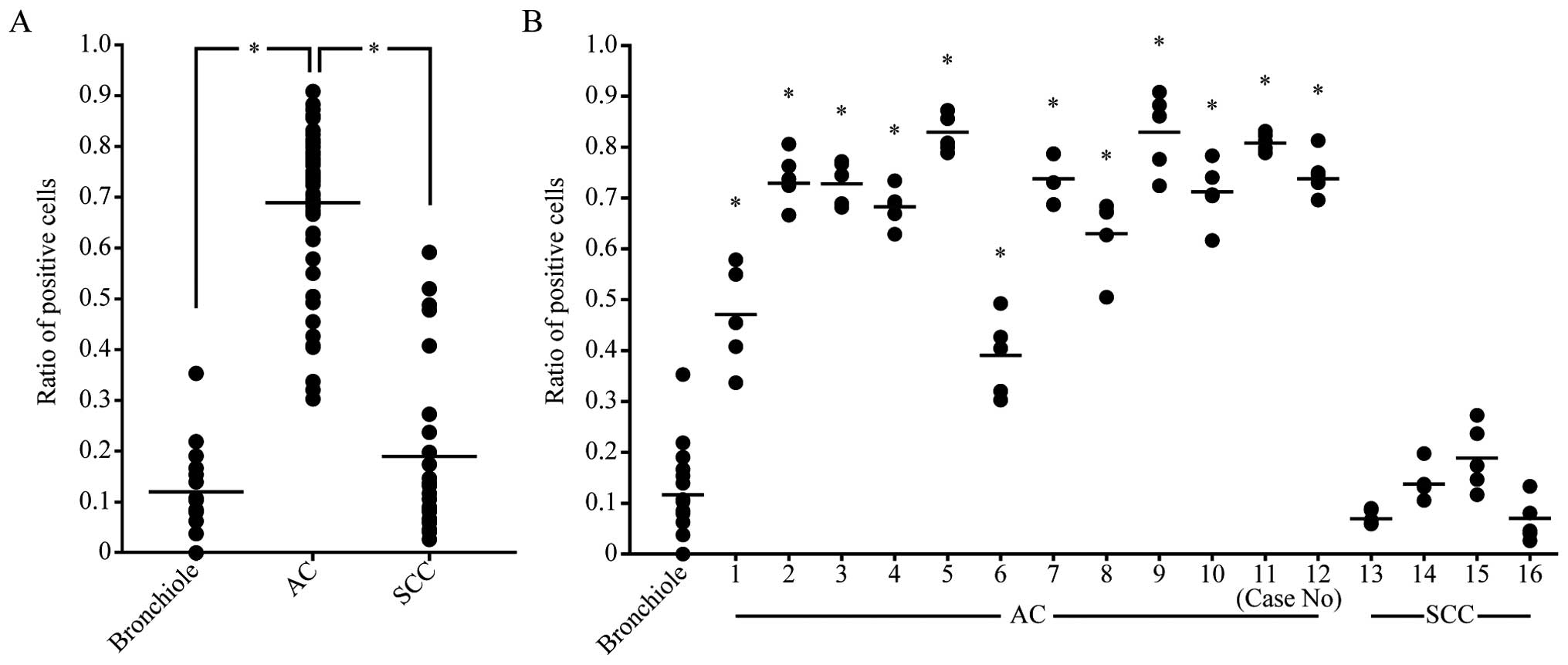
We counted positive cells in 5 fields of a
high-power microscope and calculated the ratio of IER3-positive
cells in each field. The ratios were significantly higher in
adenocarcinomas (p<0.01) than in alveolar or bronchiolar
epithelia. The average ratio of IER3-positive cells in
adenocarcinomas was 4–8 times higher than that in normal epithelia.
The ratio in squamous cell carcinomas was not significantly
different from that in bronchiolar epithelia (Fig. 2A). ANOVA followed by Dunnett's
multiple comparison showed that the ratios in all the
adenocarcinomas were significantly higher (p<0.01) than that in
normal epithelia (Fig. 2B). None of
the squamous cell carcinomas showed a significant difference from
normal epithelia.
Immunohistochemical detection of
phosphorylated ERK
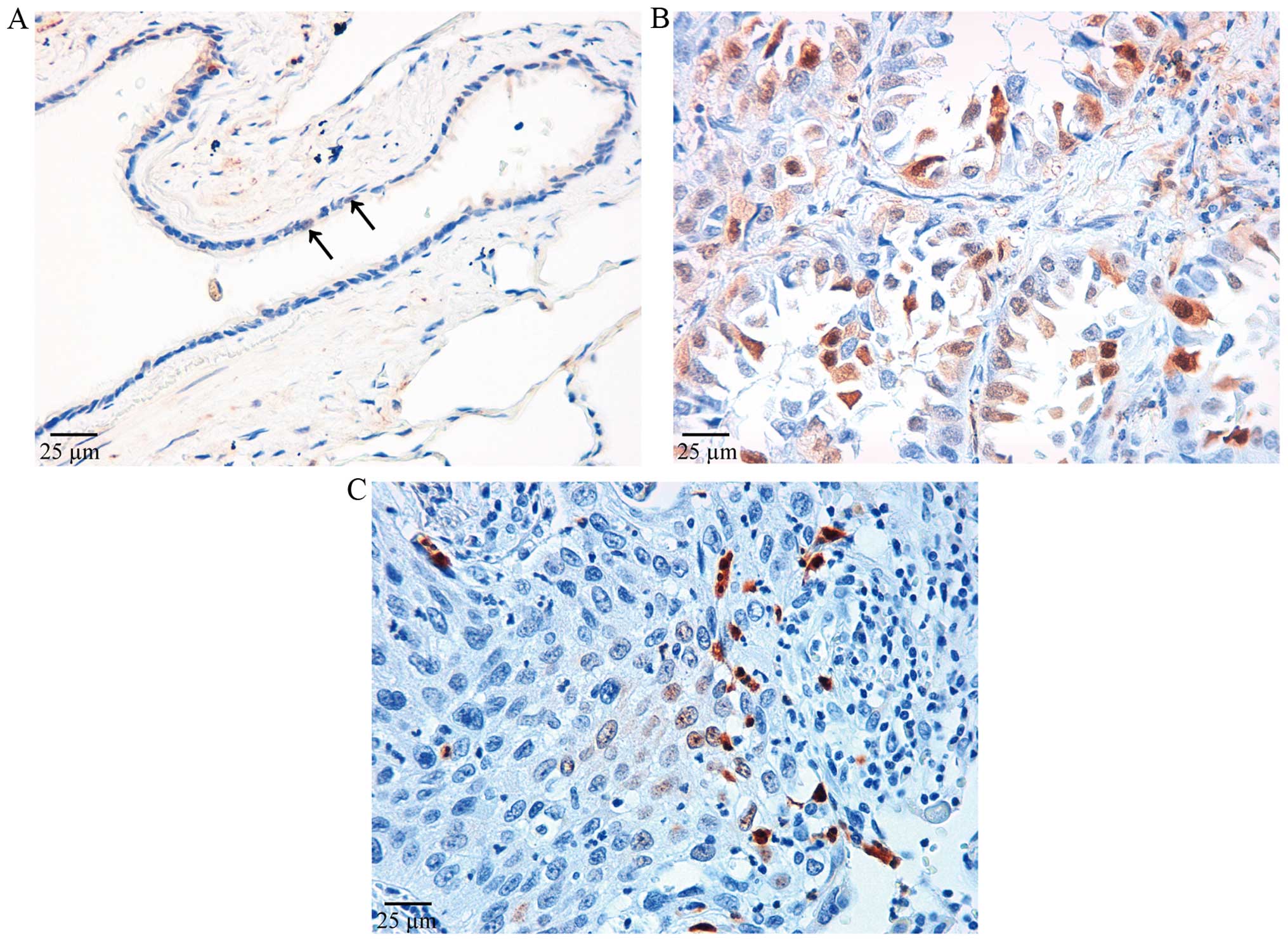
Phosphorylated ERK was immunostained in both nuclei
and cytoplasm of bronchiolar epithelia (Fig. 3A). In the present study, only cells
immunostained in the nuclei were considered positive. The pattern
of distribution of pERK-positive cells was different from that of
IER3-positive cells. pERK-positive cells were restricted to the
peripheral areas of the tumor in adenocarcinomas, and negative
areas were excluded from evaluation of the cell count. pERK was
intensely stained mainly in the nuclei (Fig. 3B). Positive cells were stained in
small clusters, which were distributed evenly throughout squamous
cell carcinoma (Fig. 3C).
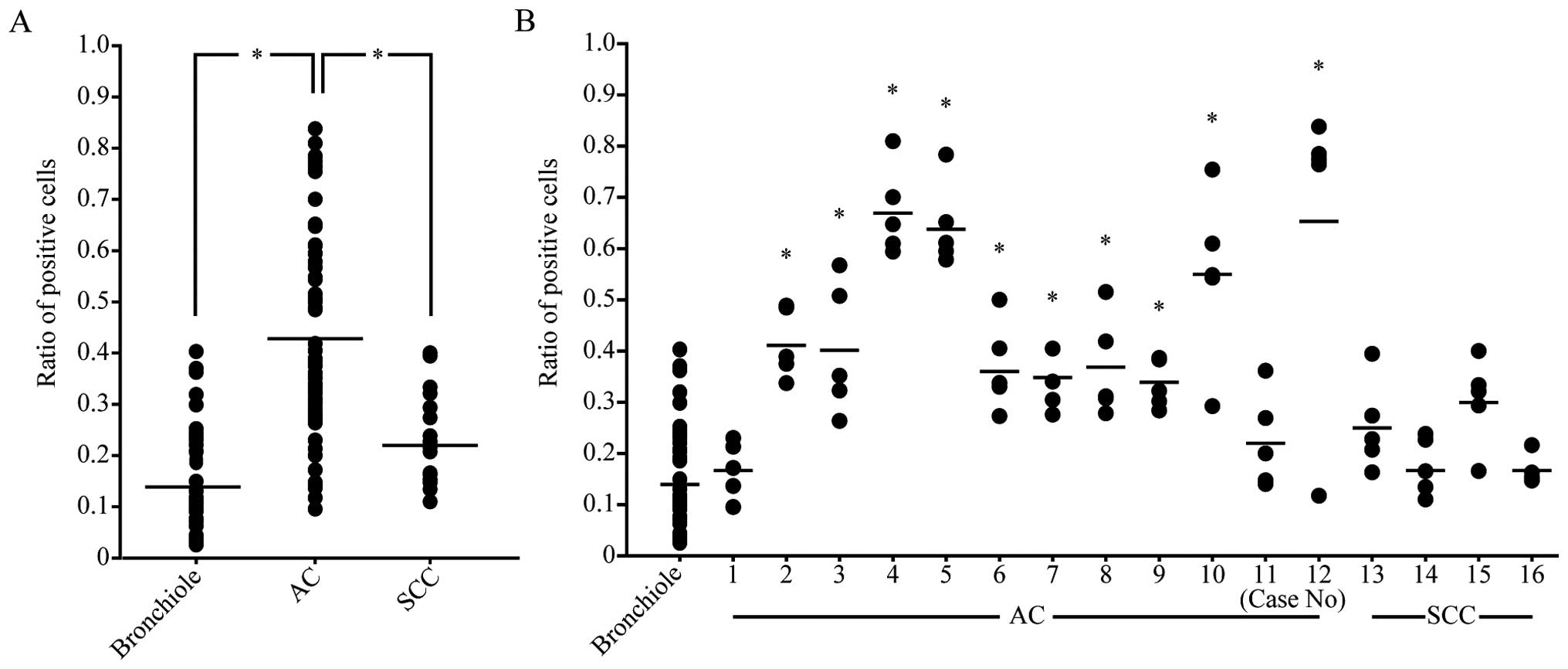
The ratio of pERK expression was significantly
higher in almost all adenocarcinomas than in normal epithelia,
except for 1 case. None of the squamous cell carcinomas showed a
significant difference from normal epithelia (Fig. 4A). When the ratios in all the cases
were compared, multiple comparison showed a significant difference
in adenocarcinoma, but not in squamous cell carcinoma (Fig. 4B).
Direct sequencing
To examine the relationship between IER3/pERK
overexpression in adenocarcinoma and alteration of the EGFR-ERK
cascade or the ERK/IER3/B56γ1 feedback loop, we looked for
mutations in the sequences of EGFR, K-RAS, PPP2R5C and
IER3. The results are summarized in Table II.
| Table IIProtein overexpression, gene
mutations and allelic deletion of PPP2R5C. |
Table II
Protein overexpression, gene
mutations and allelic deletion of PPP2R5C.
| Case | Overexpression
| Mutation
| Deletion
|
|---|
| IER3 | pERK | EGFR | KRAS
IER3 | | PPP2R5C | PPP2R5C |
|---|
| 1 | + | − | − | − | − | − | − |
| 2 | + | + | L858R | − | − | − | − |
| 3 | + | + | − | − | − | − | Not
informative |
| 4 | + | + | L858R | − | − | − | − |
| 5 | + | + | L861Q | − | − | − | − |
| 6 | + | + | 746-753del | − | − | − | Not
informative |
| 7 | + | + | 746-750del | − | − | − | + |
| 8 | + | + | − | − | − | − | Not
informative |
| 9 | + | + | − | − | − | − | + |
| 10 | + | + | − | − | − | − | + |
| 11 | + | − | − | − | − | − | Not
informative |
| 12 | + | + | − | − | − | − | + |
In 12 cases of adenocarcinoma, EGFR mutation
was found in 5 cases; deletion of EGFR exon 19 (746-750del)
was found in 2 cases. In the other 3 cases, point mutation was
found in EGFR exon 21 (L861Q or L858R). No mutation of
K-RAS exon 2 was found. No EGFR and K-RAS
mutations were found in squamous cell carcinomas. IER3 showed no
mutation in any of the cases.
In the process of sequencing of PPP2R5C, we
observed intermittent duplicated waves in the electropherograms
from samples of non-tumor lung tissues, indicating the presence of
a mixture of different sequences. Insertion or deletion of one
nucleotide in the first intron in one allele apparently generated
the duplication. We further designed a primer set spanning intron
1, and confirmed the duplication by TA cloning analysis. The
sequence of one allele is the same as the reported sequence
(accession no., NC 000014), while the sequence of the other allele
showed insertion of a single nucleotide (G) 122 bases upstream of
the end of the first intron (IVS1-122insG). The electropherogram
using cDNA and the primer sets (Table
I) amplifying the boundary of exon 1 and exon 2 showed single
waves of the reported sequence (accession no. NM 002719). Finally,
it was concluded that there was no mutation in PPP2R5C in
any of the cases.
Analysis of LOH at PPP2R5C using the
single nucleotide insertion
Heterozygous sequences in intron 1 of PPP2R5C
were found in 8 out of 12 cases (67%) in the non-tumor tissue and
in 4 out of 12 cases (33%) in the tumor. Finally, 4 out of 8
informative cases were heterozygous, and 4 were homozygous (50%),
indicating LOH (Fig. 5).
Discussion
IER3 inhibits PP2A-B56γ, resulting in inhibition of
ERK dephosphorylation (13,17), so that overexpression of IER3 could
result in sustained activation of ERK, leading to cell
proliferation. Our immunohistochemical findings that both IER3 and
pERK were overexpressed in adenocarcinomas support this idea.
However, it was not clear whether the overexpression was due
directly to changes of IER3 or ERK, or to an alteration of upstream
signaling protein, such as EGFR. EGFR exons 18, 19, 20 and
21 encode an intracellular tyrosine kinase, and mutations in these
regions cause ligand-independent EGFR activation (31), leading to overexpression of pERK
through the EGFR/RAS/RAF/MEK cascade (26,32).
We found EGFR mutation in 42% of lung adenocarcinomas, and all of
the EGFR mutation cases showed pERK overexpression. This is
consistent with the results of a previous study (33). In addition, overexpression of ERK by
IHC is reported to be positive in 89% of lung adenocarcinomas that
overexpress EGFR (34). Thus, it is
strongly suggested that overexpression of pERK/IER3 is caused by
activating mutation of EGFR. However, in the cases with no mutation
of EGFR, there is no obvious reason for pERK/IER3 overexpression.
Therefore, we considered the possible role of a positive feedback
loop of ERK/IER3/PP2A-B56γ1 in the nucleus (17). Both IER3 and pERK were overexpressed
in the nucleus in adenocarcinomas examined by us, which is
consistent with the existence of an ERK/IER3/PP2A-B56γ1-positive
feedback loop in lung adenocarcinoma. This positive feedback loop
may be involved in carcinogenesis of lung adenocarcinoma (35).
Recently, Garcia et al (36) proposed that pERK enhanced the
immediate expression of IER3 in pancreatic cancer, and they found
that IER3 is prominently expressed in pancreatic ductal
adenocarcinoma. IER3 expression sustains phosphorylation of ERK
through PP2A. Invasive breast cancer (7) and multiple myeloma (37) show high IER3 expression, but on the
contrary, IER3 expression in ovarian cancer is lower than in benign
cystadenomas (6). This might
reflect the fact that IER3 has apparently contradictory functions
of proapoptosis and cell proliferation (3,4),
possibly by inhibiting the activities of other B56 species involved
in different signaling pathways from B56γ (13). The positive correlation of IER3
expression and pERK expression may suggest that the interaction of
IER3/ERK promotes cell proliferation.
PP2A-B56γ1 dephosphorylates and inactivates ERK, and
inhibits cell proliferation and migration (15,17).
PP2A functions as a tumor-suppressor gene (38,39).
One of the mechanisms of inactivation of tumor-suppressor genes is
allelic deletion or loss of heterozygosity (LOH). PPP2R5C
(PP2A B56γ gene) is located at the tip of the long arm of
chromosome 14 (14q32.31). Deletion of 14q has been reported in lung
non-small cell carcinoma (21), and
may be relevant to the present findings. The reported 14q deletion
in lung non-small cell carcinoma occurred only in 4% of cases
(21), which is much less frequent
than 50% in the present study. However, much more frequent LOH at
shorter portions in 14q have been reported (40,41),
although LOH of 14q32 has not been examined for lung carcinoma. LOH
of 14q32 was found in 32–44% of colorectal cancers (22,42),
and is associated with metastatic recurrence (42). LOH was also found in 31% of
neuroblastomas (43), 37% of
esophageal cancers (23), and 16%
of gastric cancers (44). Breast
carcinoma with BRCA2 mutation shows 14q32 LOH in 80% of
cases, which may suggest the presence of tumor suppressor genes at
14q32 (45).
Suppressor genes including RB and
TP53, which are recessive genes, are mutated or deleted in
one allele in addition to deletion in the other allele in lung
carcinomas (46). In the present
study, LOH at PPP2R5C was found frequently, but we did not
find mutation of PPP2R5C. Therefore, some other mechanism of
cPP2A-B56γ inactivation must exist if PP2A-B56γ is involved in
carcinogenesis. PP2A consists of three subunits, A, B and C.
Deletion or inactivating mutation in the subunit A or C gene, in
addition to allelic deletion of B56γ, would further reduce function
of PP2A-B56γ. Subunit A gene, PPP1R1B, is mutated in 15% of
lung and colon cancers (47), and
expression of Aβ is lost in colon cancer (48). The Aβ gene is located at 11q22-23,
and 11q is deleted in 20% of lung cancers (21). Further, 11q22-24 is deleted in 63%
of lung adenocarcinomas (49). The
subunit C gene, PPP2CA, is located at 5q, which is deleted
in 20% of lung carcinomas (21).
Taken together, these findings suggest it is highly likely that
PP2A-B56γ function is frequently lost in lung adenocarcinoma, as
well as other carcinomas such as colorectal carcinoma, leading to
overexpression of IER3/pERK. We found LOH of PPP2R5C in 50%
of lung adenocarcinomas, and many of them did not appear to have
any other mutation that might cause IER3/pERK overexpression. Thus,
deletion of 14q32 could be an important factor in carcinogenesis of
the lung.
Acknowledgments
This study was supported by grants-in-aid for
scientific research from the ministry of Education, Culture,
Sports, Science and Technology (grant no. 26462119).
References
|
1
|
Charles CH, Yoon JK, Simske JS and Lau LF:
Genomic structure, cDNA sequence, and expression of gly96, a growth
factor-inducible immediate-early gene encoding a short-lived
glycosylated protein. Oncogene. 8:797–801. 1993.PubMed/NCBI
|
|
2
|
Kondratyev AD, Chung KN and Jung MO:
Identification and characterization of a radiation-inducible
glycosylated human early-response gene. Cancer Res. 56:1498–1502.
1996.PubMed/NCBI
|
|
3
|
Arlt A and Schäfer H: Role of the
immediate early response 3 (IER3) gene in cellular stress response,
inflammation and tumorigenesis. Eur J Cell Biol. 90:545–552. 2011.
View Article : Google Scholar
|
|
4
|
Wu MX: Roles of the stress-induced gene
IEX-1 in regulation of cell death and oncogenesis. Apoptosis.
8:11–18. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sasada T, Azuma K, Hirai T, Hashida H,
Kanai M, Yanagawa T and Takabayashi A: Prognostic significance of
the immediate early response gene X-1 (IEX-1) expression in
pancreatic cancer. Ann Surg Oncol. 15:609–617. 2008. View Article : Google Scholar
|
|
6
|
Han L, Geng L, Liu X, Shi H, He W and Wu
MX: Clinical significance of IEX-1 expression in ovarian carcinoma.
Ultrastruct Pathol. 35:260–266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang C, Trent S, Ionescu-Tiba V, Lan L,
Shioda T, Sgroi D and Schmidt EV: Identification of cyclin D1- and
estrogen-regulated genes contributing to breast carcinogenesis and
progression. Cancer Res. 66:11649–11658. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Steensma DP, Neiger JD, Porcher JC, Keats
JJ, Bergsagel PL, Dennis TR, Knudson RA, Jenkins RB, Santana-Davila
R, Kumar R, et al: Rearrangements and amplification of IER3 (IEX-1)
represent a novel and recurrent molecular abnormality in
myelodysplastic syndromes. Cancer Res. 69:7518–7523. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Janssens V and Goris J: Protein
phosphatase 2A: A highly regulated family of serine/threonine
phosphatases implicated in cell growth and signalling. Biochem J.
353:417–439. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee TY, Lai TY, Lin SC, Wu CW, Ni IF, Yang
YS, Hung LY, Law BK and Chiang CW: The B56γ3 regulatory subunit of
protein phosphatase 2A (PP2A) regulates S phase-specific nuclear
accumulation of PP2A and the G1 to S transition. J Biol Chem.
285:21567–21580. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eichhorn PJ, Creyghton MP and Bernards R:
Protein phosphatase 2A regulatory subunits and cancer. Biochim
Biophys Acta. 1795:1–15. 2009.
|
|
12
|
Westermarck J and Hahn WC: Multiple
pathways regulated by the tumor suppressor PP2A in transformation.
Trends Mol Med. 14:152–160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Letourneux C, Rocher G and Porteu F:
B56-containing PP2A dephosphorylate ERK and their activity is
controlled by the early gene IEX-1 and ERK. EMBO J. 25:727–738.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Garcia J, Ye Y, Arranz V, Letourneux C,
Pezeron G and Porteu F: IEX-1: A new ERK substrate involved in both
ERK survival activity and ERK activation. EMBO J. 21:5151–5163.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mebratu Y and Tesfaigzi Y: How ERK1/2
activation controls cell proliferation and cell death: Is
subcellular localization the answer? Cell Cycle. 8:1168–1175. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wortzel I and Seger R: The ERK cascade:
Distinct functions within various subcellular organelles. Genes
Cancer. 2:195–209. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kawahara E, Maenaka S, Shimada E,
Nishimura Y and Sakurai H: Dynamic regulation of extracellular
signal-regulated kinase (ERK) by protein phosphatase 2A regulatory
subunit B56γ1 in nuclei induces cell migration. PLoS One.
8:e637292013. View Article : Google Scholar
|
|
18
|
Traverse S, Gomez N, Paterson H, Marshall
C and Cohen P: Sustained activation of the mitogen-activated
protein (MAP) kinase cascade may be required for differentiation of
PC12 cells. Comparison of the effects of nerve growth factor and
epidermal growth factor. Biochem J. 288:351–355. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marshall CJ: Specificity of receptor
tyrosine kinase signaling: Transient versus sustained extracellular
signal-regulated kinase activation. Cell. 80:179–185. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nobumori Y, Shouse GP, Wu Y, Lee KJ, Shen
B and Liu X: Characterization of B56γ tumor-associated mutations
reveals mechanisms for B56γ-PP2A tumor suppressor activity. Mol
Cancer Res. 11:995–1003. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tepeli E, Muslumanoglu MH, Uludag A,
Buyukpinarbasili N, Ozdemir M, Oznur M, Aslan H and Artan S:
Detection of deletions and/or amplifications of genes related with
lung cancer by multiplex ligation-dependent probe amplification
(MLPA) technique. Cancer Biol Ther. 8:2160–2165. 2009. View Article : Google Scholar
|
|
22
|
Bando T, Kato Y, Ihara Y, Yamagishi F,
Tsukada K and Isobe M: Loss of heterozygosity of 14q32 in
colorectal carcinoma. Cancer Genet Cytogenet. 111:161–165. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ihara Y, Kato Y, Bando T, Yamagishi F,
Minamimura T, Sakamoto T, Tsukada K and Isobe M: Allelic imbalance
of 14q32 in esophageal carcinoma. Cancer Genet Cytogenet.
135:177–181. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ito A, Kataoka TR, Watanabe M, Nishiyama
K, Mazaki Y, Sabe H, Kitamura Y and Nojima H: A truncated isoform
of the PP2A B56 subunit promotes cell motility through paxillin
phosphorylation. EMBO J. 19:562–571. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zheng H, Chen Y, Chen S, Niu Y, Yang L, Li
B, Lu Y, Geng S, Du X and Li Y: Expression and distribution of
PPP2R5C gene in leukemia. J Hematol Oncol. 4:212011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Montagut C and Settleman J: Targeting the
RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 283:125–134.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koudelakova V, Kneblova M, Trojanec R,
Drabek J and Hajduch M: Non-small cell lung cancer - genetic
predictors. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub.
157:125–136. 2013.PubMed/NCBI
|
|
28
|
Seo JS, Ju YS, Lee WC, Shin JY, Lee JK,
Bleazard T, Lee J, Jung YJ, Kim JO, Shin JY, et al: The
transcriptional landscape and mutational profile of lung
adenocarcinoma. Genome Res. 22:2109–2119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hiramatsu M, Ninomiya H, Inamura K, Nomura
K, Takeuchi K, Satoh Y, Okumura S, Nakagawa K, Yamori T, Matsuura
M, et al: Activation status of receptor tyrosine kinase downstream
pathways in primary lung adenocarcinoma with reference of KRAS and
EGFR mutations. Lung Cancer. 70:94–102. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stella GM, Scabini R, Inghilleri S, Cemmi
F, Corso S, Pozzi E, Morbini P, Valentini A, Dore R, Ferrari S, et
al: EGFR and KRAS mutational profiling in fresh non-small cell lung
cancer (NSCLC) cells. J Cancer Res Clin Oncol. 139:1327–1335. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chung BM, Dimri M, George M, Reddi AL,
Chen G, Band V and Band H: The role of cooperativity with Src in
oncogenic transformation mediated by non-small cell lung
cancer-associated EGF receptor mutants. Oncogene. 28:1821–1832.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rinehart J, Adjei AA, Lorusso PM,
Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury
P, Kaldjian EP, et al: Multicenter phase II study of the oral MEK
inhibitor, CI-1040, in patients with advanced non-small-cell lung,
breast, colon, and pancreatic cancer. J Clin Oncol. 22:4456–4462.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Erman M, Grunenwald D, Penault-Llorca F,
Grenier J, Besse B, Validire P, Morat L, Girard P, Le Chevalier T,
Sabatier L, et al: Epidermal growth factor receptor, HER-2/neu and
related pathways in lung adenocarcinomas with bronchioloalveolar
features. Lung Cancer. 47:315–323. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu MX, Ustyugova IV, Han L and Akilov OE:
Immediate early response gene X-1, a potential prognostic biomarker
in cancers. Expert Opin Ther Targets. 17:593–606. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Garcia MN, Grasso D, Lopez-Millan MB,
Hamidi T, Loncle C, Tomasini R, Lomberk G, Porteu F, Urrutia R and
Iovanna JL: IER3 supports KRASG12D-dependent pancreatic cancer
development by sustaining ERK1/2 phosphorylation. J Clin Invest.
124:4709–4722. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ria R, Todoerti K, Berardi S, Coluccia AM,
De Luisi A, Mattioli M, Ronchetti D, Morabito F, Guarini A,
Petrucci MT, et al: Gene expression profiling of bone marrow
endothelial cells in patients with multiple myeloma. Clin Cancer
Res. 15:5369–5378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shi Y: Serine/threonine phosphatases:
Mechanism through structure. Cell. 139:468–484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Janssens V, Goris J and Van Hoof C: PP2A:
The expected tumor suppressor. Curr Opin Genet Dev. 15:34–41. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Abujiang P, Mori TJ, Takahashi T, Tanaka
F, Kasyu I, Hitomi S and Hiai H: Loss of heterozygosity (LOH) at
17q and 14q in human lung cancers. Oncogene. 17:3029–3033. 1998.
View Article : Google Scholar
|
|
41
|
Kwong FM, Wong PS and Lung ML: Genetic
alterations detected on chromosomes 13 and 14 in Chinese non-small
cell lung carcinomas. Cancer Lett. 192:189–198. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Al-Mulla F, AlFadhli S, Al-Hakim AH, Going
JJ and Bitar MS: Metastatic recurrence of early-stage colorectal
cancer is linked to loss of heterozygosity on chromosomes 4 and
14q. J Clin Pathol. 59:624–630. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hoshi M, Shiwaku HO, Hayashi Y, Kaneko Y
and Horii A: Deletion mapping of 14q32 in human neuroblastoma
defines an 1,100-kb region of common allelic loss. Med Pediatr
Oncol. 35:522–525. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dai YC, Ho CL, Tsai YC, Hsu YH, Chang YC,
Liu HS, Chen HH and Chow NH: Allelic loss of 14q32 in the
pathogenesis of gastrointestinal and ampullary malignancies:
Mapping of the target region to a 17 cM interval. J Cancer Res Clin
Oncol. 131:94–100. 2005. View Article : Google Scholar
|
|
45
|
Pécuchet N, Popova T, Manié E, Lucchesi C,
Battistella A, Vincent-Salomon A, Caux-Moncoutier V, Bollet M,
Sigal-Zafrani B, Sastre-Garau X, et al: Loss of heterozygosity at
13q13 and 14q32 predicts BRCA2 inactivation in luminal breast
carcinomas. Int J Cancer. 133:2834–2842. 2013.PubMed/NCBI
|
|
46
|
Sekido Y, Fong KM and Minna JD: Progress
in understanding the molecular pathogenesis of human lung cancer.
Biochim Biophys Acta. 1378:F21–F59. 1998.PubMed/NCBI
|
|
47
|
Wang SS, Esplin ED, Li JL, Huang L, Gazdar
A, Minna J and Evans GA: Alterations of the PPP2R1B gene in human
lung and colon cancer. Science. 282:284–287. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Carmen Figueroa-Aldariz M,
Castañeda-Patlán MC, Santoyo-Ramos P, Zentella A and Robles-Flores
M: Protein phosphatase 2A is essential to maintain active Wnt
signaling and its Aβ tumor suppressor subunit is not expressed in
colon cancer cells. Mol Carcinog. 54:1430–1441. 2015. View Article : Google Scholar
|
|
49
|
Rasio D, Negrini M, Manenti G, Dragani TA
and Croce CM: Loss of heterozygosity at chromosome 11q in lung
adenocarcinoma: Identification of three independent regions. Cancer
Res. 55:3988–3991. 1995.PubMed/NCBI
|