Introduction
Cisplatin is widely considered as one of the most
effective chemotherapeutic drugs for the treatment of cancers
including lung cancer (1). However,
cisplatin resistance of cancer cells remains an obstacle to
successful chemotherapy (2). The
mechanisms of cisplatin cytotoxicity include DNA damage and
inhibition of DNA synthesis in cancer cells, which subsequently
prevent or enhance cancer cell death through activation of various
signaling pathways including endoplasmic reticulum (ER) stress and
autophagy (3).
Recent studies have shown that ER stress has a dual
role, either promoting cell survival or triggering cell death
depending on an imbalance between ER protein folding load and
capacity (4,5). ER is responsible for two key functions
in eukaryotic cells, namely protein processing and intracellular
calcium storage. ER stress is triggered under various physiological
and pathological conditions, such as exposure to chemotherapeutic
agents and accumulation of misfolded proteins (6,7).
However, accumulation of misfolded proteins in ER lumen resulting
in ER stress causes the unfolded protein response (UPR) to initiate
the expression of chaperones and proteins, such as
glucose-regulated proteins [e.g. 78 kDa glucose-regulated protein
(GRP78)], calreticulin, calnexin and several folding enzymes [e.g.
the thioredoxin-like protein disulfide isomerase (PDI)] (8). Moderate ER stress promotes cancer cell
survival and enhances chemotherapeutic resistance, however, severe
ER stress leads to cancer cell apoptosis (9). Moreover, to alleviate ER stress, the
UPR signaling may activate autophagy to clear the accumulated
misfolded proteins from the ER lumen (10).
Autophagy plays an important role in cell
metabolism, and it can degrade intracellular macromolecules and
damaged organelles to maintain cell homeostasis (11). Besides its cytoprotective role in
regulating protein homeostasis, autophagy is associated with cell
apoptosis as a form of programmed cell death (12). Recent research shows that cisplatin
triggers autophagic cell death by damaging DNA replication
(13), but chemotherapeutic drugs,
including cisplatin, could induce autophagy to promote drug
resistance in cancers (14). The
effect of autophagy on apoptosis induced by cisplatin in lung
cancer cells has not been fully understood.
In the present study, we found that cisplatin
induced apoptosis, autophagy and ER stress in lung cancer cell
lines. Interfering ER stress by exposure to 4-phenylbutyric acid
(4-PBA) or tauroursodeoxycholic acid sodium (TUDC) enhanced the
cytotoxicity induced by cisplatin in human lung cancer cells.
Similarly, the autophagic inhibitor 3-methylade-nine (3-MA) or
chloroquine (CQ) promoted cisplatin-induced apoptosis in human lung
cancer cells via inhibition of autophagy. Therefore, our data
suggest that ER stress and autophagy may play a protective role in
the apoptosis induced by cisplatin in human lung cancer cells.
Materials and methods
Cell lines and cell culture
The human lung cancer cell lines A549 and H460 were
cultured at 37°C in a 5% CO2 and 95% air atmosphere, in
Roswell Park Memorial Institute (RPMI)-1640 medium (Gibco,
Carlsbad, CA, USA), supplemented with 10% fetal bovine serum
(Invitrogen, Carlsbad, CA, USA), 100 U/ml penicillin and 100 U/ml
streptomycin.
Cell viability assays
Cell growth inhibition (GI) was assessed by the MTT
assay. Cells were seeded at densities of 8,000/well in 96-well
plates. After 24 h, various concentrations of cisplatin
[cis-diamminedichloroplatinum II (CDDP)] were added to the
wells and incubated for 24 h. Each treatment was repeated in
triplicate wells. Following 24 h of cisplatin treatment, each well
received 20 µl MTT (5 mg/ml; Sigma-Aldrich, St. Louis, MO,
USA) and was kept for 4 h in the dark. Then, 150 µl dimethyl
sulphoxide was added to dissolve the formazan crystals. Absorbance
was measured with an enzyme-linked immunosorbent assay reader
(Infinite M200; Tecan Group Ltd., Männedorf, Switzerland) at a
wavelength of 570 nm.
Lactate dehydrogenase (LDH) assays
The level of LDH, as an indicator of cell injury,
was measured in human lung cancer A549 and H460 cells treated with
cisplatin using a standard method. The activity of the enzyme was
measured with a cytotoxicity detection kit (LDH; Roche). All
experiments were performed at least in triplicate.
Mitochondrial transmembrane potential
(MMP) assay
The disruption of MMP was measured using
fluorochrome dye 1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine
iodide (JC-1) staining according to previously reported procedures
(15). A549 and H460 cells were
treated with or without different concentrations of cisplatin for
24 h. Then, the cells were harvested and stained with 20 µM
JC-1 for 30 min at 37°C under dark conditions. Next, the cells were
washed and resuspended with cold phosphate-buffered saline (PBS)
for fluorescence. In addition, the wavelength was measured using a
fluorescence plate reader with an excitation wavelength at 590 nm
and an emission wavelength at 540 nm. According to the ratio of
fluorescence intensities at 590 and 540 nm, the loss of
mitochondrial membrane potential (MMP) was assessed.
Western blotting
After treatment with cisplatin alone or in
combination, the cells were washed twice with cold PBS and then 120
µl of RIPA buffer was added. Cell lysates were sonicated for
5 sec on ice and then static at 4°C for 45 min. After centrifuging
at 3,000 × g for 15 min, protein concentrations were measured using
the BCA protein assay (Pierce, Rockford, IL, USA). Samples of
extracted proteins were boiled for 10 min at 100°C, and equivalent
amounts of proteins (30–90 µg) were separated by
electrophoresis using Criterion TGX Precast 12% gels and
transferred onto Trans-Blot Turbo Midi PVDF Transfer Packs
(Whatman, Maidstone, UK). The membranes were blocked with 5% (w/v)
skimmed milk for 1 h at room temperature, and then proteins were
detected by specific primary antibodies (Santa Cruz Biotechnology,
Santa Cruz, CA, USA): activated caspase-3, PARP, GRP78, cytochrome
c, PERK and ERE1 overnight at 4°C, followed by incubation
with horseradish peroxidase-conjugated secondary antibodies
(1:2,000; Sigma) for 2 h at room temperature. The levels of
proteins were quantified using Quantity One software (Bio-Rad,
Hercules, CA, USA).
Flow cytometric analysis
After the A549 and H460 cells were treated and
incubated with the drugs, the cells were digested by 0.25% trypsin,
and then cell death was determined using propidium iodide (PI; 1
µg/ml) and Annexin V-FITC (1 µg/ml; Invitrogen).
After a 15-min incubation at 37°C, the samples were detected using
a FACScan flow cytometer (BD Biosystems, San Jose, CA, USA). All
experiments in the present study were performed in triplicate.
Statistical analysis
All data were analyzed using the t-test. P<0.05
was considered to indicate a statistically significant difference.
Data in the present study are representative of three independent
experiments performed in triplicate.
Results
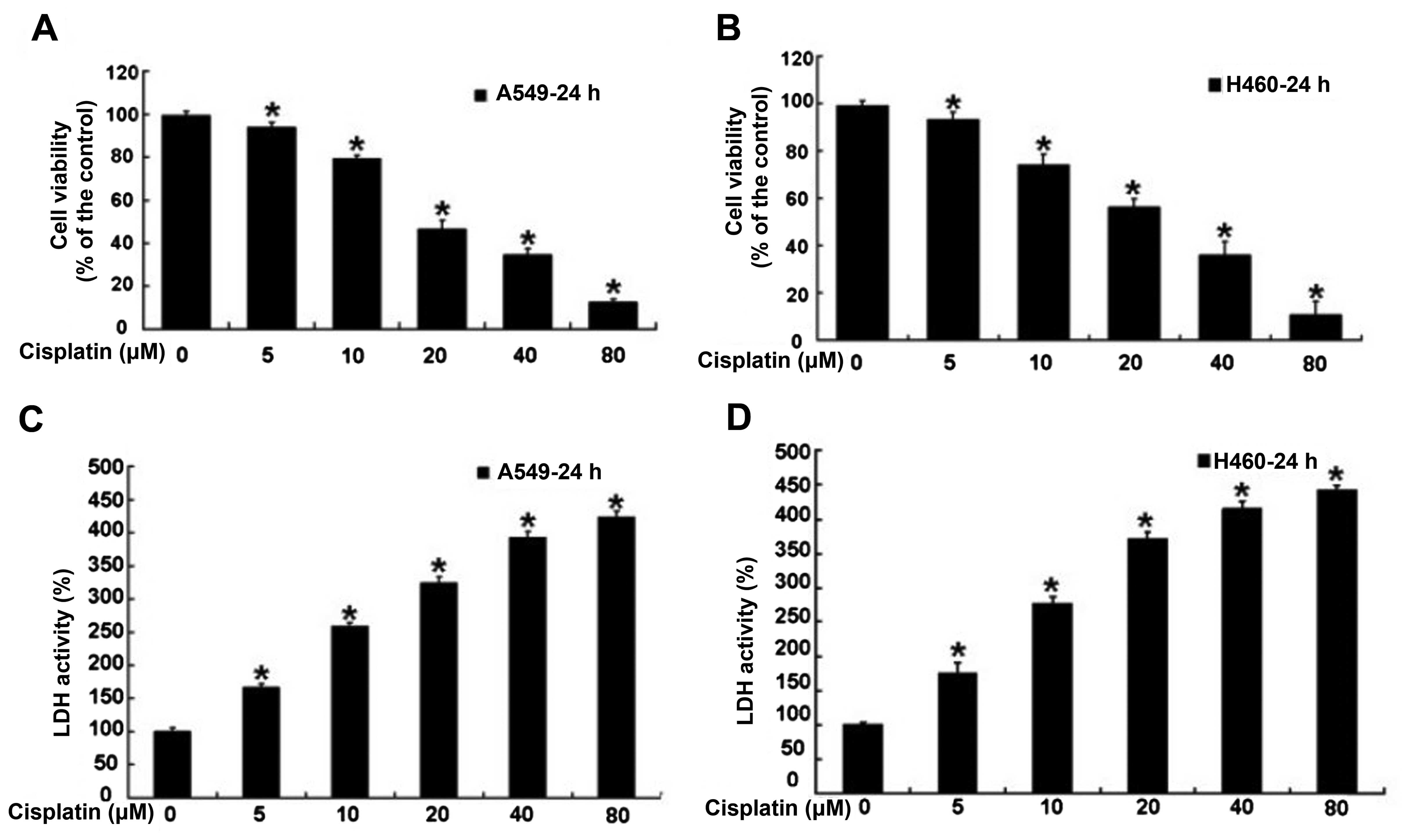
Cisplatin inhibits the cell proliferation
of human lung cancer A549 and H460 cells
Human lung cancer A549 and H460 cells were treated
with varying doses of cisplatin for 24 h, and then the cell
viability rate was examined by MTT assay. According to the results,
we found that cisplatin inhibited A549 and H460 cell proliferation
in a dose-dependent manner (Fig. 1A and
B). Meanwhile, the cell death of the treated A549 and H460
cells was assessed via measuring the leakage of LDH. The result
showed that the level of LDH was obviously increased in the A549
and H460 cells treated with cisplatin, and cisplatin significantly
increased the release of LDH in a dose-dependent manner (Fig. 1C and D). These results indicate that
cisplatin inhibited the cell growth of human lung cancer A549 and
H460 cells.
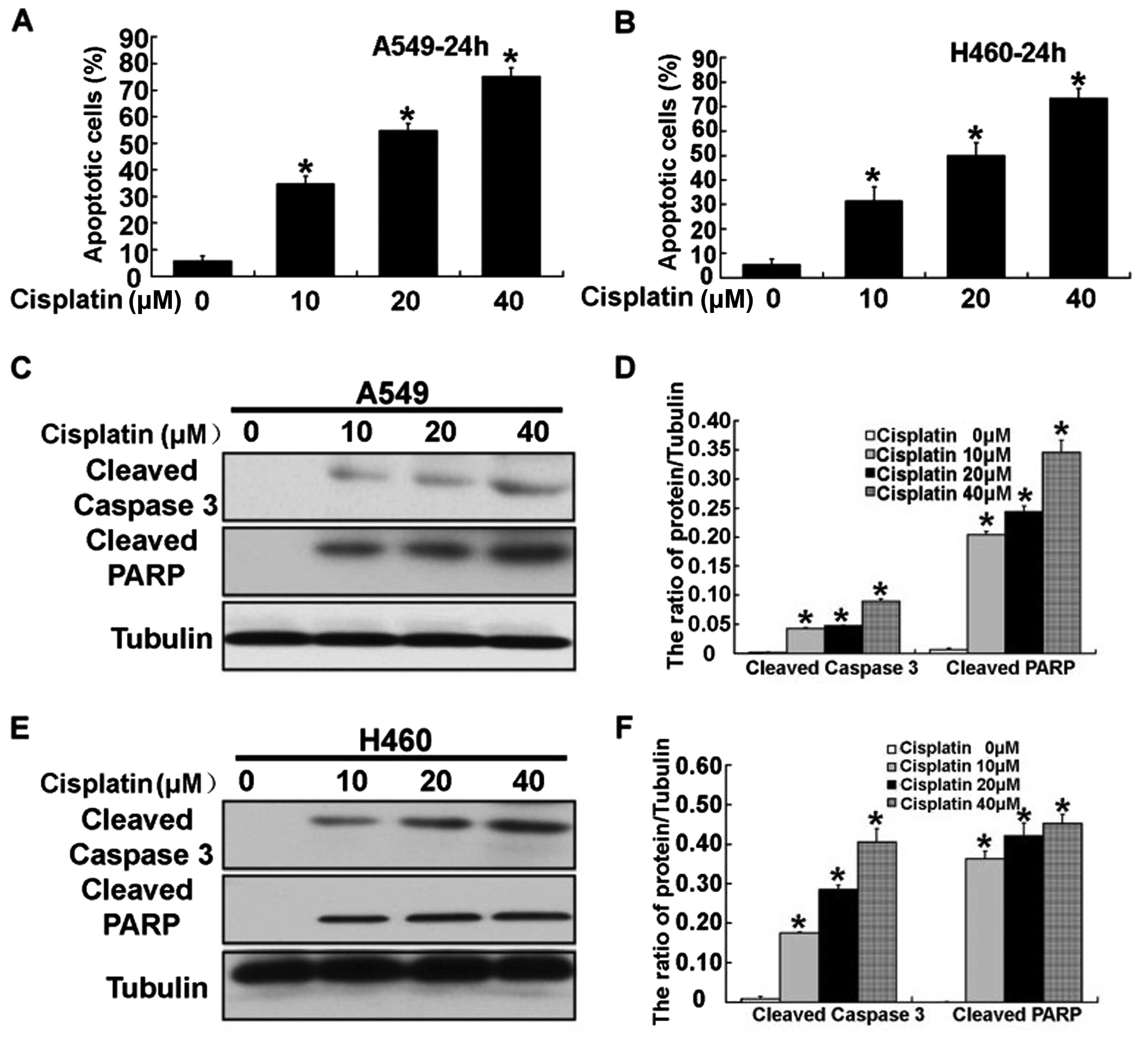
Cisplatin induces human lung cancer A549
and H460 cell death by mitochondrial-pathway apoptosis
After treatment with cisplatin, we next aimed to
ascertain the mechanism involved in the cisplatin-induced apoptosis
of human lung cancer A549 and H460 cells. Compared with the
control, the apoptotic rate was obviously increased in the A549 and
H460 cells following treatment with 10, 20 and 40 µM
cisplatin which was detected by flow cytometric analysis (Fig. 2A and B). Meanwhile, we assessed the
apoptotic effects of cisplatin on A549 and H460 cells, through
examination of the activation of caspase-3 and PARP using western
blotting. The levels of activation of caspase-3 and PARP were
significantly increased in the A549 and H460 cells following
treatment with cisplatin (Fig.
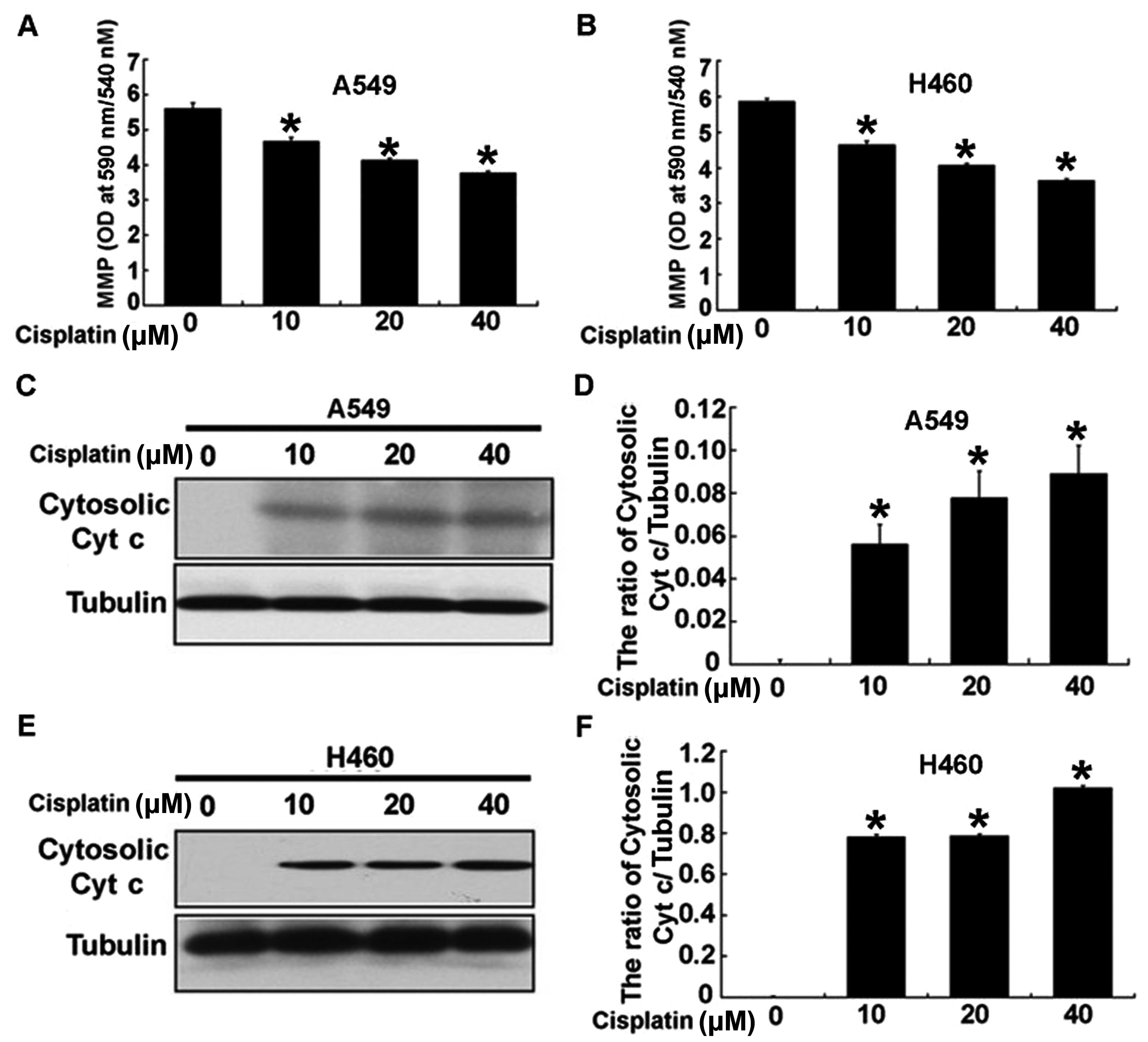
2C–F). To assess the exact mechanism of cisplatin-induced
apoptosis, we detected the intrinsic mitochondrial-mediated
apoptotic pathway in the A549 and H460 cells. As shown in Fig. 3A and B, cisplatin obviously reduced
the mitochondrial potential in the A549 and H460 cells treated with
cisplatin. In addition, cisplatin upregulated the level of
cytosolic cytochrome c in the A549 and H460 cells (Fig. 3C–F). These results indicated that
cisplatin significantly induced the apoptosis of A549 and H460
cells through the intrinsic mitochondrial apoptotic pathway.
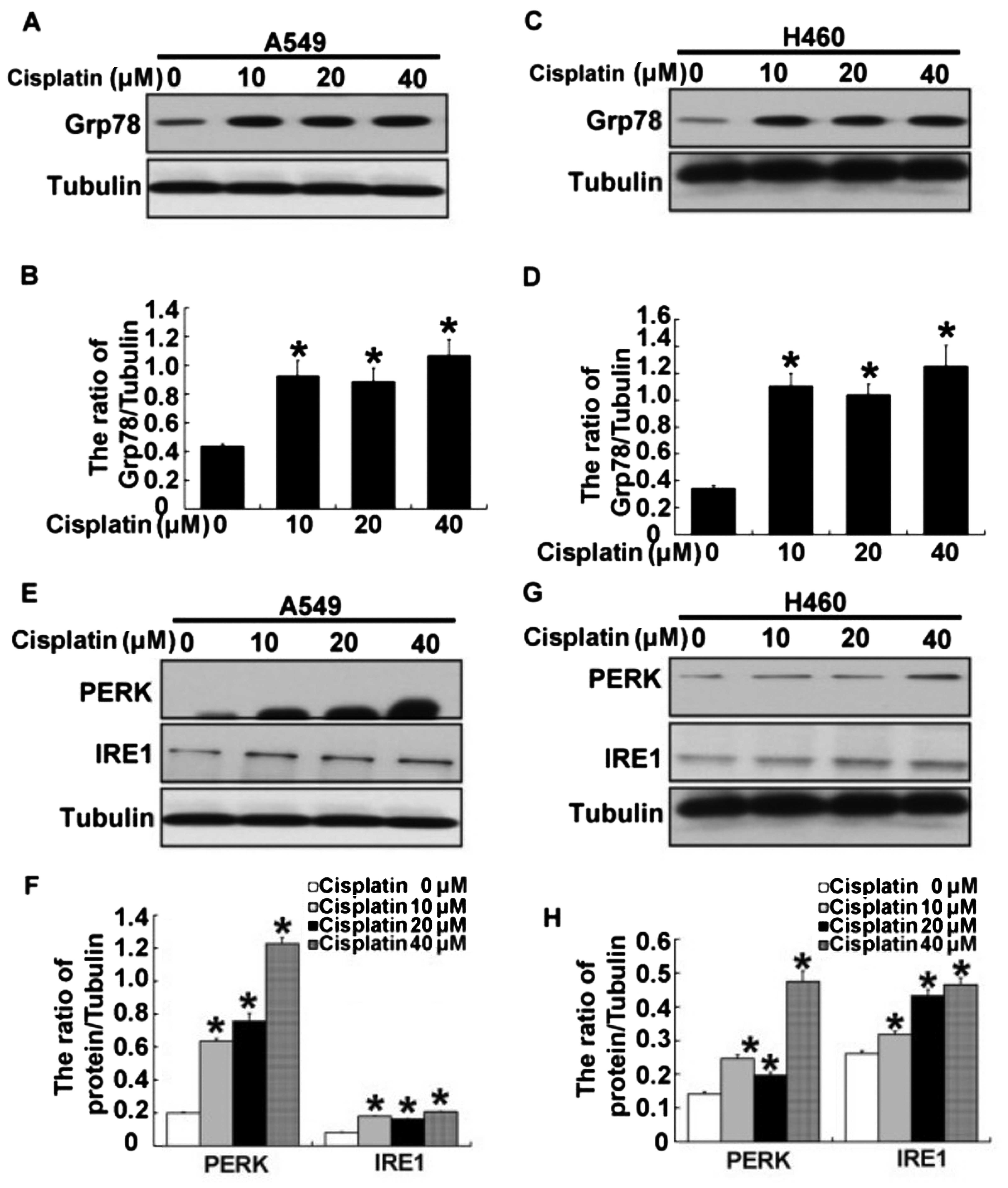
Cisplatin induces ER stress in human lung
cancer A549 and H460 cells
To further assess whether ER stress is associated
with the mechanism of cisplatin-induced apoptosis in human lung
cancer A549 and H460 cells, we examined expression of ER stress
proteins in the cells following treatment with cisplatin. After
A549 and H460 cells were treated with 10, 20 and 40 µM
cisplatin for 24 h, we detected the expression of Grp78, an ER
stress marker. Using western blotting, we found that the expression
of Grp78 was increased in the A549 and H460 cells treated with
cisplatin (Fig. 4A–D). When ER
stress is induced, it triggers the UPR to respond to environmental
factors. Thus UPR regulates the expression of the ER stress sensor
protein PKR-like ER kinase (PERK), and the inositol-requiring
enzyme 1 (IRE-1). As shown in Fig.
4E–H, expression of ER stress-associated proteins PERE/IRE1
were increased in the A549 and H460 cells following treatment with
cisplatin. These results demonstrated that ER stress was induced in
the A549 and H460 cells following treatment with cisplatin, and
that this was regulated by the PERK and IRE1 pathway.
ER stress is involved in
cisplatin-induced apoptosis in human lung cancer A549 and H460
cells
We next wanted to examine the relevance of ER stress
in cisplatin-induced apoptosis in human lung cancer A549 and H460
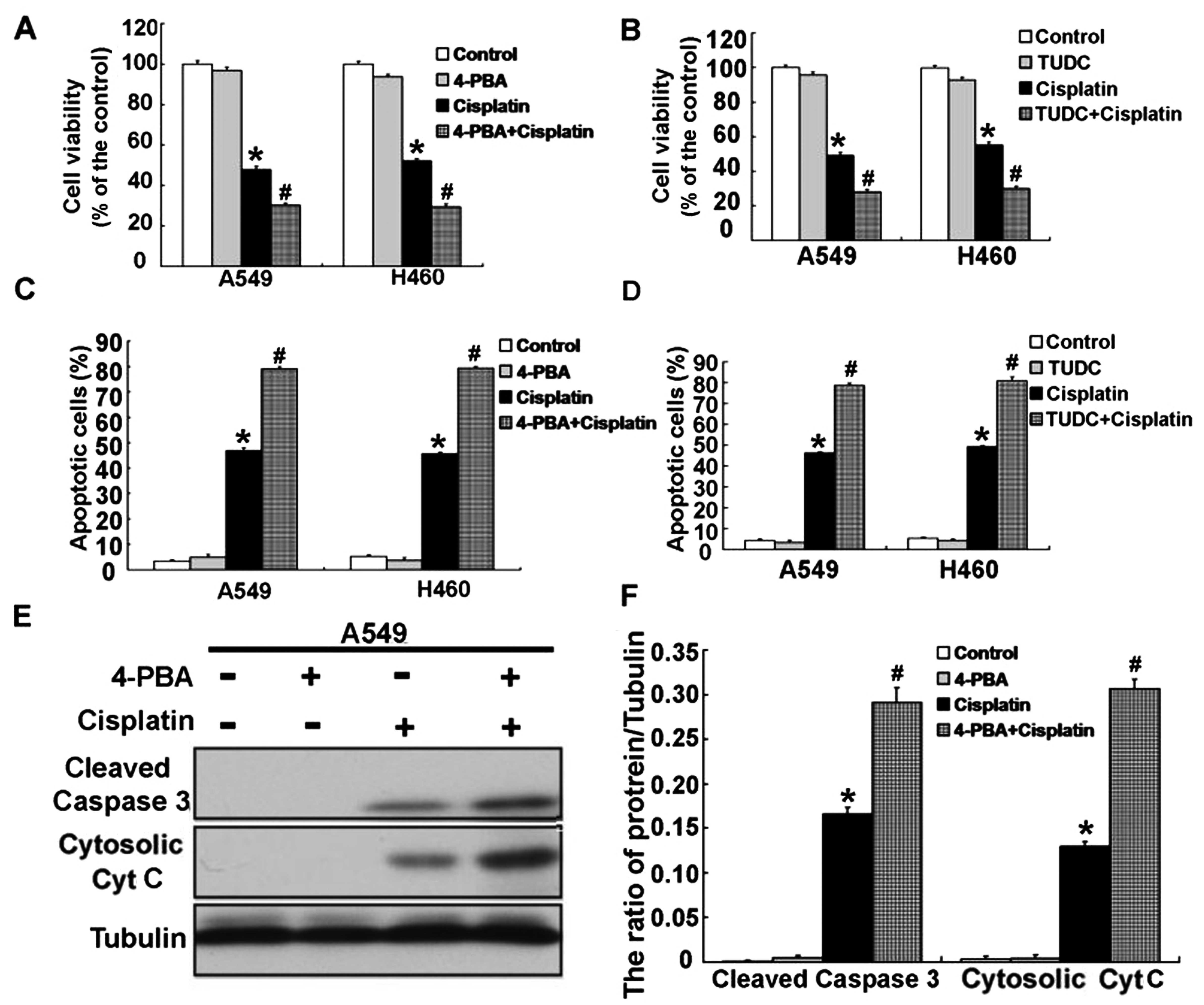
cells. We used ER stress inhibitors 4-PBA and TUDC to alleviate ER
stress in the A549 and H460 cells treated with cisplatin. Using MTT
assay, we found that inhibition of ER stress effectively decreased
the cell viability of the cisplatin-treated A549 and H460 cells at
24 h (Fig. 5A and B). Consistent
with the inhibition of cell viability, inhibition of ER stress
significantly enhanced the cell apoptosis of the cisplatin-treated
A549 and H460 cells at 24 h (Fig. 5C
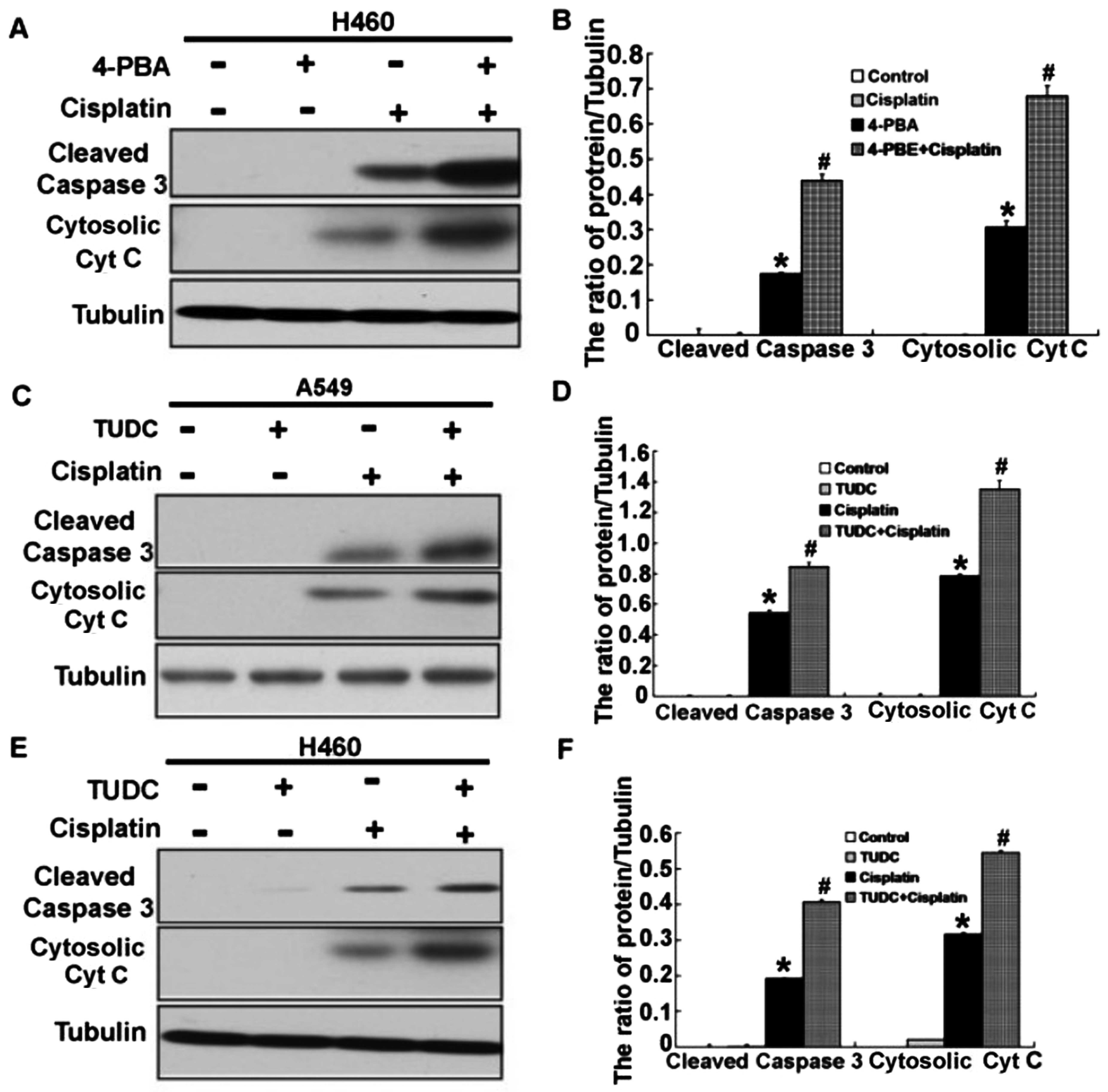
and D). Then, we examined the expression of
apoptosis-associated proteins, cleaved caspase-3 and cytosolic
cytochrome c. According to the results, activation of
caspase-3 was obviously upregulated, and the level of cytosolic
cytochrome c was also significantly increased (Figs. 5E and F and 6A–F). These results demonstrated that
cisplatin can induce ER stress, and inhibition of ER stress
increased the apoptosis in the A549 and H460 cells treated with
cisplatin.
Cisplatin induces the activation of
autophagy in human lung cancer A549 and H460 cells
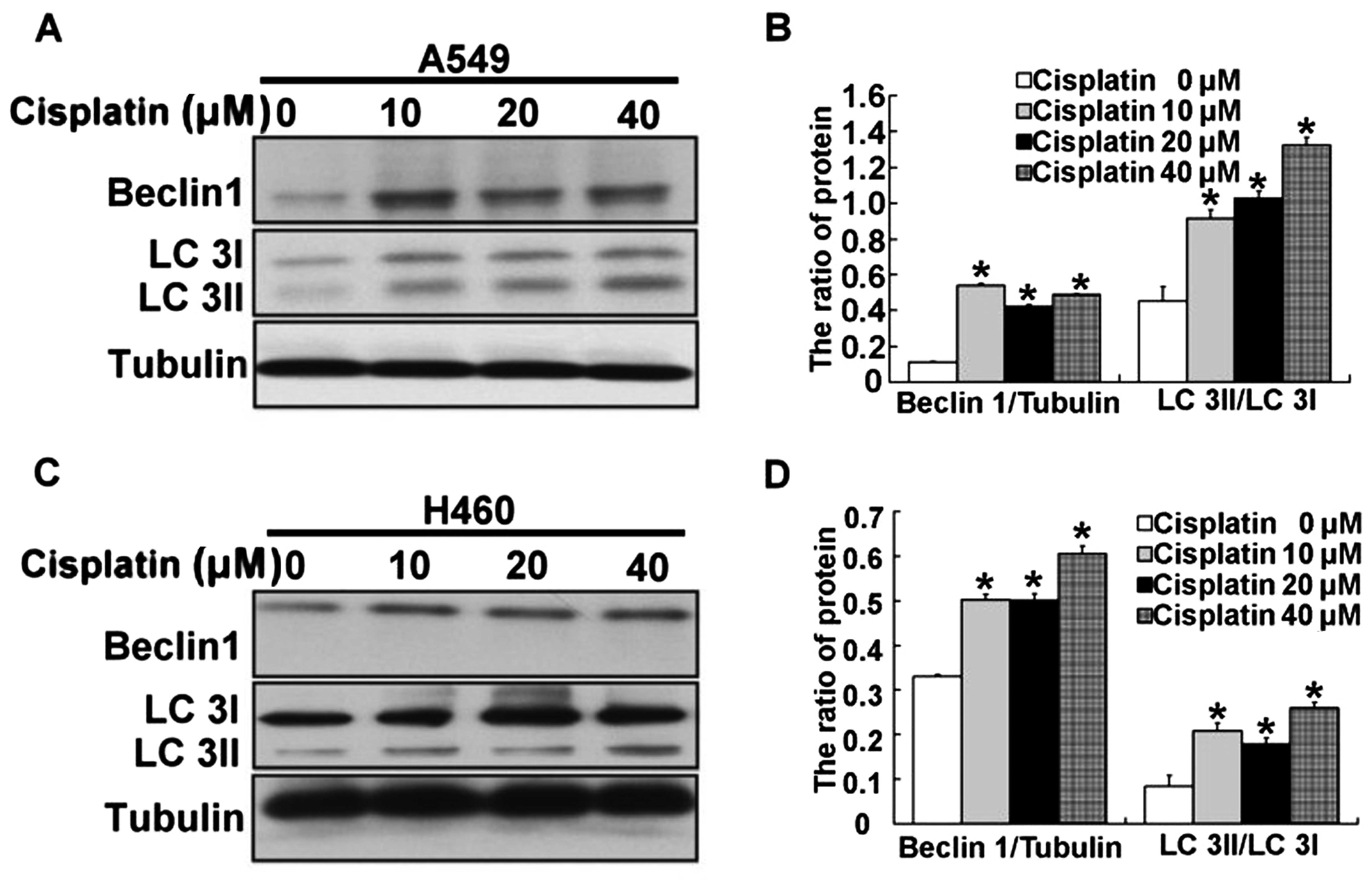
In addition to regulating ER stress, we also found
activation of autophagy in the A549 and H460 cells treated with
cisplatin. Thus, we examined the expression of the protein Beclin
1, which is required for autophagy. After A549 and H460 cells were
treated with 10, 20 and 40 µM cisplatin for 24 h, we found
that expression of Beclin 1 was significantly upregulated in the
A549 and H460 cells (Fig. 7A–D).
Next, LC3, a molecular marker of autophagy, was assessed. We found
that the ratio of LC3 II/LC3 I was high in the A549 and H460 cells
treated by cisplatin (Fig. 7A–D).
These results demonstrated that autophagy was induced by cisplatin
in the A549 and H460 cells.
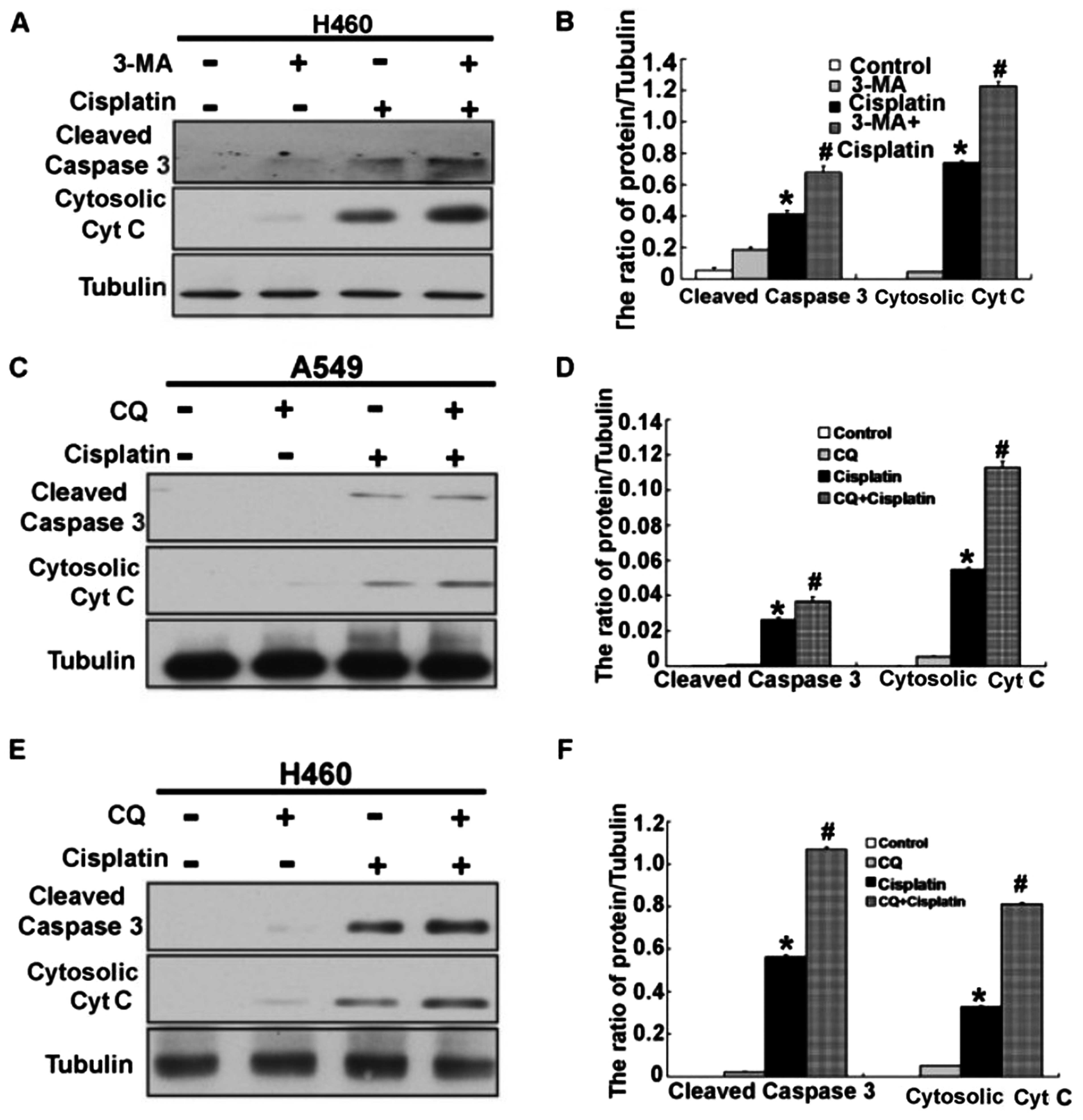
Inhibition of autophagy enhances
cisplatin-induced cytotoxicity in human lung cancer A549 and H460
cells
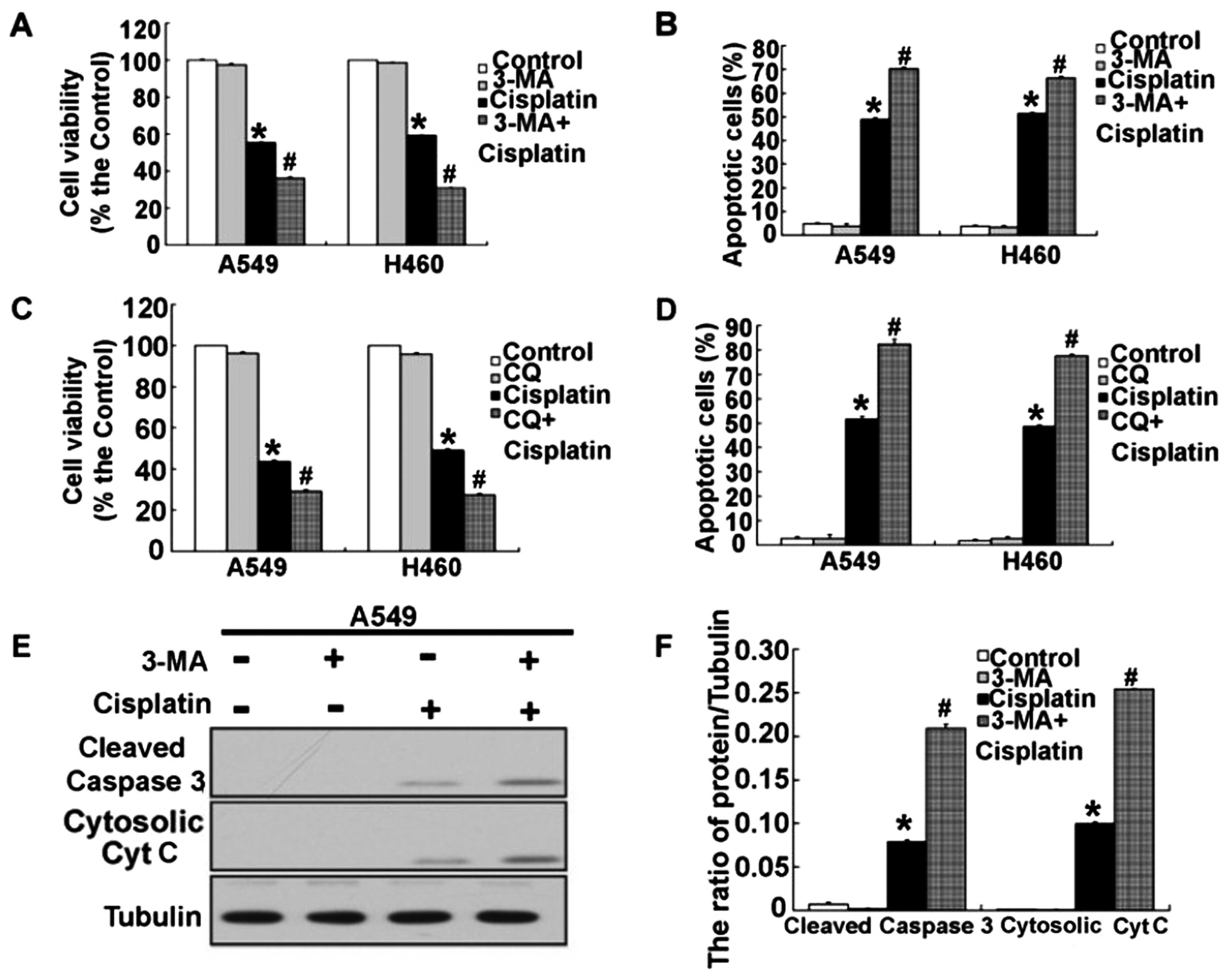
The previous results demonstrated that cisplatin
induced activation of autophagy and apoptosis. However, the
mechanisms of autophagy and apoptosis are still unclear, but
important in cancer treatment. Next, we explored whether there was
a relationship between autophagy and apoptosis in the A549 and H460
cells treated with cisplatin. To clarify the action of autophagy in
the A549 and H460 cells treated with cisplatin, we used two
different autophagy inhibitors, 3-MA (which inhibits the initiation
of autophagy) and CQ (which blocks the fusion of autophagosomes and
lysosomes). As shown in Fig. 8A–D,
we found that combination of 3-MA or CQ and cisplatin further
decreased the cell viability and induced the cell apoptosis of A549
and H460 cells. When we inhibited autophagy by 3-MA and CQ, we
found that the level of apoptosis-associated proteins, activated
caspase-3 and cytosolic cytochrome c, were obviously
increased in the A549 and H460 cells treated with cisplatin
(Figs. 8E and F and 9A–F). These results suggest that
inhibition of autophagy enhanced cisplatin-induced cytotoxicity and
apoptosis in the human lung cancer cells.
Discussion
Lung cancer is one of the most common malignancies,
and cisplatin is often used as a first-line chemotherapeutic agent
in cancers including lung cancer. However, lung cancer cells become
resistant to cisplatin and it is an obstacle in clinical treatment
(16–18). Numerous studies have shown that
cisplatin induces the mitochondrial apoptosis pathway by disturbing
mitochondrial potential, thus resulting in a caspase-dependent
intrinsic pathway (19–21). Recent studies show that cisplatin
induces ER stress as well, and sustained and severe ER stress
results in activation of caspase-mediated apoptosis in cancers
(22,23). However, the exact mechanisms of
cisplatin-induced cell death in human lung cancer cells are not
fully understood. Consistently in the present study, we found that
cisplatin triggered the apoptosis of human lung cancer A549 and
H460 cells through classic caspase-mediated apoptosis. Cisplatin
also altered mitochondrial potential, thus leading to activation of
mitochondrial apoptotic pathways through classic cytochrome
c and caspase-3.
ER is a key organelle with protein processing,
intracellular calcium storage, as well as crucial biosynthetic and
signaling regulation functions in eukaryotic cells (24). When destroying an imbalance between
ER protein folding load and capacity in different physiological and
pathological conditions, such as cisplatin, this results in
accumulated unfolded proteins of ER lumen, known as 'ER stress'. ER
stress can initiate cell UPR, which regulates the expression of ER
molecular chaperone GRP78/Bip, the ER stress sensor protein PERK,
the inositol-requiring enzyme 1 (IRE-1). Mild to moderate ER stress
promotes cell survival through UPR to alleviate ER stress (25). In addition, it has been proven that
human lung cancer cells can acquire cisplatin-resistance via ER
stress (26). However sustained and
severe ER stress or inhibition of ER stress leads to cell death in
some cancers including lung cancer (27). However, the exact function of
cisplatin-induced ER stress is not fully defined. In the present
study, we demonstrated that cisplatin induced ER stress through the
expression of GRP78, IRE1 and PERK. However, we could not detect
the expression of ER stress associated protein CHOP. It may be due
to the low level of the ER stress induced by cisplatin which could
not induce ER stress-associated apoptosis. When we blocked ER
stress by ER stress inhibitors 4-PBA or TUDC, cytotoxicity and
apoptosis were obviously increased in the human lung cancer A549
and H460 cells treated with cisplatin. Besides ER stress, it is
reported that some antitumor agents can induced autophagy (28,29).
Meanwhile, some studies showed that UPR signaling may activate
autophagy to alleviate ER stress through the PERK signaling pathway
(30).
Autophagy is an intracellular metabolic system in
eukaryotic cells, in which autophagosomes fuse with the lysosome,
and degrade intracellular materials to maintain cell homeostasis
(31). Various studies have shown
that activation of autophagy protects cells from cisplatin-induced
apoptosis, allowing cells to alleviate ER stress, consequently
causing cisplatin resistance (14,32).
However, the role of autophagy in human lung cancer cells treated
with cisplatin is still not clear. According to the expression of
LC3 and Beclin 1, we demonstrated that cisplatin induced autophagy
in human lung cancer A549 and H460 cells. However, it is unclear
whether activation of autophagy protects cells from
cisplatin-induced apoptosis or leads to cell death in human lung
cancer cells. Our results showed that inhibition of autophagy by
3-MA or CQ enhanced cisplatin-induced apoptosis in human lung
cancer A549 and H460 cells by triggering the
mitochondrial-apoptosis pathway. These data indicate that autophagy
plays a protective role and is involved in cisplatin resistance in
human lung cancer A549 and H460 cells.
In summary, we found that cisplatin can induce
apoptosis, ER stress and autophagy, and ER stress and autophagy are
involved in the mechanism of cisplatin resistance in human lung
cancer cells. Inhibition of ER stress or autophagy can further
increase the apoptosis induced by cisplatin. Therefore, our data
suggest that cisplatin-induced ER stress and autophagy may play a
protective role in apoptosis induced by cisplatin in human lung
cancer cells.
References
|
1
|
Lee HY, Mohammed KA, Goldberg EP, Kaye F
and Nasreen N: Cisplatin loaded albumin mesospheres for lung cancer
treatment. Am J Cancer Res. 5:603–615. 2015.PubMed/NCBI
|
|
2
|
Wu DW, Lee MC, Hsu NY, Wu TC, Wu JY, Wang
YC, Cheng YW, Chen CY and Lee H: FHIT loss confers cisplatin
resistance in lung cancer via the AKT/NF-κB/Slug-mediated PUMA
reduction. Oncogene. 34:25462015. View Article : Google Scholar
|
|
3
|
Zhang R, Wang R, Chen Q and Chang H:
Inhibition of autophagy using 3-methyladenine increases
cisplatin-induced apoptosis by increasing endoplasmic reticulum
stress in U251 human glioma cells. Mol Med Rep. 12:1727–1732.
2015.PubMed/NCBI
|
|
4
|
Jang JH, Kim YJ, Kim H, Kim SC and Cho JH:
Buforin IIb induces endoplasmic reticulum stress-mediated apoptosis
in HeLa cells. Peptides. 69:144–149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shen Y, Yang J, Zhao J, Xiao C, Xu C and
Xiang Y: The switch from ER stress-induced apoptosis to autophagy
via ROS-mediated JNK/p62 signals: A survival mechanism in
methotrexate-resistant choriocarcinoma cells. Exp Cell Res.
334:207–218. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin YD, Chen S, Yue P, Zou W, Benbrook DM,
Liu S, Le TC, Berlin KD, Khuri FR and Sun SY: CAAT/enhancer binding
protein homologous protein-dependent death receptor 5 induction is
a major component of SHetA2-induced apoptosis in lung cancer cells.
Cancer Res. 68:5335–5344. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ron D and Hubbard SR: How IRE1 reacts to
ER stress. Cell. 132:24–26. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dong D, Ni M, Li J, Xiong S, Ye W, Virrey
JJ, Mao C, Ye R, Wang M, Pen L, et al: Critical role of the stress
chaperone GRP78/BiP in tumor proliferation, survival, and tumor
angiogenesis in transgene-induced mammary tumor development. Cancer
Res. 68:498–505. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kouroku Y, Fujita E, Tanida I, Ueno T,
Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E and Momoi T: ER
stress (PERK/eIF2alpha phosphorylation) mediates the
polyglutamine–induced LC3 conversion, an essential step for
autophagy formation. Cell Death Differ. 14:230–239. 2007.
View Article : Google Scholar
|
|
11
|
Li MQ and Liu ZG: Dual role of autophagy
in chronic myeloid leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi.
23:583–586. 2015.In Chinese. PubMed/NCBI
|
|
12
|
Kim MO, Lee HS, Chin YW, Moon DO and Ahn
JS: Gartanin induces autophagy through JNK activation which
extenuates caspase-dependent apoptosis. Oncol Rep. 34:139–146.
2015.PubMed/NCBI
|
|
13
|
He J, Yu JJ, Xu Q, Wang L, Zheng JZ, Liu
LZ and Jiang BH: Downregulation of ATG14 by EGR1-MIR152 sensitizes
ovarian cancer cells to cisplatin-induced apoptosis by inhibiting
cyto–protective autophagy. Autophagy. 11:373–384. 2015. View Article : Google Scholar
|
|
14
|
Bao L, Jaramillo MC, Zhang Z, Zheng Y, Yao
M, Zhang DD and Yi X: Induction of autophagy contributes to
cisplatin resistance in human ovarian cancer cells. Mol Med Rep.
11:91–98. 2015.
|
|
15
|
Shen H, Zeng G, Sun B, Cai X, Bi L, Tang G
and Yang Y: A polysaccharide from Glycyrrhiza inflata Licorice
inhibits proliferation of human oral cancer cells by inducing
apoptosis via mitochondrial pathway. Tumour Biol. 36:4825–4831.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Köberle B, Tomicic MT, Usanova S and Kaina
B: Cisplatin resistance: Preclinical findings and clinical
implications. Biochim Biophys Acta. 1806:172–182. 2010.PubMed/NCBI
|
|
17
|
Chen J, Solomides C, Parekh H, Simpkins F
and Simpkins H: Cisplatin resistance in human cervical, ovarian and
lung cancer cells. Cancer Chemother Pharmacol. 75:1217–1227. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Horibe S, Matsuda A, Tanahashi T, Inoue J,
Kawauchi S, Mizuno S, Ueno M, Takahashi K, Maeda Y, Maegouchi T, et
al: Cisplatin resistance in human lung cancer cells is linked with
dysregulation of cell cycle associated proteins. Life Sci.
124:31–40. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu J, Yang Y and Wu J: Bcl-2 cleavages at
two adjacent sites by different caspases promote cisplatin-induced
apoptosis. Cell Res. 17:441–448. 2007.PubMed/NCBI
|
|
20
|
Sharma H, Sen S and Singh N: Molecular
pathways in the chemosensitization of cisplatin by quercetin in
human head and neck cancer. Cancer Biol Ther. 4:949–955. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wei Q, Dong G, Franklin J and Dong Z: The
pathological role of Bax in cisplatin nephrotoxicity. Kidney Int.
72:53–62. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Z, Sun Y, Ren L, Huang Y, Cai Y, Weng
Q, Shen X, Li X, Liang G and Wang Y: Evaluation of a curcumin
analog as an anti-cancer agent inducing ER stress-mediated
apoptosis in non-small cell lung cancer cells. BMC Cancer.
13:4942013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu SH, Hang LW, Yang JS, Chen HY, Lin HY,
Chiang JH, Lu CC, Yang JL, Lai TY, Ko YC, et al: Curcumin induces
apoptosis in human non-small cell lung cancer NCI-H460 cells
through ER stress and caspase cascade- and mitochondria-dependent
pathways. Anticancer Res. 30:2125–2133. 2010.PubMed/NCBI
|
|
24
|
Moir RD, Gross DA, Silver DL and Willis
IM: SCS3 and YFT2 link transcription of phospholipid biosynthetic
genes to ER stress and the UPR. PLoS Genet. 8:e10028902012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hetz C: The UPR as a survival factor of
cancer cells: More than folding proteins? Leuk Res. 33:880–882.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin Y, Wang Z, Liu L and Chen L: Akt is
the downstream target of GRP78 in mediating cisplatin resistance in
ER stress-tolerant human lung cancer cells. Lung Cancer.
71:291–297. 2011. View Article : Google Scholar
|
|
27
|
Cui WY, Liu Y, Zhu YQ, Song T and Wang QS:
Propofol induces endoplasmic reticulum (ER) stress and apoptosis in
lung cancer cell H460. Tumour Biol. 35:5213–5217. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lan D, Wang W, Zhuang J and Zhao Z:
Proteasome inhibitor-induced autophagy in PC12 cells overexpressing
A53T mutant α-synuclein. Mol Med Rep. 11:1655–1660. 2015.
|
|
29
|
Zang Y, Thomas SM, Chan ET, Kirk CJ,
Freilino ML, DeLancey HM, Grandis JR, Li C and Johnson DE: The next
generation proteasome inhibitors carfilzomib and oprozomib activate
prosurvival autophagy via induction of the unfolded protein
response and ATF4. Autophagy. 8:1873–1874. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tallóczy Z, Jiang W, Virgin HW IV, Leib
DA, Scheuner D, Kaufman RJ, Eskelinen EL and Levine B: Regulation
of starvation- and virus-induced autophagy by the eIF2alpha kinase
signaling pathway. Proc Natl Acad Sci USA. 99:190–195. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu M, Ma S, Liu M, Hou Y, Liang B, Su X
and Liu X: Synergistic killing of lung cancer cells by cisplatin
and radiation via autophagy and apoptosis. Oncol Lett. 7:1903–1910.
2014.PubMed/NCBI
|
|
32
|
Bao LJ, Jaramillo MC, Zhang ZB, Zheng YX,
Yao M, Zhang DD and Yi XF: Nrf2 induces cisplatin resistance
through activation of autophagy in ovarian carcinoma. Int J Clin
Exp Pathol. 7:1502–1513. 2014.PubMed/NCBI
|