Introduction
Clear cell sarcoma (CCS), regarded as malignant
melanoma of soft parts, is a rare aggressive tumor that accounts
for less than 1% of all soft tissue sarcomas (1,2). CCS
is characterized by its poor prognosis due to late diagnosis,
multiple local recurrence, propensity to late metastases, and a
high rate of tumor-related mortality (3,4). As
one of the few sarcomas with a high propensity for lymph node
metastases (5), CCS is a locally
aggressive neoplasm with a high rate of recurrence and metastasis
(more than 50%) (2,6,7). The
5-year disease-specific survival rates have been reported to be
approximately 50–67%, but these values are not representative of
long-term survival since many patients develop lung and bone
metastases more than five years after initial resection (7,8). This
tumor is observed most frequently in young adults and predominantly
affects the soft tissues of the distal extremities, with the
majority being deep seated and involving tendons and aponeuroses
(4,9). Occasionally, it can arise in visceral
organs including the gastrointestinal tract (10). It is rarely seen in the head or
neck, with for example only 1.2% of the approximately 500 reported
cases of CCS currently involving the head or neck (1).
Clear cell sarcoma was first described by Dr Franz
M. Enzinger in 1965 (11). CCS is
characterized by a nested or fascicular growth pattern of spindled
and epithelioid cells with clear or lightly eosinophilic cytoplasm
surrounded by fibrous septa (12).
The tumor cells have elongated oval nuclei with prominent nucleoli
and occasional nuclear pseudo-inclusions (13). Multinucleated giant cells are
identified in more than half of the reported cases (7). Uniquely among primary soft tissue
tumors, pre-melanosomes are present in almost all cases of CCS,
detectable by electron microscopy (14). As a result, immunohistochemistry
(IHC)-based tests of CCS cells are almost always positive for the
melanoma markers S-100, HMB45, MelanA, and microphthalmia
transcription factor (MITF), although melanin staining is not
always observed (12,15). Adverse prognostic factors for CCS
identified to date include large tumor size and any microscopic
tumor necrosis. Surgery is the mainstay of treatment for this high
grade sarcoma, with chemotherapy having little effect. Although the
melanocytic differentiation of CCS is indisputable, its precise
lineage remains unclear. Thus, CCS maintains the status of a unique
yet enigmatic clinicopathological entity (4).
As a rare type of soft tissue sarcoma, CCS exhibits
morphological, immunohistochemical and ultrastructural similarity
with malignant melanoma (5). CCSs
share many features with malignant melanoma, including expression
of melanoma markers (16). However,
in contrast to most melanomas, CCSs lack BRAF mutations (9). In addition, molecular analysis has
revealed that CCSs are distinct tumors; they present the specific
t(12;22)(q13;q12) translocation that results in the chimeric gene
EWSR1/ATF1, which is not observed in melanomas (5). The genetic cause for CCS is considered
to be this defining gene translocation. Previous cytogenetic
studies have established the specificity of the recurrent t(12;22)
(q13;q12) translocation, resulting in an EWSR1-ATF1 fusion for CCS
(17). However, EWSR1-ATF1 fusion
derived from cytogenetic rearrangements is characteristic but not
entirely unique for CCS, as similar fusion genes are also present
in angiomatoid fibrous histiocytoma (18). Generally, detection of this fusion
gene and the absence of BRAF gene mutations easily distinguish CCS
from cutaneous melanoma (4).
Having said that, it must be noted that not all CCSs
present with EWSR1 rearrangements. According to a previous study,
approximately 70% of CCS cases harbored a rearrangement (EWSR1-ATF1
or EWSR1-CREB1) in the EWSR1 locus with a mean of 81.6% positive
cells/sample (range, 60–95%) (19).
The more prevalent fusion event in CCS, EWSR1-ATF1, also occurs in
both hyalinizing clear cell carcinoma (20) and angiomatoid fibrous histiocytoma
(18). However, the exons involved
are different, with most CCS tumors involving EWSR1 exons 7, 8, or
10 fused to ATF1 exons 4, 5, or 7 (21), but with HCC tumors harboring EWSR1
exon 11 fused in-frame to exon 3 of ATF1 (20). Therefore, the specific EWSR1-ATF1
fusion in CCS can typically be used to distinguish CCS from its
mimics such as spindle cell melanoma, spindle cell squamous
carcinoma, cutaneous leiomyosarcoma and atypical fibroxanthoma, as
well as from other tumors with melanocytic differentiation
(12).
To date, the pathogenesis of CCS lacking an EWSR1
rearrangement remains poorly characterized. No somatic mutations
involved in CCS have been identified, but single nucleotide
variants are known to play a significant role in tumorigenesis
(22). In the present study, we
identified a somatic missense mutation c.1061C>T (p.P354L) in
exon 9 of the Nibrin (NBN) gene in a patient with CCS of the
salivary gland via a combination of exome sequencing and Sanger
sequencing. It is known that although mutations of NBN do
not play a major role in predisposition to melanoma of the skin,
alterations in this gene may contribute to the risk for breast
cancer (23–25).
NBN is a protein associated with the repair of
double-strand breaks (DSBs), which cause serious damage to genomes.
NBN is a 754 amino acid protein known to be a member of the
NBS1/hMre11/RAD50 double-strand DNA break repair complex (referred
to as MRN) (26). This complex
recognizes DNA damage and rapidly relocates to DSB sites, forming
nuclear foci. NBN also has a role in the regulation of MRN protein
complex activity, including involvement in end-processing of both
physiological and mutagenic DNA DSBs. The mutations within exons
6–10 of the NBN gene in patients suffering Nijmegen breakage
syndrome (NBS) result in a truncated protein (27). Patients with NBS are predisposed to
cancers. This predisposition to cancer may be linked to the DSBs
that occur during the development of lymphoid cells. Moreover,
mutations of NBN have been reported to be associated with many
types of cancers, including gastrointestinal lymphoma (28), childhood acute leukaemia (29), glioblastomas (30), and breast cancer (25). In this study, we report the first
known somatic mutation of NBN that was found to be involved in CCS
of the salivary gland, and provide molecular insight into future
clinical genetic diagnosis for CCS by broadening the genotypic
spectrum of CCS.
Materials and methods
Subject and statement of ethics
The subject was a 20-year-old Chinese male that
presented with a gradual mass of duration 3–4 months located on the
left parotid gland. The mass was diagnosed as clear cell sarcoma
(CCS) of the salivary gland by incisional biopsy.
The experimental methods used in this study were
carried out in accordance with the relevant guidelines and
regulations. This study was approved by the Ethics Committee of the
General Hospital of the Chinese People's Liberation Army. Relevant
informed consent was obtained from all participants, including the
CCS patient and the healthy control subjects.
All sequencing data for the tumor and venous blood
samples have been deposited in the Sequence Read Archive (SRA,
http://www.ncbi.nlm.nih.gov/sra/) with
accession nos. BioSample SAMN03384326 and SAMN03384334,
respectively, in BioProject SRP055838.
Exome sequencing
The tumor and venous blood from the Chinese male CCS
patient was selected for exome sequencing. Exome sequencing was
carried out using an Agilent SureSelect Human All Exon v5.0 (51M)
kit, according to the instructions from Illumina's TruSeq Exome
Enrichment Guide (SureSelectXT Target Enrichment System for
Illumina Paired-End Sequencing Library, Agilent). Genomic DNA
libraries were prepared according to the manufacturer's
instructions (Illumina Inc., USA). Briefly, 3 µg of genomic
DNA was randomly fragmented into pieces of 100–500 bp in size using
a Diagenode Bioruptor® system (Diagenode). DNA fragments
between 150 and 250 bp were recovered by gel extraction. An end
repair and size selection procedure was then performed with T4 DNA
polymerase and Klenow polymerase cleavage 3′. An 'A' base was added
to the 3′ end of the fragments using Klenow 3′ to 5′ exo minus. The
DNA fragments were then ligated to the Illumina multi-PE-adaptor.
The adapter-ligated templates were purified using Agencourt AMPure
SPRI beads and amplified by four-cycle ligation-mediated polymerase
chain reaction (LM-PCR), under the following PCR conditions: 2 min
at 94°C, four cycles of 10 sec at 94°C, 30 sec at 62°C, and 30 sec
at 72°C, and then 5 min at 72°C. The LM-PCR products were
hybridized to the Agilent Oligo pool for 24 h at 65°C for
enrichment. The hybridized fragments were bound to streptavidin
beads and non-hybridized fragments were washed out. Captured LM-PCR
products were amplified by PCR (2 min at 98°C; 10–12 cycles for 10
sec at 98°C, 30 sec at 60°C, 30 sec at 72°C, followed by 5 min at
72°C). The magnitude of enrichment of the samples was estimated
with an Agilent 2100 bioanalyzer. The captured library was then
sequenced on Illumina HiSeq 2000 analyzers with 126 cycles/read, to
generate paired-end reads and 8 bp of index tag (following the
manufacturer's standard sequencing instructions).
Read mapping and variant analysis
Image analysis and base calling were performed with
Illumina Basecaller program (v1.8). Indexed primers were used for
data fidelity surveillance. The sequence reads were aligned to the
human genome reference obtained from the UCSC database (http://genome.ucsc.edu/), version hg19 (GRCh37), using
the SOAP aligner program (v2.21). Single nuclotide polymorphisms
(SNPs) were called using SOAPsnp (v1.03) with the default
parameters, after the duplicated reads (produced mainly in the PCR
step) had been removed using Picard (v1.63) (31). Short insertions or deletions
(InDels) altering coding sequence or splicing sites were identified
by GATK (v1.4-33-g051b450) through realignment analysis of
insertions and deletions, quality recalibration, and InDels calling
(UnifiedGenotyper in GATK). We then filtered candidate SNPs with
the following criteria: SNP quality, ≥20; sequencing depth, ≥10;
the estimated copy number, ≤2; and the distance between two SNPs,
>5. Subsequently, we used VarScan2 v2.3.7 (http://varscan.source-forge.net/) to detect the
somatic mutations in the exome data from tumor-normal pairs. We
applied varElect (http://varelect.genecards.org/) to select mutations
associated with the function of salivary glands. The effect of
candidate mutations to protein features were predicted with the
GERP++ program (May 22, 2011). Potential rejected substitutions
were evaluated by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT
prediction (http://sit.jcvi.org/).
PCR and Sanger sequencing
Following the analysis of the exome sequencing data,
Sanger sequencing was performed to verify the detected genetic
variants. Primers flanking the mutation area of NBN (NM_002485)
were designed based on the reference genomic sequences of the human
genome from NCBI GenBank, and synthesized in Shanghai, China, by
Thermo Scientific. The sequence of the forward primer was
5′-CCCTACCTCATTGGCTTTGTG-3′, and that of the reverse primer was
5′-TATCACGGTCCCTGCTTCC-3′. All PCR amplification was carried out
using an Applied Biosystems Life Technologies (ABI) 9700 thermal
cycler. PCR products were directly sequenced on an ABI PRISM 3730
automated sequencer (Applied Biosystems Life Technologies).
Sequence comparisons and analyses were performed using the Jalview
program (v2.8.2; http://www.jalview.org/).
Results
Clinical phenotype
A 20-year-old male presented with a gradual mass of
duration of 3–4 months on the left parotid gland. He occasionally
suffered from a short period of needle pain. On palpation, a
sub-mucosal mass measuring 3×2×2 cm in size was confirmed. The mass
was non-tender and varied in consistency from soft to firm. On
general examination, the patient appeared apparently healthy with
neither submandibular nor cervical lymph node metastases. The mass
was diagnosed as CCS of the salivary gland by incisional biopsy
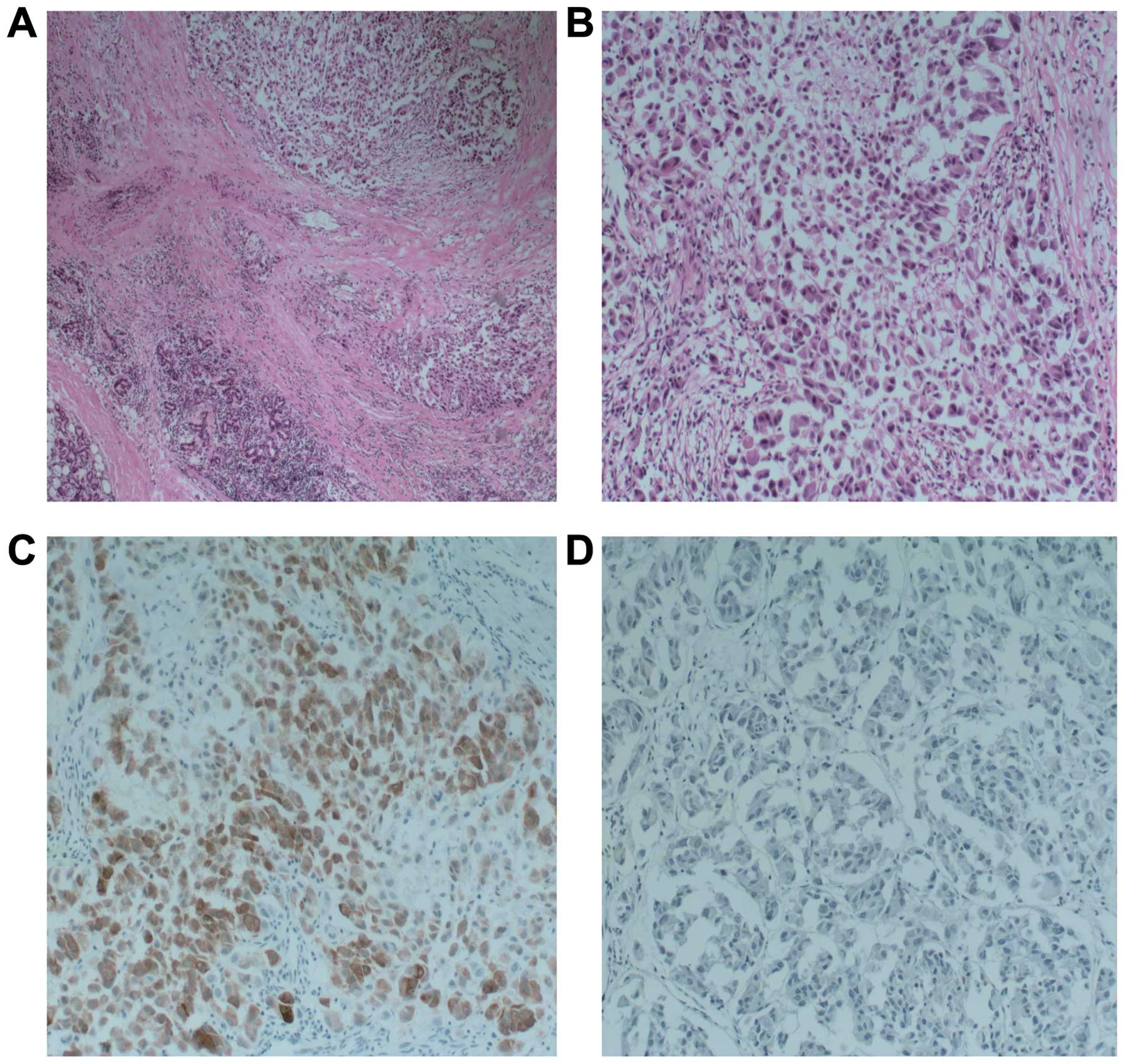
(Fig. 1A–D). Histologically, the
tumor had typical features including a combination of cords and
nests of clear and eosinophilic cells in a hyalinized background
(Fig. 1B). However, there was a
relatively wide range of features in the tumor cells. Most of the
tumor cells actually had a pale eosinophilic cytoplasm rather than
a clear cytoplasm, or they may have had a mixture of both (Fig. 1A). Focal squamous differentiation
was also noted occasionally (Fig.
1C). There was a tendency for the cells in the center of the
mass to be admixed with, or to be surrounded by, a hyalinized
basement membrane-like material (Fig.
1D). Tumor cells at the periphery of the mass had a greater
tendency for nest formation and for wide infiltration without a
desmoplastic response or stromal deposition (Fig. 1A). Moreover, immunohistochemical
staining revealed that the tumors were HMB45-positive (Fig. 1C) and CK-negative (Fig. 1D). Based on these observations and
lines of evidence, we diagnosed this tumor as CCS of the salivary
gland.
Mutation analysis
We performed exome sequencing of the tumor tissue
from the CCS patient. We generated 19.28 and 7.95 billion bases of
125-bp paired-end read sequences from the tumor and from the venous
blood samples, respectively. Billion bases [19.09 (99%) and 7.88
(99%)] passed the quality assessment, and 19.01 (98.61%) and 7.91
(99.46%) billion bases aligned to the human reference for the tumor
and venous blood samples, respectively. For tumor tissue, 11.39
billion bases (59.9%) mapped to the targeted regions with a mean
coverage of 80.47 X 21,077 genetic variants, including 9,295
non-synonymous variants, were identified in either the coding
regions or the splice sites. For venous blood, 4.61 billion bases
(58.3%) mapped to the targeted regions with a mean coverage of
63.42 X; 19,930 genetic variants, including 8,404 non-synonymous
variants, were identified in either the coding regions or the
splice sites. A prioritization scheme was applied to identify the
pathogenic mutation in the tumor tissue, using methods similar to
those reported in recent studies (32,33).
We excluded the known variants that had been identified in the
dbSNP141, 1000 genomes, HapMap, and ESP-6500 datasets. After
filtering the variants displayed in venous blood, we obtained 542
candidate somatic mutations in the tumor tissue.
Subsequently, we used varElect software to select
mutations associated with the function of the salivary gland and
acquired four candidate causal mutations. A somatic mutation,
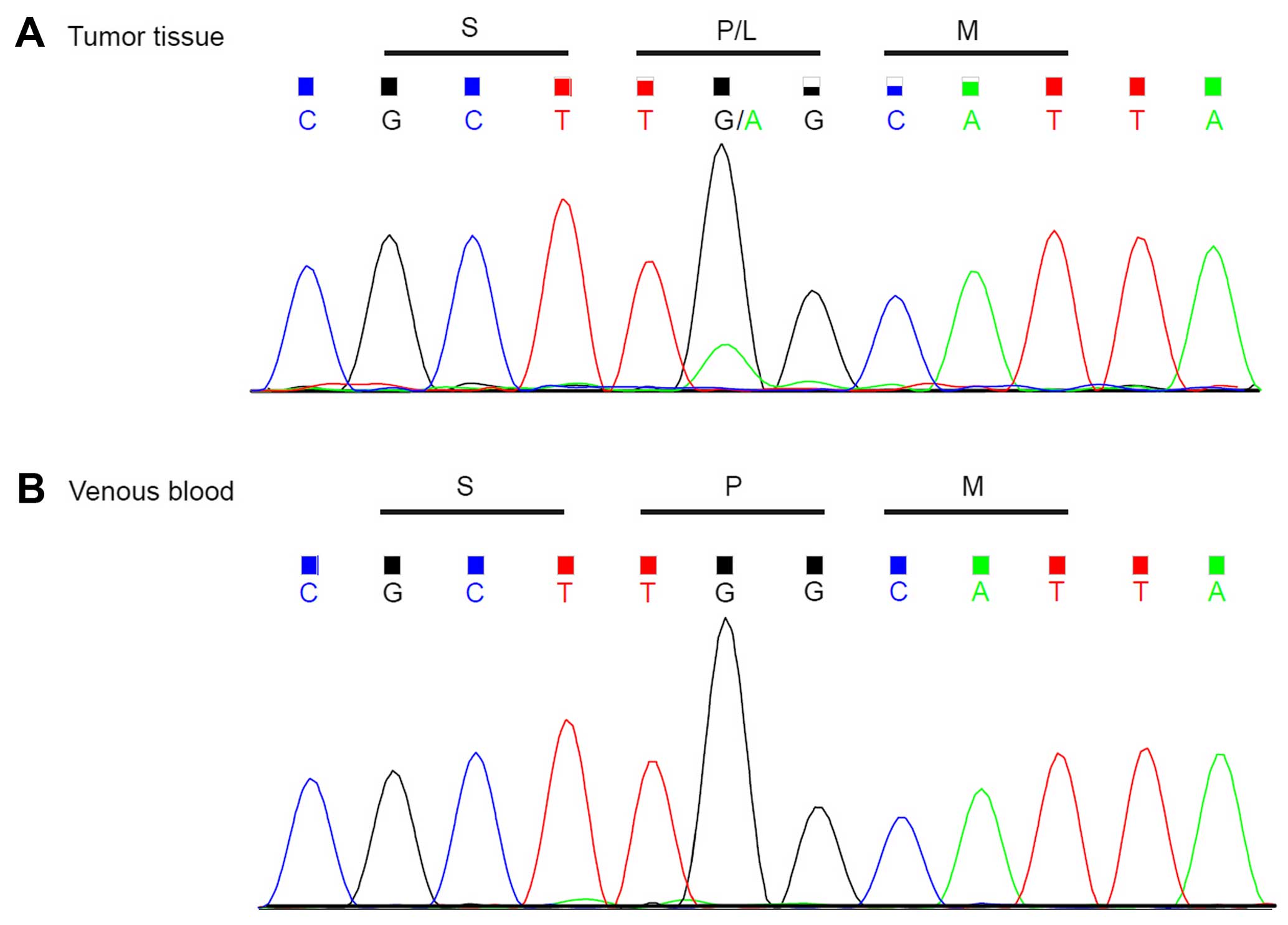
c.1061C>T (p.P354L), was eventually identified in exon 9 of the
NBN gene in the tumor and this was validated using Sanger
sequencing. This mutation results in a missense variant. The same
mutation was absent in peritumoral tissue of the patient (Fig. 2) and venous blood samples from 30
ethnically matched normal control individuals. This mutation was
also absent in the dbSNP141, 1000 genomes, HapMap, and ESP-6500
datasets. To evaluate whether or not there were EWSR1-ATF1 or
EWSR1-CREB1 gene translocations, RT-PCR (reverse transcription
polymerase chain reaction) was performed to validate the fusion
site; the negative result was thus confirmed.
Bioinformatic analysis of NBN mutations
in CCS
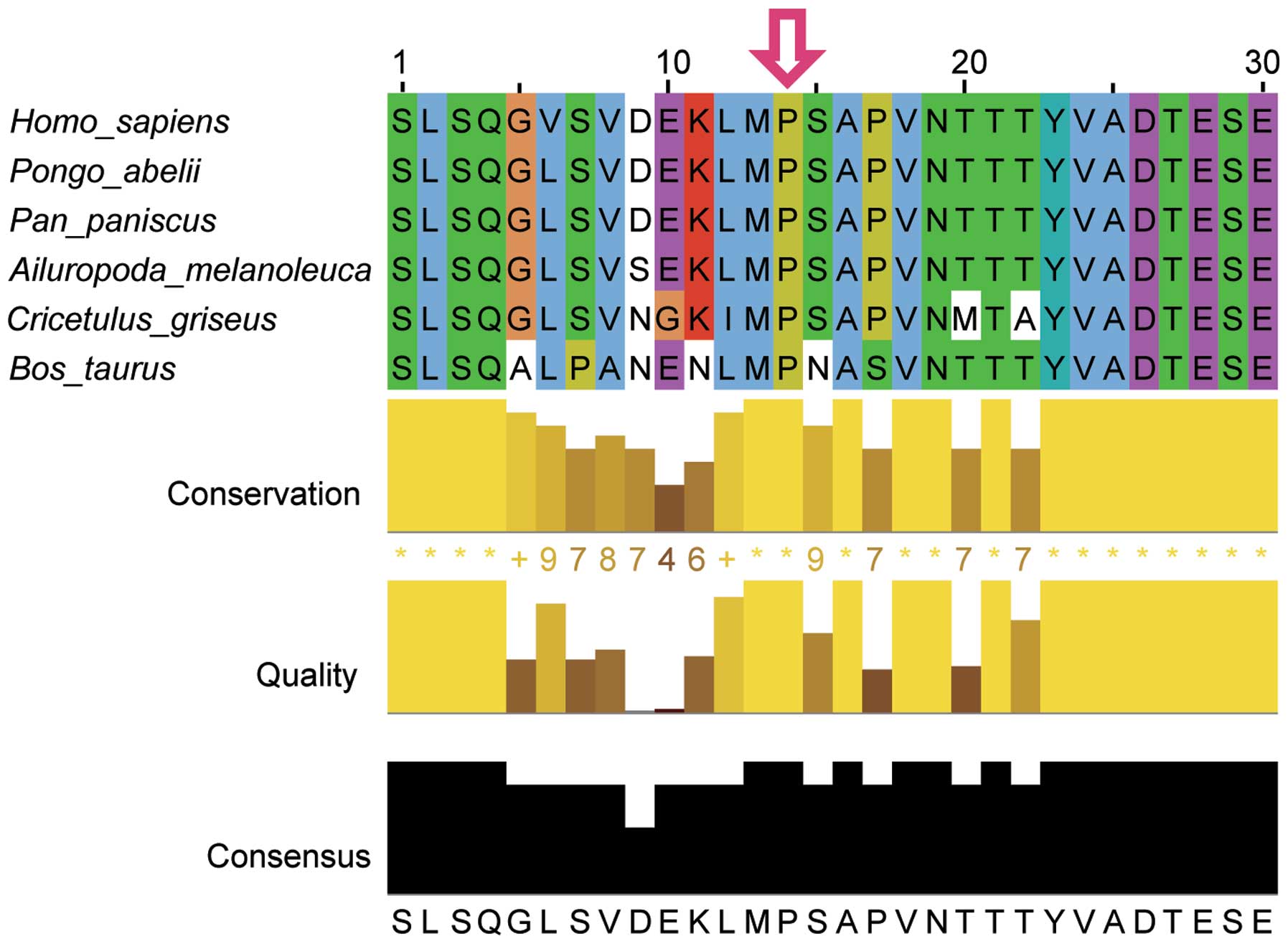
We obtained sequences for NBN family proteins using
the BLAST tools of the NCBI databases and performed
multiple-sequence alignments using Jalview in various animal
species, including Homo sapiens, Pongo abelii, Pan paniscus,
Ailuropoda melanoleuca, Cricetulus griseus and Bos taurus
(Fig. 3). The p.Pro354Leu variant
was found to be located in a highly conserved region of the NBN
protein, suggesting its likely structural and functional
importance. This mutation was predicted to affect the protein
features and be rejected substitutions predicted by GERP++ with a
score 3.07. SIFT prediction indicated a deleterious effect for this
mutation, with a score of 0. In addition, PolyPhen-2 prediction
also suggested that this mutation probably conferred a damaging
effect, with a confident score of 0.998.
GO annotations related to NBN include damaged DNA
binding and transcription factor binding. However, it remains
unclear which histone modifications regulate the expression of NBN
and it is not yet clear which transcriptional factors are
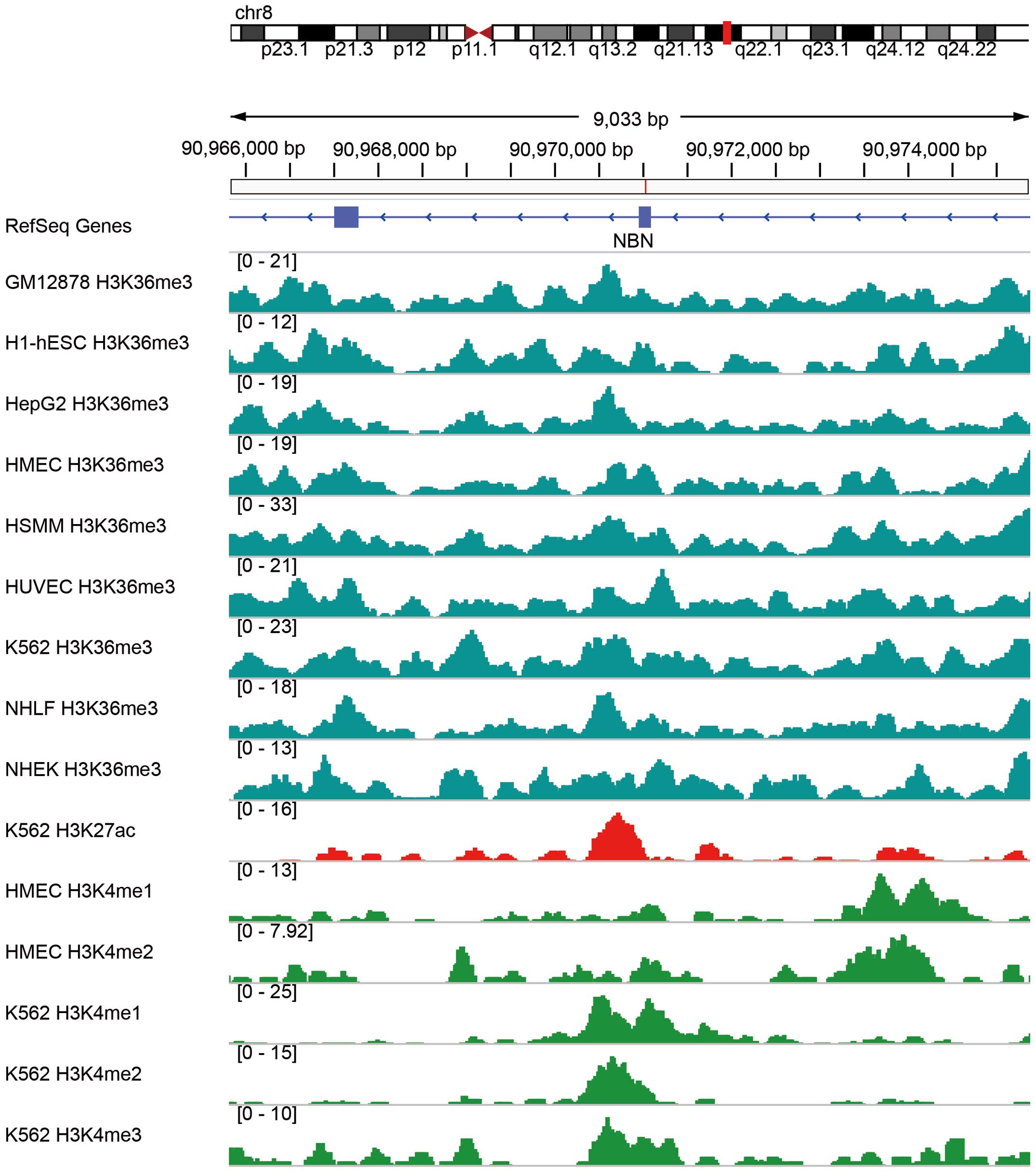
associated with the mutation locus of NBN. Using integrated
analysis of the data for histone modifications deposited in the
Encyclopedia of DNA Elements (ENCODE) project (34), we found that histone modifications
at the NBN locus and the c.1061C>T mutant variant include
H3K36me3, H3K27ac, and H3K4me1/2/3 in multiple cell lines such as
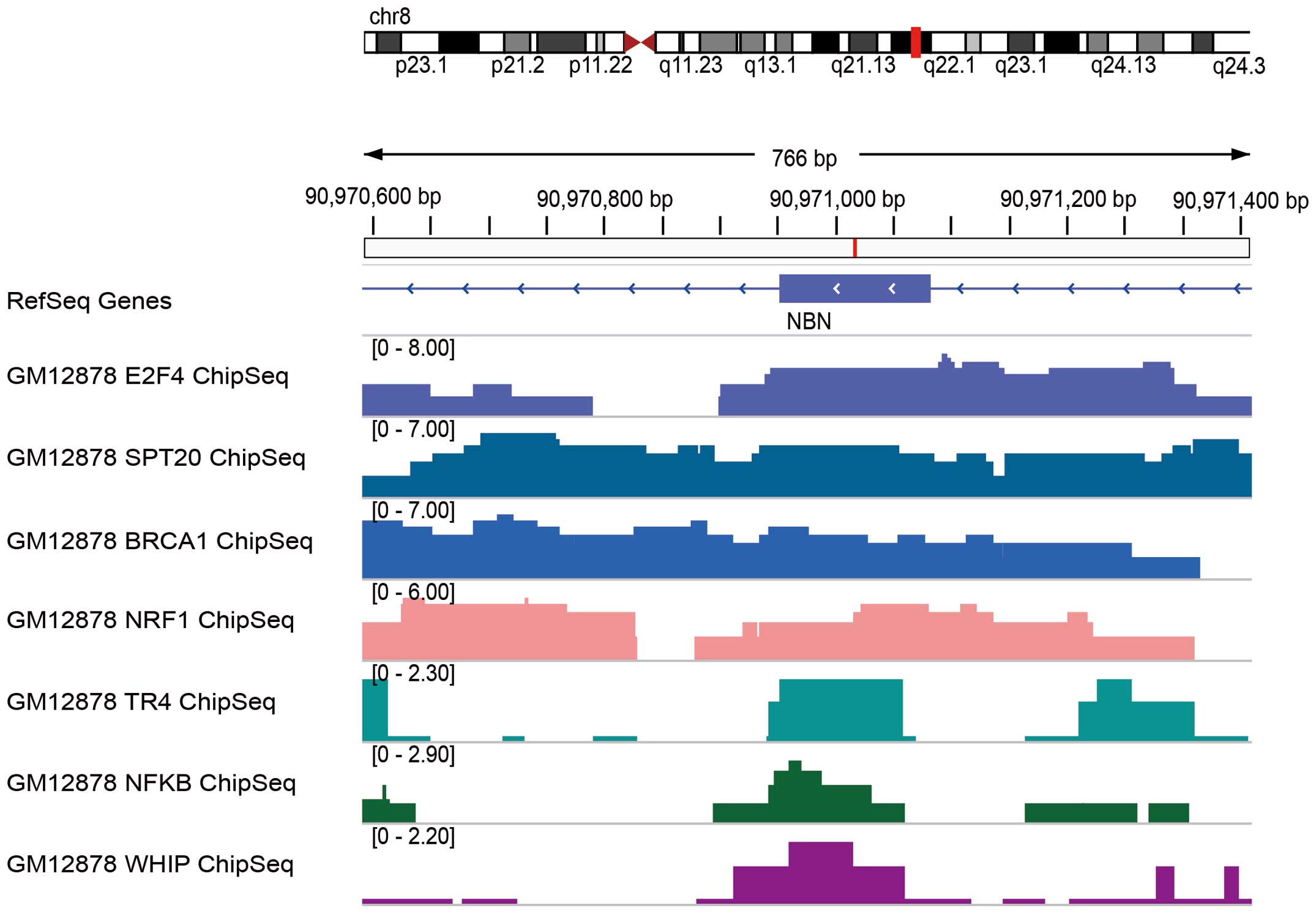
GM12878, H1-hESC and K562 (Fig. 4).
As these histone modifications are canonically active molecule
markers (35), we speculated that
the c.1061C>T variant may influence histone modification and
affect the expression of NBN, although it must be noted that the
histone modification data are from cell lines rather than from CCS
tumors. Indeed, according to our PCR validation of expression
analysis, the expression of NBN was decreased in the CCS tumor
compared with its expression level in peritumoral tissue.
Similarly, several transcriptional factors deposited in the ENCODE
project were found to be enriched by ChIP-seq in the locus where
the c.1061C>T variant occurred in the NBN gene for cell
line GM12878 (Fig. 5). These
transcriptional factors were identified as E2F4, SPT20, BRCA1,
NRF1, TR4, NFKB and WHIP; these may function to regulate the
transcription of the NBN gene.
Discussion
Clear cell sarcoma (CCS) of soft tissue, formerly
referred to as malignant melanoma of soft parts, is a neoplasm with
poor prognosis that primarily affects young adults between the ages
of 20 and 40 years (36). The tumor
has a high propensity for lymph node metastasis and local
recurrence. CCS typically involves tendons and aponeuroses. Primary
CCS of the salivary glands is exceedingly rare. Only a few cases of
primary CCS arising in the ulna, metatarsals, ribs, radius, sacrum,
humerus (37) and jejunum (10) have been reported, and to the best of
our knowledge, our case of CCS arising in the salivary glands is
only the second such study to date.
CCS is a translocation-associated sarcoma. In
chromosomal translocations, the pieces of two chromosomes are
swapped; this can result in an abnormal fusion of genes. Most cases
of CCS harbor a fusion of EWSR1/ATF1 resulting from translocation
(21). The genetic cause for CCS is
thought to be its defining gene translocation. However, 30% of
cases of CCS have no observed EWSR1 translocation (10). A CCS lacking a translocation may
have other, as yet uncharacterized, genetic mutations that can
cause the same pathological effect.
Here, we report a somatic missense mutation
c.1061C>T (p.P354L) in the NBN gene of a Chinese patient
with CCS that did not harbor the typically expected EWSR1-ATF1
fusion. The pathogenesis of the ~30% of CCS cases lacking an EWSR1
rearrangement has always been a mystery (19). The EWSR1-ATF1 fusion is one of the
characteristics used to distinguish CCS from cutaneous melanoma
(4). In contrast to most melanomas,
CCS cells lack BRAF mutations (9)
and show immunohistochemical positive staining for HMB45 and
negative staining for CK (12,15).
In our case study, the immunohistochemical results were consistent
with the diagnostic criteria of CCS. Moreover, we observed a
combination of cords and nests of clear and eosinophilic cells in a
hyalinized background, which is a typical feature of CCS in
histological diagnoses. Therefore, the finding of this novel
NBN mutation provides a novel genotypic feature for the
clinical genetic diagnosis of CCS.
This mutation in NBN was located within a
phylogenetically conserved region, suggesting that it has a role of
structural and/or functional importance. Previous studies have
indicated that mutations in NBN are associated with Nijmegen
breakage syndrome, an autosomal recessive chromosomal instability
syndrome characterized by microcephaly, growth retardation,
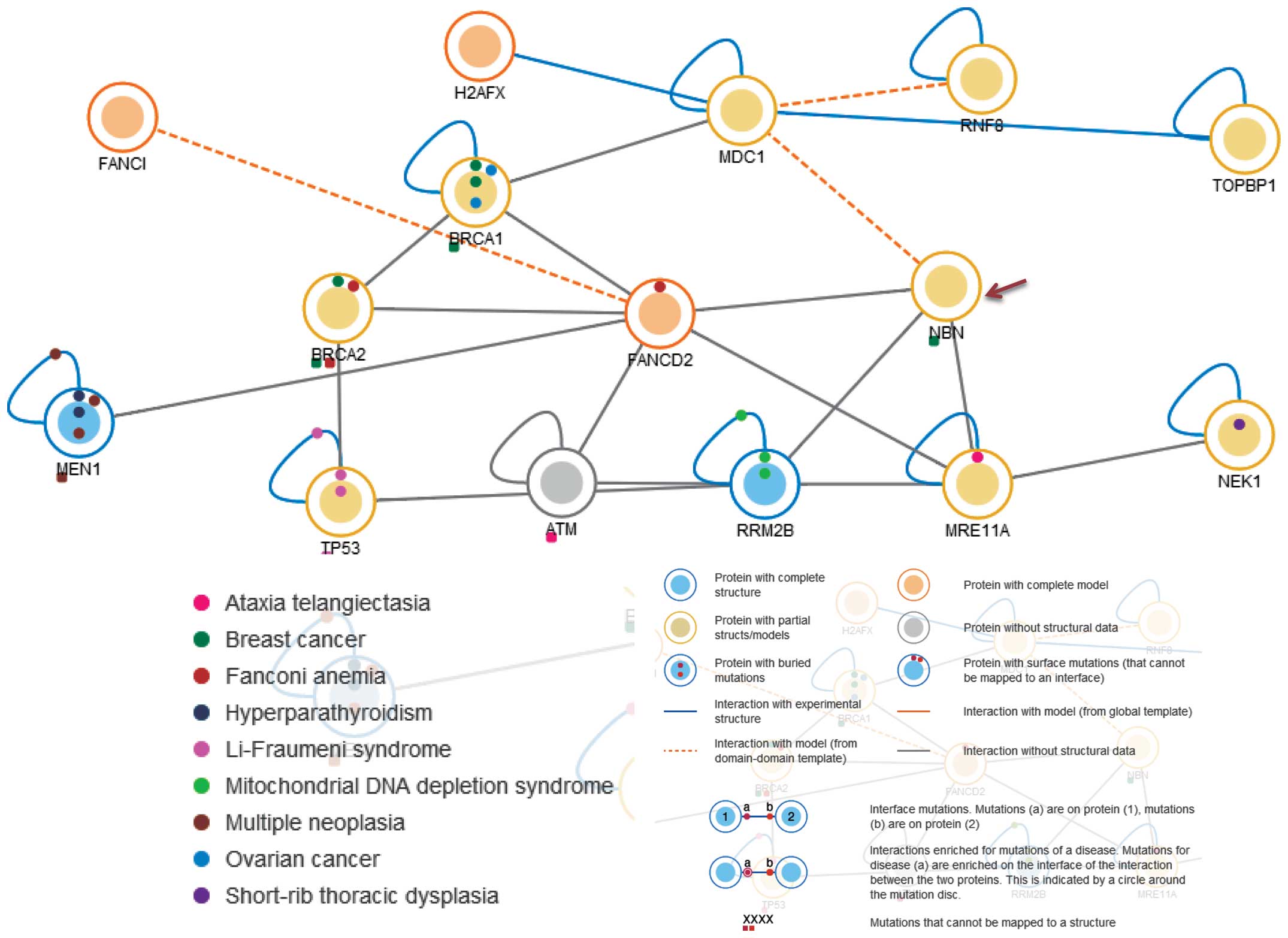
immunodeficiency and cancer predisposition. The NBN protein is a
member of the MRE11/RAD50 double-strand break (DSB) repair complex,
which consists of 5 proteins including NBN, MDC1, FANCD2, RRM2B and
MRE11A (Fig. 6). This NBN protein
is thought to be involved in DNA DSB repair and DNA damage-induced
checkpoint activation (38).
Cancers associated with NBN include gastrointestinal lymphoma
(28), childhood acute leukemia
(29), glioblastomas (30), breast cancer (25), prostate cancer (39) and ovarian cancer (40). Given these known associations, the
mechanism of carcinogenesis for CCS may be related to or share
similar pathway(s) associated with other NBN-related cancers. This
intriguing supposition will require further investigation. In
conclusion, our findings broaden the genotypic spectrum of CCS and
provide new molecular insight that should prove valuable for the
future clinical genetic diagnosis for CCS.
Acknowledgments
We thank all subjects for their participation. We
also thank the scientists responsible for various web sources,
including the 1000 Genomes Project (http://www.1000genomes.org), dbSNP (http://www.ncbi.nlm.nih.gov/SNP), ESP-6500
(http://evs.gs.washington.edu/EVS/),
HapMap (http://hapmap.ncbi.nlm.nih.gov/) and the various DNA
damage prediction tools including SIFT (http://sift.jcvi.org) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2),
among others.
References
|
1
|
Manoel EM, Reiser R, Brodskyn F, Franco M,
Abrahão M and Cervantes O: Clear cell sarcoma of the parotid
region. Rev Braz Otorrinolaringol. 78:1352012. View Article : Google Scholar
|
|
2
|
Ipach I, Mittag F, Kopp HG, Kunze B, Wolf
P and Kluba T: Clear-cell sarcoma of the soft tissue - a rare
diagnosis with a fatal outcome. Eur J Cancer Care (Engl).
21:412–420. 2012. View Article : Google Scholar
|
|
3
|
Nwanyanwu KH, Comer G and Demirci H:
Presumed choroidal metastasis secondary to clear cell sarcoma of
the right knee. Int Ophthalmol. 33:163–165. 2013. View Article : Google Scholar
|
|
4
|
Meis-Kindblom JM: Clear cell sarcoma of
tendons and aponeuroses: a historical perspective and tribute to
the man behind the entity. Adv Anat Pathol. 13:286–292. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang WL, Mayordomo E, Zhang W, Hernandez
VS, Tuvin D, Garcia L, Lev DC, Lazar AJ and López-Terrada D:
Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1
chimeric transcripts in clear cell sarcoma (melanoma of soft
parts). Mod Pathol. 22:1201–1209. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Malchau SS, Hayden J, Hornicek F and
Mankin HJ: Clear cell sarcoma of soft tissues. J Surg Oncol.
95:519–522. 2007. View Article : Google Scholar
|
|
7
|
Mavrogenis A, Bianchi G, Stavropoulos N,
Papagelopoulos P and Ruggieri P: Clinicopathological features,
diagnosis and treatment of clear cell sarcoma/melanoma of soft
parts. Hippokratia. 17:298–302. 2013.
|
|
8
|
Singh M, Ieremia E, Debiec-Rychter M,
Connolly G and Calonje JE: Clear cell sarcoma of the tongue.
Histopathology. 64:750–751. 2014. View Article : Google Scholar
|
|
9
|
Kraft S, Antonescu CR, Rosenberg AE,
Deschler DG and Nielsen GP: Primary clear cell sarcoma of the
tongue. Arch Pathol Lab Med. 137:1680–1683. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lasithiotakis K, Protonotarios A, Lazarou
V, Tzardi M and Chalkiadakis G: Clear cell sarcoma of the jejunum:
a case report. World J Surg Oncol. 11:172013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang W, Shen Y, Wan R and Zhu Y: Primary
clear cell sarcoma of the sacrum: a case report. Skeletal Radiol.
40:633–639. 2011. View Article : Google Scholar
|
|
12
|
Sidiropoulos M, Busam K, Guitart J, Laskin
WB, Wagner AM and Gerami P: Superficial paramucosal clear cell
sarcoma of the soft parts resembling melanoma in a 13-year-old boy.
J Cutan Pathol. 40:265–268. 2013. View Article : Google Scholar
|
|
13
|
Curry CV, Dishop MK, Hicks MJ, Naeem R,
Reed JA and López-Terrada DH: Clear cell sarcoma of soft tissue:
diagnostic utility of fluorescence in situ hybridization and
reverse transcriptase polymerase chain reaction. J Cutan Pathol.
35:411–417. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hisaoka M, Ishida T, Kuo TT, Matsuyama A,
Imamura T, Nishida K, Kuroda H, Inayama Y, Oshiro H, Kobayashi H,
et al: Clear cell sarcoma of soft tissue: a clinicopathologic,
immunohistochemical, and molecular analysis of 33 cases. Am J Surg
Pathol. 32:452–460. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Davis IJ, Kim JJ, Ozsolak F, Widlund HR,
Rozenblatt-Rosen O, Granter SR, Du J, Fletcher JA, Denny CT,
Lessnick SL, et al: Oncogenic MITF dysregulation in clear cell
sarcoma: defining the MiT family of human cancers. Cancer Cell.
9:473–484. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang L, Chen Y, Cui T, Knösel T, Zhang Q,
Geier C, Katenkamp D and Petersen I: Identification of biomarkers
to distinguish clear cell sarcoma from malignant melanoma. Hum
Pathol. 43:1463–1470. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Antonescu CR, Tschernyavsky SJ, Woodruff
JM, Jungbluth AA, Brennan MF and Ladanyi M: Molecular diagnosis of
clear cell sarcoma: detection of EWS-ATF1 and MITF-M transcripts
and histopathological and ultrastructural analysis of 12 cases. J
Mol Diagn. 4:44–52. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Antonescu CR, Dal Cin P, Nafa K, Teot LA,
Surti U, Fletcher CD and Ladanyi M: EWSR1-CREB1 is the predominant
gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes
Cancer. 46:1051–1060. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Patel RM, Downs-Kelly E, Weiss SW, Folpe
AL, Tubbs RR, Tuthill RJ, Goldblum JR and Skacel M: Dual-color,
break-apart fluorescence in situ hybridization for EWS gene
rearrangement distinguishes clear cell sarcoma of soft tissue from
malignant melanoma. Mod Pathol. 18:1585–1590. 2005.PubMed/NCBI
|
|
20
|
Antonescu CR, Katabi N, Zhang L, Sung YS,
Seethala RR, Jordan RC, Perez-Ordoñez B, Have C, Asa SL, Leong IT,
et al: EWSR1-ATF1 fusion is a novel and consistent finding in
hyalinizing clear-cell carcinoma of salivary gland. Genes
Chromosomes Cancer. 50:559–570. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang WL, Mayordomo E, Zhang W, Hernandez
VS, Tuvin D, Garcia L, Lev DC, Lazar AJ and López-Terrada D:
Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1
chimeric transcripts in clear cell sarcoma (melanoma of soft
parts). Mod Pathol. 22:1201–1209. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gerlinger M, Rowan AJ, Horswell S, Larkin
J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A,
Tarpey P, et al: Intratumor heterogeneity and branched evolution
revealed by multiregion sequencing. N Engl J Med. 366:883–892.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Debniak T, Górski B, Cybulski C,
Jakubowska A, Kurzawski G, Lener M, Mierzejewski M, Masojć B,
Medrek K, Kładny J, et al: Germline 657del5 mutation in the NBS1
gene in patients with malignant melanoma of the skin. Melanoma Res.
13:365–370. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meyer P, Stapelmann H, Frank B, Varon R,
Burwinkel B, Schmitt C, Boettger MB, Klaes R, Sperling K, Hemminki
K, et al: Molecular genetic analysis of NBS1 in German melanoma
patients. Melanoma Res. 17:109–116. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Desjardins S, Beauparlant JC, Labrie Y,
Ouellette G and Durocher F; INHERIT BRCAs: Variations in the
NBN/NBS1 gene and the risk of breast cancer in non-BRCA1/2 French
Canadian families with high risk of breast cancer. BMC Cancer.
9:1812009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jazayeri A, Balestrini A, Garner E, Haber
JE and Costanzo V: Mre11-Rad50-Nbs1-dependent processing of DNA
breaks generates oligonucleotides that stimulate ATM activity. EMBO
J. 27:1953–1962. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Varon R, Vissinga C, Platzer M,
Cerosaletti KM, Chrzanowska KH, Saar K, Beckmann G, Seemanová E,
Cooper PR, Nowak NJ, et al: Nibrin, a novel DNA double-strand break
repair protein, is mutated in Nijmegen breakage syndrome. Cell.
93:467–476. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Steffen J, Maneva G, Popławska L, Varon R,
Mioduszewska O and Sperling K: Increased risk of gastrointestinal
lymphoma in carriers of the 657del5 NBS1 gene mutation. Int J
Cancer. 119:2970–2973. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mosor M, Ziółkowska I,
Januszkiewicz-Lewandowska D and Nowak J: Polymorphisms and
haplotypes of the NBS1 gene in childhood acute leukaemia. Eur J
Cancer. 44:2226–2232. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Watanabe T, Nobusawa S, Lu S, Huang J,
Mittelbronn M and Ohgaki H: Mutational inactivation of the nijmegen
breakage syndrome gene (NBS1) in glioblastomas is associated with
multiple TP53 mutations. J Neuropathol Exp Neurol. 68:210–215.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xiu X, Yuan J, Deng X, Xiao J, Xu H, Zeng
Z, Guan L, Xu F and Deng S: A novel COL4A5 mutation identified in a
Chinese Han family using exome sequencing. BioMed Res Int.
2014:1860482014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gilissen C, Arts HH, Hoischen A, Spruijt
L, Mans DA, Arts P, van Lier B, Steehouwer M, van Reeuwijk J, Kant
SG, et al: Exome sequencing identifies WDR35 variants involved in
Sensenbrenner syndrome. Am J Hum Genet. 87:418–423. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo Y, Yuan J, Liang H, Xiao J, Xu H, Yuan
L, Gao K, Wu B, Tang Y, Li X, et al: Identification of a novel
COL4A5 mutation in a Chinese family with X-linked Alport syndrome
using exome sequencing. Mol Biol Rep. 41:3631–3635. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
ENCODE Project Consortium: The ENCODE
(ENCyclopedia Of DNA Elements) Project. Science. 306:636–640. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou VW, Goren A and Bernstein BE:
Charting histone modifications and the functional organization of
mammalian genomes. Nat Rev Genet. 12:7–18. 2011. View Article : Google Scholar
|
|
36
|
Bianchi G, Charoenlap C, Cocchi S, Rani N,
Campagnoni S, Righi A, Frisoni T and Donati DM: Clear cell sarcoma
of soft tissue: a retrospective review and analysis of 31 cases
treated at Istituto Ortopedico Rizzoli. Eur J Surg Oncol.
40:505–510. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nakayama S, Yokote T, Iwaki K, Akioka T,
Miyoshi T, Hirata Y, Takayama A, Nishiwaki U, Masuda Y, Tsuji M, et
al: A rare case of primary clear cell sarcoma of the pubic bone
resembling small round cell tumor: an unusual morphological
variant. BMC Cancer. 12:5382012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang J, Grotzer MA, Watanabe T, Hewer E,
Pietsch T, Rutkowski S and Ohgaki H: Mutations in the Nijmegen
breakage syndrome gene in medulloblastomas. Clin Cancer Res.
14:4053–4058. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zuhlke KA, Johnson AM, Okoth LA, Stoffel
EM, Robbins CM, Tembe WA, Salinas CA, Zheng SL, Xu J, Carpten JD,
et al: Identification of a novel NBN truncating mutation in a
family with hereditary prostate cancer. Fam Cancer. 11:595–600.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Porhanova NV, Sokolenko AP, Sherina NY,
Ponomariova DN, Tkachenko NN, Matsko DE and Imyanitov EN: Ovarian
cancer patient with germline mutations in both BRCA1 and NBN genes.
Cancer Genet Cytogenet. 186:122–124. 2008. View Article : Google Scholar : PubMed/NCBI
|