Introduction
Lung cancer is the most common malignant tumor in
the world and seriously affects human health due to the poor
prognosis of these patients (1).
Non-small cell lung cancer (NSCLC) is the main pathological type of
lung cancer. Approximately 10–15% of NSCLC patients harbor
epithelial growth factor receptor (EGFR) mutations while, it is as
high as 30–40% in East Asian patients (2–4). The
most common EGFR mutations are exon 19 deletions (19 del) and exon
21 L858R point mutations (L858R), together representing 90% of all
mutations (2,4). EGFR tyrosine kinase inhibitors
(EGFR-TKIs) have been approved for NSCLC patients harboring EGFR
activating mutations as first-line therapy, and the response rate
is as high as ~75% (3,5). Yet, almost all of the patients present
with TKI resistance. Drug resistance mechanisms include exon 20
point mutation (T790M), c-met amplification, PI3K persistent
activation and hepatocyte growth factor (HGF) overexpression
(6). T790M mutations and c-met
amplifications account for approximately 75% of all TKI resistant
cases (6–8). To date, there are a lack of treatment
methods to overcome TKI resistance in the clinic.
Zoledronic acid (ZA) is a third-generation nitrogen-
containing bisphosphonate and is widely used in the treatment of
malignant tumors with bone metastases. The mechanism involves
inhibition of farnesyl pyrophosphate (FPP) synthase function in the
mevalonate pathway, and it inhibits the activity of osteoclasts and
induces osteoclast apoptosis (9,10). ZA
has shown antitumor effects in many tumor cells, including NSCLC
(11,12). Meanwhile, it can increase the
antitumor effects in combination with chemotherapy and radiotherapy
(13,14). The antitumor mechanisms include
inhibition of the mevalonate pathway, affecting tumor signaling
pathways, regulation of immune response and anti-angiogenesis
(12,13).
NSCLC patients with EGFR mutations and bone
metastases are often concurrently administered ZA and EGFR-TKI. Our
retrospective clinical study found that bisphosphonates (BPs) can
improve the progression-free survival and overall survival of
advanced NSCLC patients with EGFR mutations and bone metastases
undergoing EGFR-TKI treatment (15). A preclinical study also found that
ZA can increase the antitumor effect of gefitinib treatment for
NSCLC with EGFR activating mutations (16). Yet the mechanism needs further
research. In addition, the effects and mechanisms of ZA and
EGFR-TKI treatment in TKI-resistant NSCLC are unclear. Therefore,
we explored whether ZA could increase the antitumor effects of
gefitinib treatment for NSCLC with EGFR activating mutations and
TKI resistance and the related mechanisms.
Materials and methods
Reagents
Gefitinib was purchased from Biochempartner
(Shanghai, China) and ZA was kindly provided by Novartis Pharma
Stein AG (Basel, Switzerland). The Annexin V-FITC Apoptosis
Detection kit was purchased from BD Pharmingen (San Diego, CA,
USA). Primary antibodies, EGFR, p-EGFR (Tyr1173), p44/42 MAPK
(ERK1/2), p-p44/42 MAPK (Thr202/Tyr204), Akt, p-Akt (Ser473),
STAT3, p-STAT3 (Tyr705), p-STAT3 (Ser727), caspase-3, β-actin and
β-tubulin were purchased from Cell Signaling Technology, Inc.
(Beverly, MA, USA). The horseradish peroxidase (HRP)-conjugated
secondary antibody and immunohistochemical detection kit were
purchased from ZSGB-BIO (Beijing, China).
Cell lines
The human lung adenocarcinoma cell lines HCC827,
HCC827 GR and H1975 were used in the present study. HCC827 harbors
an EGFR exon 19 in-frame deletion (E746-A750). HCC827 GR is a
gefitinib-acquired resistant cell line that was established by
chronic exposure of HCC827 cells to medium with increasing
concentrations of gefitinib. H1975 harbors an EGFR exon 21 point
mutation of L858R and an EGFR exon 20 mutation of T790M.
Cell cytotoxicity assay
The effects of gefitinib and ZA on the proliferation
of the NSCLC cells were detected by MTT assay. Briefly,
5×103 cells per well were seeded in 96-well plates and
treated with gefitinib and/or ZA. After 48 h, 20 µl of MTT
reagent (5 mg/ml) was added to each well and incubated at 37°C for
3 h. The medium was removed and 150 µl DMSO was added to
each well. Cell proliferation was evaluated by a microplate reader
at a wavelength of 570 and 630 nm. Six replicates were prepared for
each sample, and experiments were conducted in triplicate.
Detection of cell apoptosis
Cells were seeded at 2×105 per well in
6-well plates and treated with gefitinib and/or ZA. After 48 h, the
cells were harvested. The method of staining was in accordance with
the manufacturer's instructions. Cell apoptosis was analyzed by a
Becton Dickinson FACSCalibur flow cytometer, and data were analyzed
by FlowJO software. All of the experiments were repeated thrice
independently.
Analysis of cell cycle distribution
Cells were seeded at 2×105 per well in
6-well plates and treated with gefitinib and/or ZA. After 24 or 48
h, the cells were collected and washed with ice-cold
phosphate-buffered saline (PBS), fixed in 75% ethanol at 4°C for 12
h and then washed twice with PBS. Cells were stained at 4°C for 30
min with PI (50 µg/ml in PBS), and 2.0×104 cells
were analyzed using a FACSCalibur flow cytometer. DNA histograms
were analyzed using ModFit LT software.
Western blot assay
Cells were pretreated with gefitinib and/or ZA for 4
or 24 h, and protein expression was analyzed by western blot
analysis as previously described (17).
In vivo study
Six- to eight-week-old female athymic (BALB/c,
nu/nu) mice were used in this experiment. The animal experiment was
carried out according to protocols approved by Sichuan University's
Institutional Animal Care and Use Committee. HCC827 GR and H1975
xenografts were established in athymic mice by subcutaneous
injection of 7×106 cancer cells into the right flank of
the nude mice. When the volume of the tumor reached ~50
mm3, the mice were divided into control, gefitinib, ZA
and gefitinib combined with ZA groups (9–10 mice per group) and
treatment continued for 6 or 7 weeks. Tumor weight, tumor size and
body weight were measured every 3 days during the treatment period.
Tumor volume was calculated by the formula: V = 0.52 × (length ×
width2). Gefitinib suspended in 50% polyethylene glycol
(PEG) 400 in water was administered daily at 50 mg/kg/day by oral
gavage and ZA stock solution diluted in PBS was administered twice
per week at 80 µg/kg by i.p. injection. Mice in the combined
treatment group received gefitinib and ZA treatment at the same
time. Mice in the control group were given the same volumes of 50%
PEG 400 by oral gavage. At the end of the experiment, all mice were
sacrificed.
Immunohistochemistry
The protein expression of tumor tissues was
evaluated by immunohistochemistry (IHC) as previously described
(18). As negative control, PBS was
used to replace the primary antibody. The protein expression in the
cytoplasm was analyzed by Image Pro Plus 6 software and was
expressed as mean intensity optical density (IOD). The protein
expression in nuclei was analyzed by counting the proportion of
positively stained nuclei.
Statistical analysis
The results of continuous data were expressed as the
mean ± standard deviation (SD). Differences between multiple groups
were detected through an one-way ANOVA test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Zoledronic acid and gefitinib inhibit the
proliferation of NSCLC cell lines with EGFR mutations
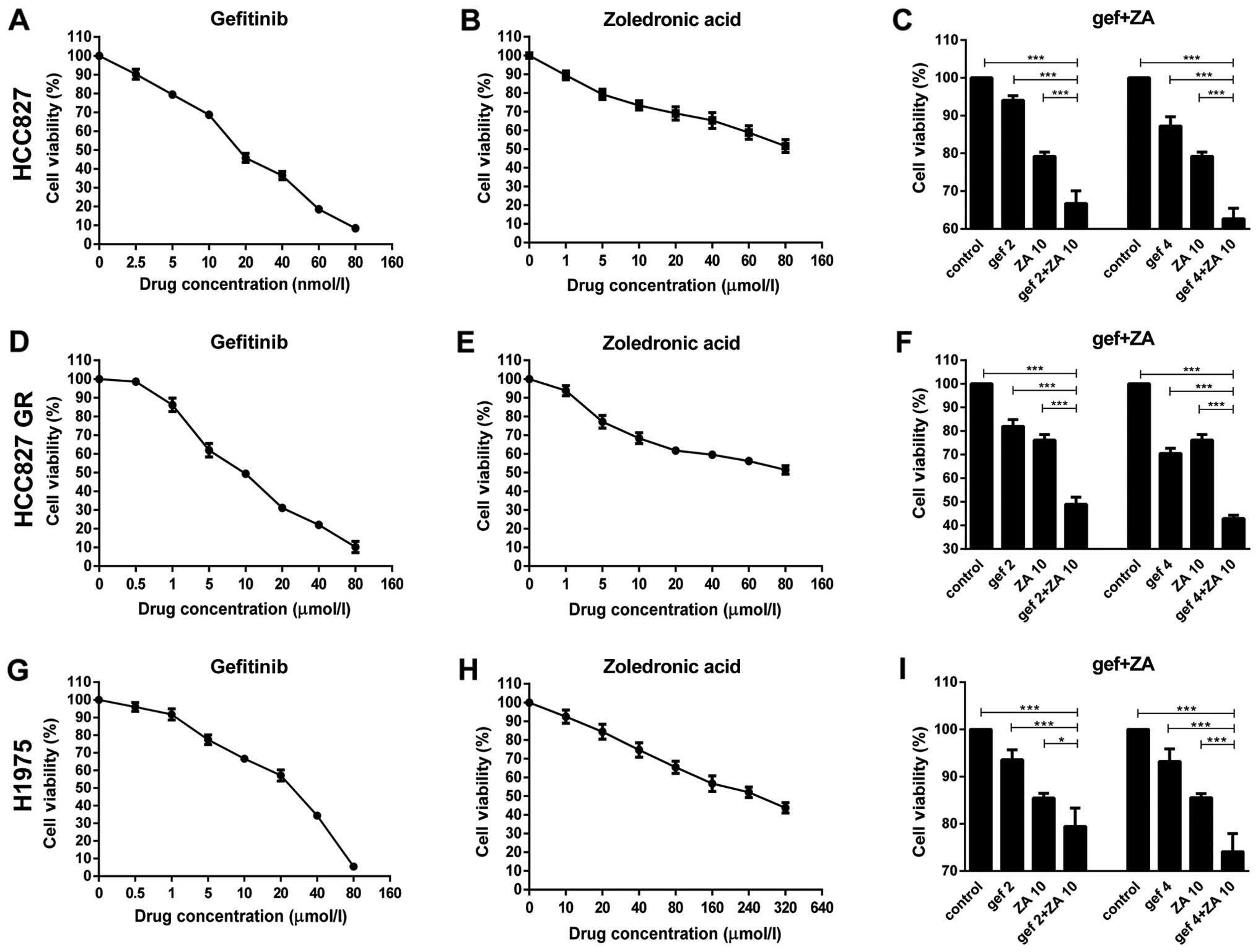
First, the antitumor effects of gefitinib and ZA on
NSCLC with EGFR mutations were detected by MTT assay. The
IC50 values of gefitinib treatment in the HCC827, HCC827
GR and H1975 cells at 48 h were 18.22±0.45 nM, 8.40±1.22 µM
and 17.94±1.77 µM, respectively (Fig. 1A, D and G). The IC50
value for ZA treatment in the H1975 cells at 48 h was 286.89±74.91
µM. When the concentrations of ZA reached 80 nM and 80
µM, it failed to reach the IC50 in HCC827 and
HCC827 GR cells, respectively (Fig. 1B,
E and H). Therefore, gefitinib or ZA alone inhibited the
proliferation of the HCC827, HCC827 GR and H1975 cells, and the
effects were more obvious with increasing drug concentrations.
Then, we detected whether ZA could increase the
antitumor effect of gefitinib in these cells. The results showed
that the cytotoxic effect of gefitinib in all three cell lines was
enhanced by co-incubation with a low concentration of ZA
(P<0.05). Higher cytotoxic effect on the tumor cells was noted
with increasing drug concentrations (Fig. 1C, F and I).
ZA increases the apoptosis induced by
gefitinib treatment on NSCLC cells with EGFR mutations
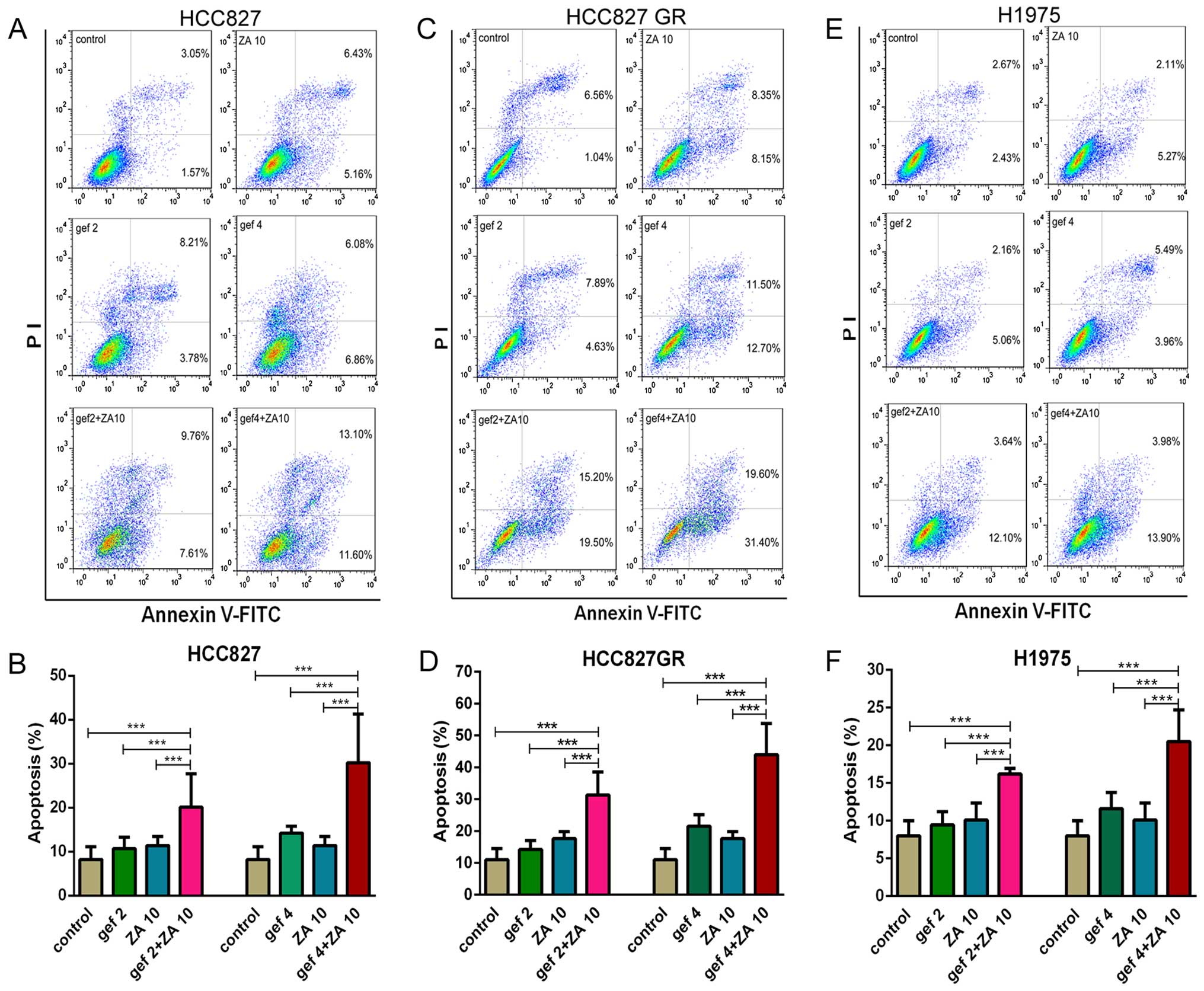
First, the effects of gefitinib or ZA on the
induction of apoptosis in NSCLC cell lines with EGFR mutations were
detected by flow cytometry. Fig. 2
shows that both gefitinib and ZA slightly induced apoptosis in the
HCC827, HCC827 GR and H1975 cells. The percentage of apoptotic
cells was significantly increased following combined treatment with
gefitinib and ZA compared with gefitinib or ZA monotherapy
(P<0.01). Thus ZA increased the apoptosis induced by gefitinib
treatment in NSCLC cell lines with EGFR activating mutations and
TKI resistance.
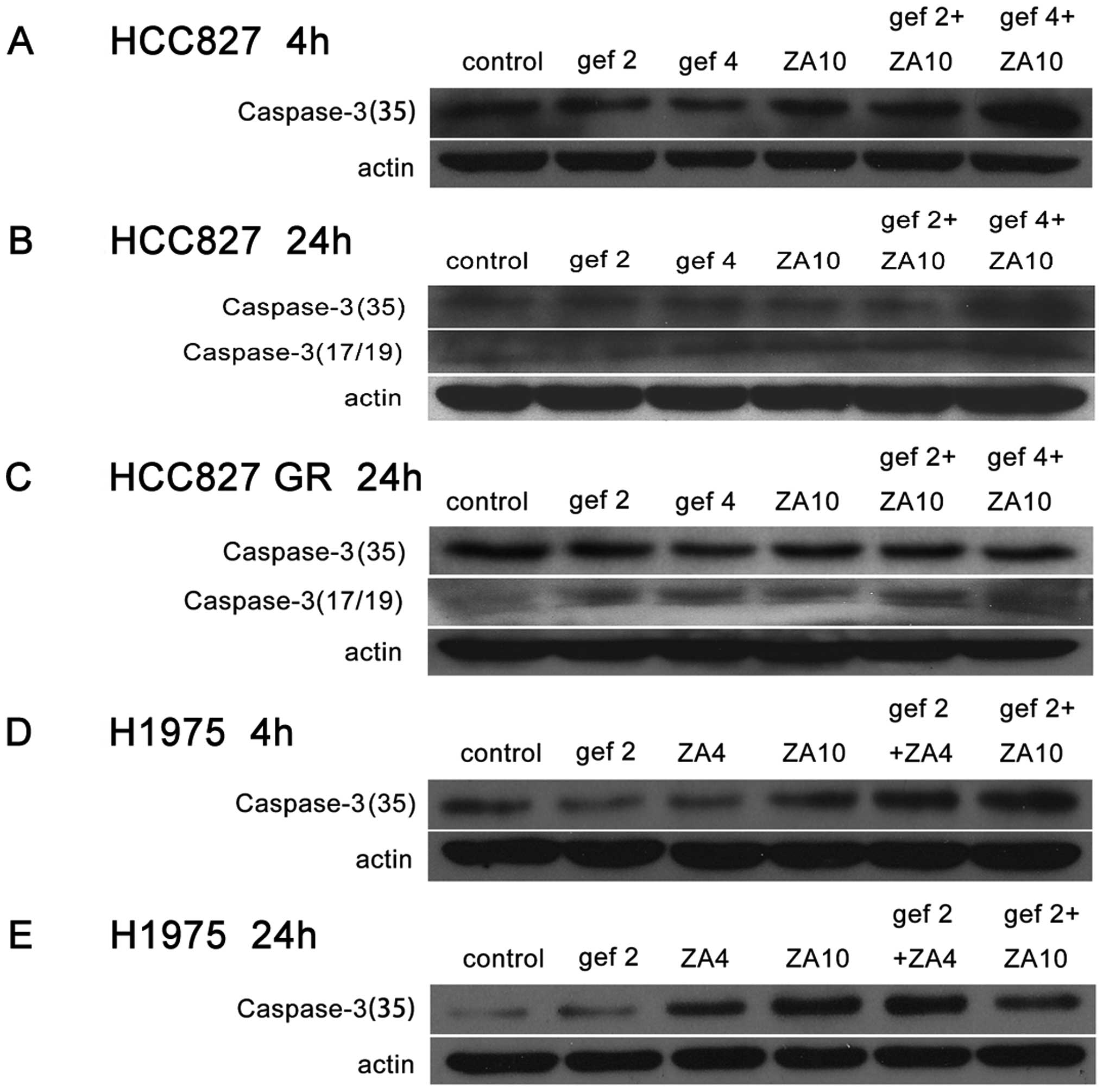
Then, we detected the effects of gefitinib and ZA on
caspase-3 protein expression on NSCLC cell lines with EGFR
mutations by western blot analysis. The results showed that the
combined treatment with gefitinib and ZA could obviously increase
caspase-3 protein expression in the HCC827, HCC827 GR and H1975
cells compared with gefitinib or ZA monotherapy (Fig. 3).
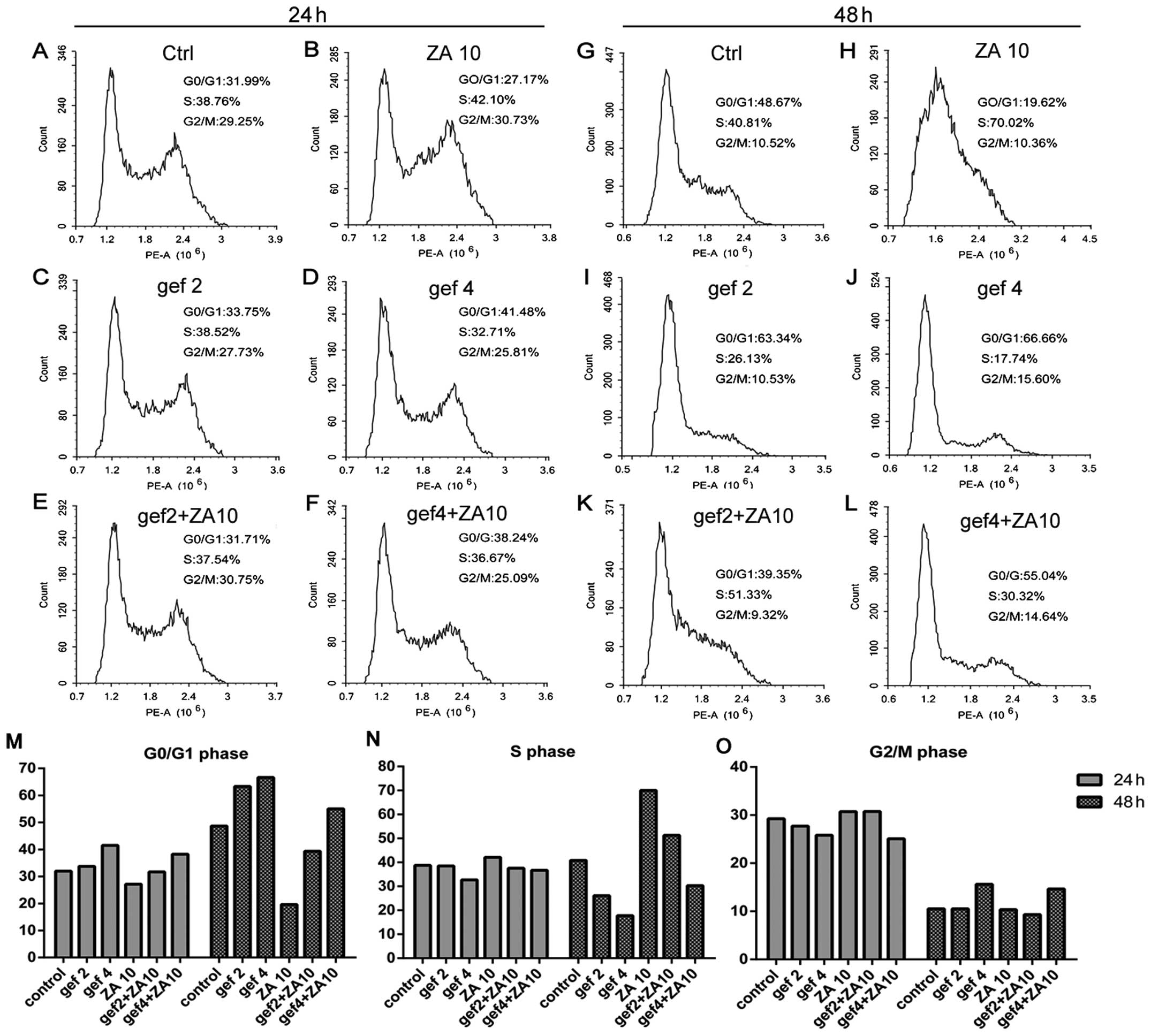
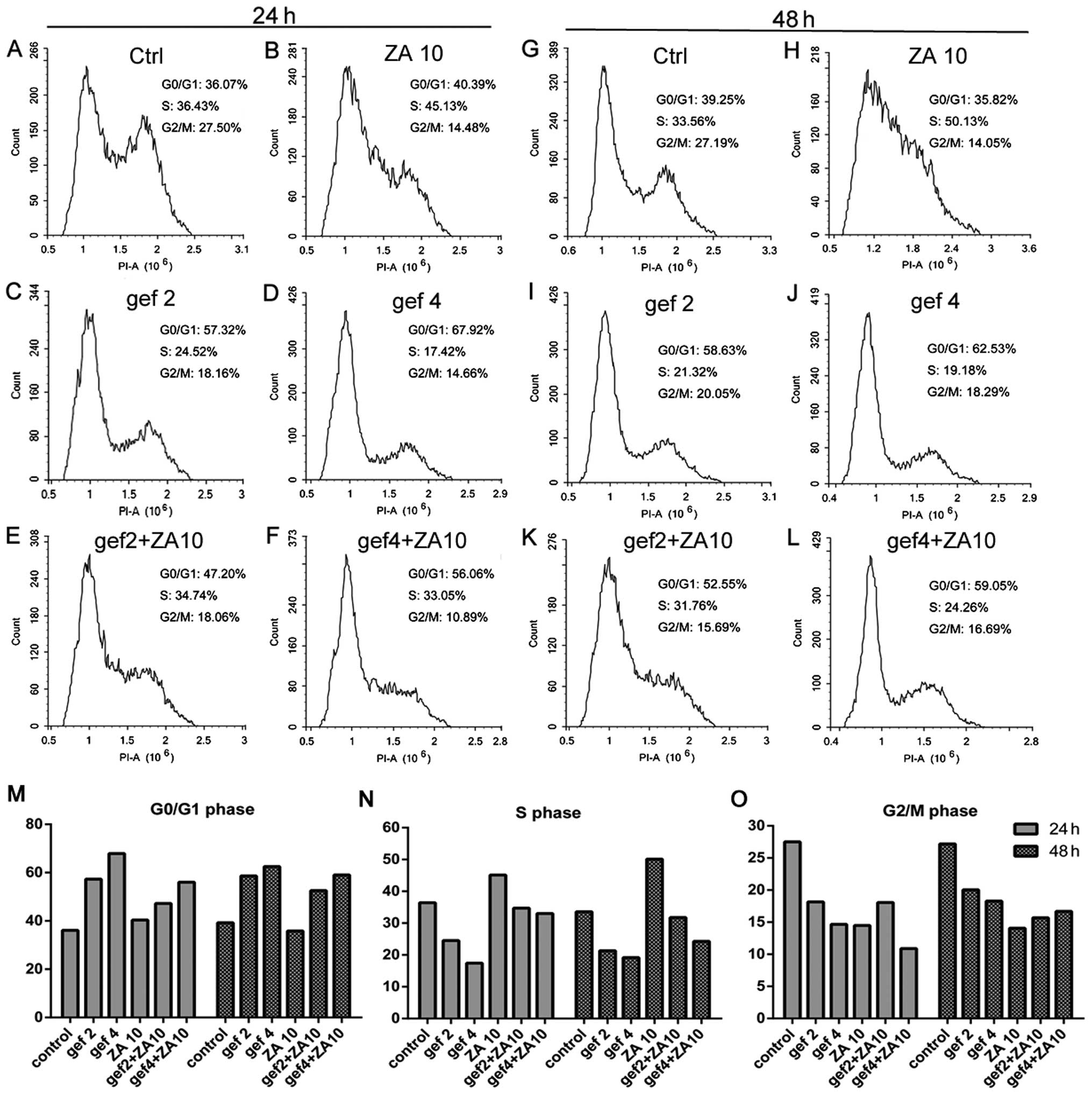
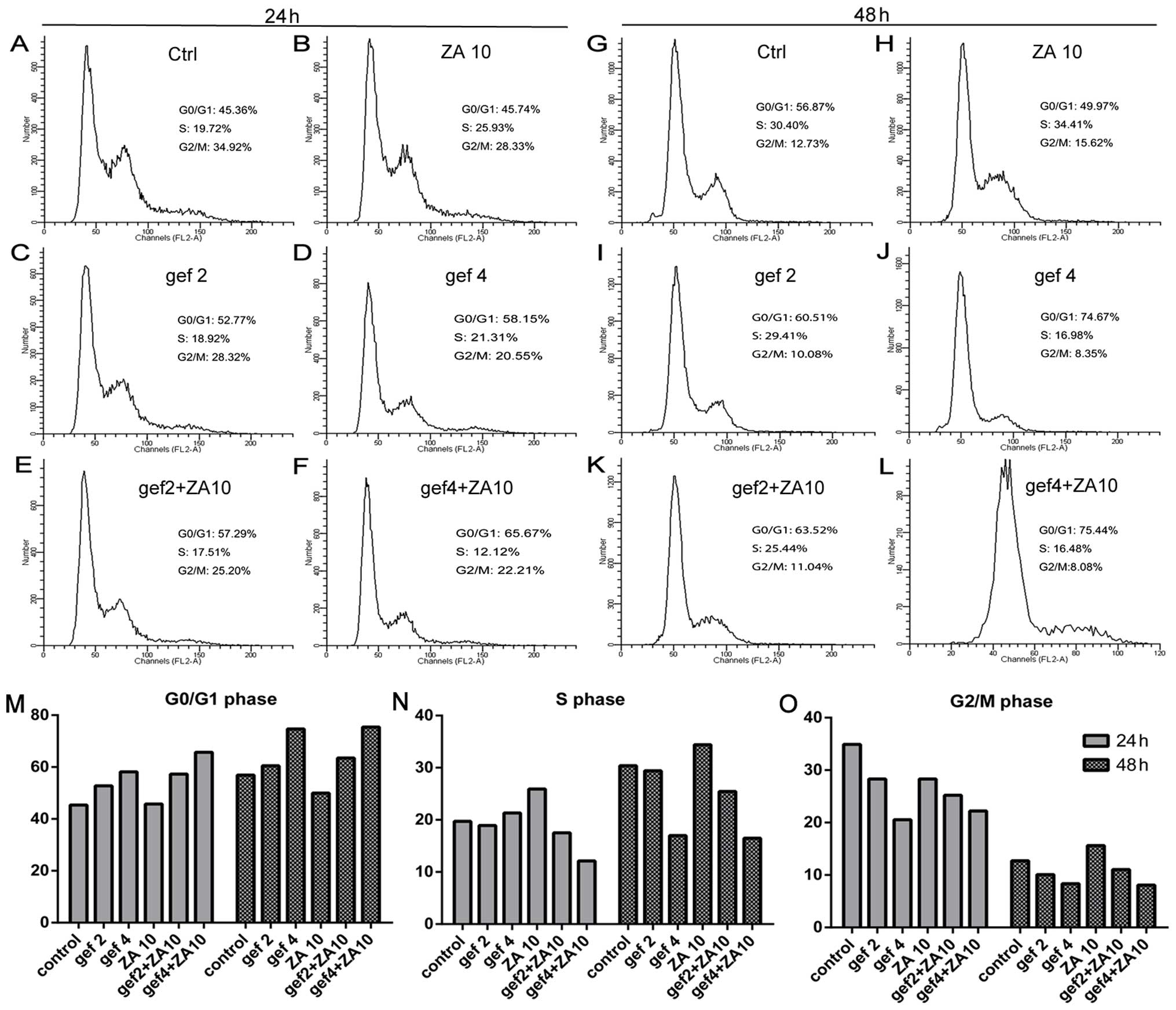
Effect of ZA and gefitinib on cell cycle
progression in the NSCLC cells with EGFR mutations
We confirmed that ZA increased the antitumor effect
of gefitinib treatment in NSCLC with EGFR mutations in
vitro. We next detected the effects of ZA and/or gefitinib on
the cell cycle. In the HCC827 GR cells (Fig. 4), gefitinib treatment for 24 and 48
h increased the percentage of cells in the G0/G1 phase and
decreased the percentage of cells in the S phase in a dose- and
time-dependent manner. Gefitinib had no effect on the G2/M phase.
ZA treatment at 24 and 48 h obviously induced S phase accumulation
and decreased G0/G1 phase accumulation of tumor cells, with
increasing treatment time. More tumor cells were blocked in the S
phase, while G2/M phase had no change. The combined treatment
increased the percentage of cells in the G0/G1 phase and decreased
the percentage of cells in the S phase compared with ZA in a dose-
and time-dependent manner. The results for gefitinib and ZA on the
cell cycle distribution of HCC827 (Fig.
5) and H1975 (Fig. 6) cells
were similar to the results found for the HCC827 GR cells.
Therefore, gefitinib caused HCC827, HCC827 GR and
H1975 cell cycle arrest in the G0/G1 phase. ZA arrested the cells
in the S phase and the effects of the combined therapy on the cell
cycle were neutralization.
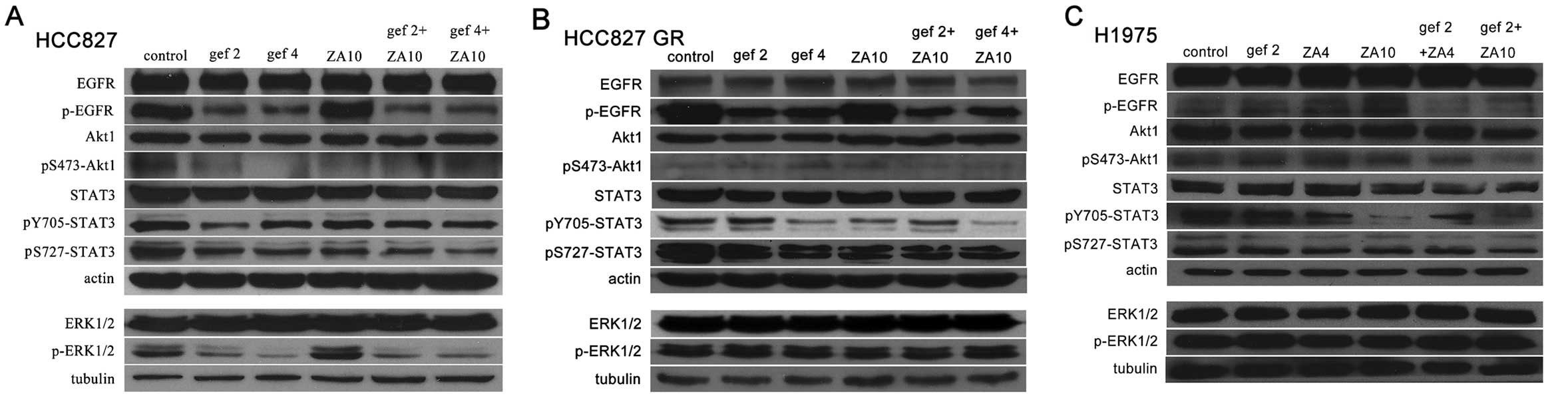
Effects of gefitinib and/or ZA on EGFR
and downstream signaling molecules in the NSCLC tumor cells
To further reveal the mechanisms of action, we
detected protein expression of EGFR and downstream signaling
molecules in the NSCLC cells with EGFR mutations following
treatment with gefitinib and/or ZA by western blot analysis. In the
HCC827 cells (Fig. 7A), gefitinib
obviously inhibited EGFR and downstream molecules Akt, STAT3 and
ERK protein phosphorylation. ZA inhibited STAT3 protein expression
and phosporylated Akt, STAT3 protein. The combined treatment also
inhibited EGFR and downstream molecules Akt, STAT3 and ERK protein
phosphorylation and slightly inhibited STAT3 protein expression.
Expression of pS727-STAT3 following the combined treatment was
lower than the level in the other treatment groups. In the HCC827
GR cells (Fig. 7B), ZA inhibited
STAT3 protein phosphorylation. The combined treatment inhibited
EGFR and downstream molecules Akt and STAT3 protein
phosphorylation; among them, pS473-Akt1 and pY705-STAT3 levels were
lower in the combined treatment group that levels noted in the
other treatment groups. In the H1975 cells (Fig. 7C), ZA inhibited STAT3 protein
expression and phosphorylation. The combined treatment inhibited
EGFR and downstream molecules Akt and STAT3 protein phosphorylation
and STAT3 protein expression. The expression levels of these
proteins were lower following combined treatment when compared with
gefitinib or ZA monotherapy. Therefore, ZA enhanced the inhibitory
effects of gefitinib on STAT3 and/or p-STAT3 protein expression in
the HCC827, HCC827 GR and H1975 cells.
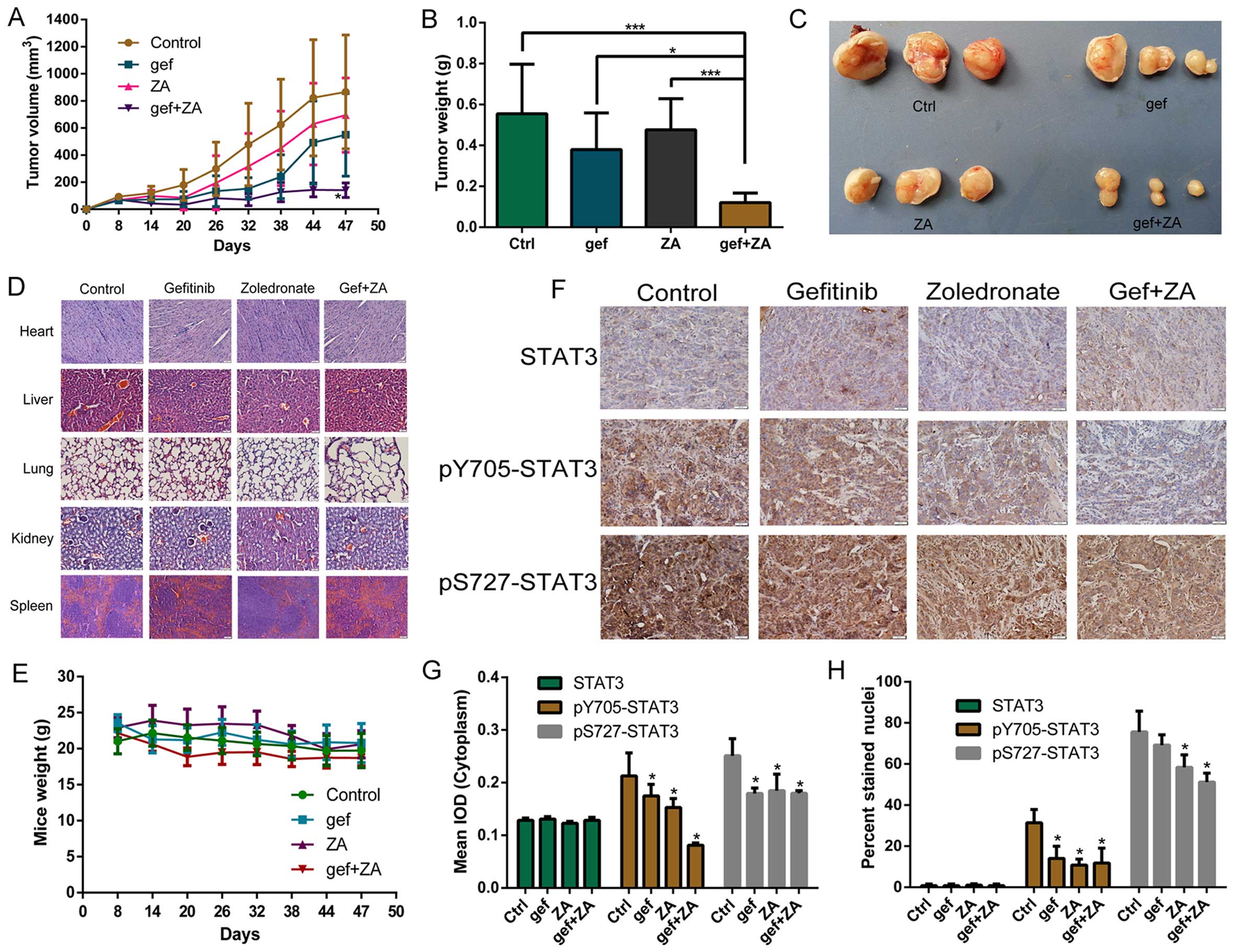
ZA enhances the antitumor effect of
gefitinib treatment in NSCLC cells with TKI resistance in vivo
We confirmed that ZA increased the antitumor effects
of gefitinib treatment in NSCLC with EGFR mutations in vitro
and revealed the mechanisms. We further aimed to ascertain the
effects of ZA and gefitinib in vivo.
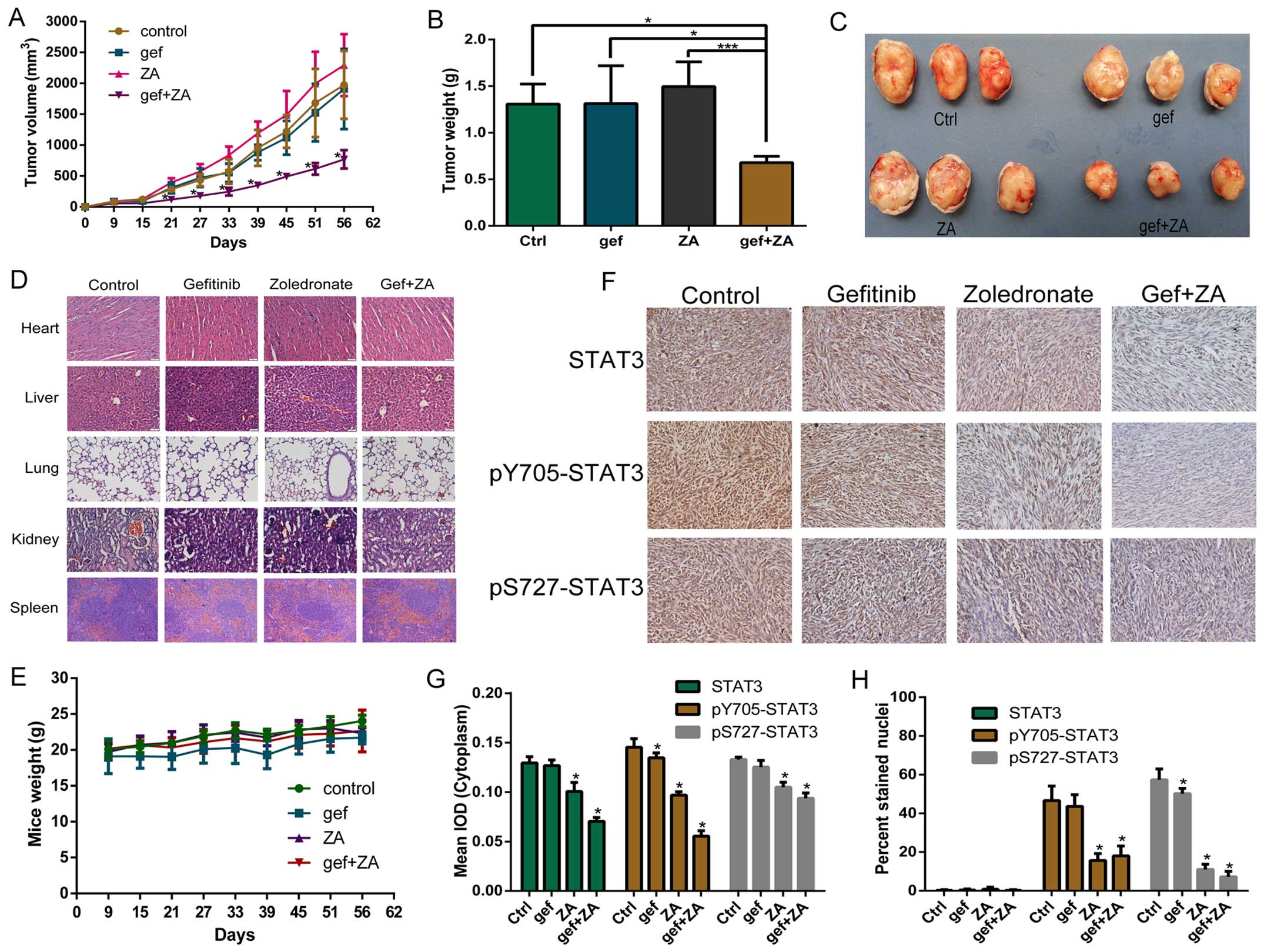
Fig. 8 shows the
effects of ZA and/or gefitinib on HCC827 GR cell-derived tumors in
nude mice. We found that ZA or gefitinib slightly suppressed the
growth of tumors, but they had no statistical difference compared
with the control group (P>0.05). The combined treatment
significantly reduced tumor volume compared with the other tumor
groups (P<0.05) (Fig. 8A). The
tumor weight of the combined group was also lower than that in the
other groups (P<0.05) (Fig. 8B).
Fig. 8C directly shows that the
tumor size of the combined group was smaller than that of the other
groups. The drug had no adverse effect on mouse organs and body
weight (Fig. 8D and E). The result
of IHC showed that ZA and the combined treatment inhibited STAT3
protein phosphorylation in the cytoplasm and nuclei of the tumor
cells, and the effect was more obvious in the combined treatment
group (Fig. 8F–H).
The results of gefitinib and ZA on H1975
cell-derived tumors were similar to those derived from the HCC827
GR cells (Fig. 9). One difference
was that ZA and the combined treatment inhibited STAT3 protein
expression in the cytoplasm and inhibited STAT3 protein
phosphorylation in the cytoplasm and nuclei of the tumor cells.
Discussion
Our study investigated whether ZA could enhance the
antitumor effects of gefitinib treatment in NSCLC cells with EGFR
mutations and the mechanisms of action. We demonstrated that ZA
enhanced the antitumor effects of gefitinib treatment in NSCLC
cells with EGFR activating mutation and TKI resistance by
regulating the cell cycle, inducing caspase-3 protein expression
and inhibiting STAT3 protein expression.
Previous studies found that ZA has antitumor effects
on many tumors and the effects are in a dose- and time-dependent
manner (12,19). Meanwhile, ZA was also found to have
antitumor effects on lung cancer cells (11,20).
In contrast, Li et al found that ZA could not induce tumor
cell apoptosis in line-1 lung cancer cells even though it could
inhibit tumor cell proliferation (21). We considered that the reason was
associated with the different lung cancer cell lines used in the
different studies. In our study, we found that even a low dose of
ZA inhibited tumor cell proliferation and induced apoptosis in
NSCLC cells with EGFR activating mutation and TKI resistance and
the effects were in a dose-dependent manner.
Studies also suggest that ZA can increase the
antitumor effects of targeted therapy. Zhang et al found
that ZA combined with sorafenib treatment of primary liver cancer
inhibited the growth of tumors and lung metastasis compared with
sorafenib (22). Tivantinib (a
c-met tyrosine kinase inhibitor) combined with ZA prolonged the
time of breast cancer metastasis to bone and improved the survival
of breast cancer patients with bone metastasis in vivo
(23). Chang et al found
that ZA enhanced the antitumor effect of gefitinib treatment in
HCC827 cells with EGFR mutations in vitro and in vivo
(16). Our study also found that ZA
increased the antitumor effect of gefitinib treatment in HCC827
cells with EGFR activating mutations in vitro, and we
further confirmed that ZA increased the antitumor effect of
gefitinib treatment in TKI-resistant NSCLC cells (HCC827 GR and
H1975) in vitro and in vivo.
Clinical studies also found that BPs can enhance the
antitumor effects combined with targeted therapy. One retrospective
clinical study conducted by us assessed the efficacy of BPs
combined with EGFR-TKI first-line treatment of advanced NSCLC
harboring EGFR mutations. Among 114 patients, 62 with bone
metastases (19 patients treated with TKI alone and 43 patients
treated with TKI plus BPs) were studied. The combined group had
significantly improved mPFS and mOS compared with the TKI group
(mPFS: 15 vs. 7 months, P=0.001; mOS: 25 vs. 10 months, P=0.001).
In addition, in 52 patients without bone metastases treated with a
TKI alone, and 43 patients with bone metastases treated with TKI +
BPs, the mPFS was 15 vs. 12 months (P=0.02) and the mOS was 25 vs.
21 months (P=0.119) in the combined group and TKI group,
respectively (15). Another two
retrospective clinical studies also found that BPs combined with
sorafenib/sunitinib treatment in patients with bone metastases from
renal cell carcinoma improved the clinical response rate and
significantly prolong PFS and OS (P<0.05) (24,25).
Our findings found that ZA combined with gefitinib
induced caspase-3 protein expression and induced apoptosis of NSCLC
with EGFR mutations. A previous study demonstrated that erlotinib
induced the apoptosis of H3255 cells (L858R mutation) by the
mitochondrial-mediated apoptosis pathway, by activating Bax and Bak
which are dependent on mitochondrial oxidative phosphorylation
(26). In addition, EGFR-TKIs also
inhibited PI3K/AKT/survivin and MEK/ERK, and eventually induced BIM
expression (27,28). ZA also induced tumor cell apoptosis
by inhibiting Bcl-2, inducing BAX and activating caspase-3,
caspase-9, and PARP protein expression (29,30).
Therefore, gefitinib and ZA may act through different mechanisms of
apoptosis, resulting in activated caspase-3, the critical protein
of the apoptosis signaling pathway thus inducing tumor cell
apoptosis.
EGFR-TKI and ZA affect the tumor cell cycle.
EGFR-TKI arrests TKI-sensitive and -resistance lung cancer cells in
the G0/G1 phase (31–33). ZA can induce NSCLC cancer cell S
phase accumulation (16,34). We also found that gefitinib arrested
the HCC827, HCC827 GR and H1975 cells in the G0/G1 phase and ZA
blocked the three tumor cell lines in the S phase, while the effect
of the combined therapy on the cell cycle in the three cell lines
was neutralization. Therefore, gefitinib and ZA had different
effects on the cell cycle of the three NSCLC cell lines resulting
in inhibition of tumor cell proliferation.
EGFR is overexpressed in 60% of NSCLC tumor tissues.
Activation of EGFR causes activation of downstream signaling of
PI3K/AKT, MAPK/ERK and JAK/STAT3, promoting tumor proliferation,
survival and inhibition of apoptosis (35,36).
STAT3 protein is activated at 705 location of tyrosine
phosphorylation and induces dimerization, translocation to the cell
nucleus and binding of specific DNA elements. The 727 location of
serine phosphorylation activation is associated with
transcriptional activity (37).
EGFR-TKIs inhibit phosphorylation of EGFR tyrosine kinase region,
thus inhibiting downstream signaling pathways. Studies have found
that the antitumor effect of ZA is associated with inhibition of
the activation of Akt and STAT3 (38–40).
Our study found that ZA can inhibit Akt and STAT3 protein
phosphorylation and STAT3 protein expression in HCC827 cells, and
the combined treatment obviously inhibited EGFR and downstream
signaling of Akt, ERK1/2 and STAT3 protein phosphorylation and
slightly inhibited STAT3 protein expression.
Although NSCLC with EGFR mutations is sensitive to
EGFR-TKIs, the majority of patients develop drug resistance. The
most common mechanism is the T790M mutation, which accounts for
~50% of acquired resistance (7).
The T790M mutation blocks the combining of TKI to the EGFR tyrosine
kinase region and continuously activates downstream signaling
pathways (41). We found that ZA
inhibits STAT3 protein expression and phosphorylation in H1975
cells, and the combined treatment inhibited EGFR and downstream
molecules Akt and STAT3 protein phosphorylation and STAT3 protein
expression in vitro. We also confirmed that the combined
treatment inhibited STAT3 protein expression and phosphorylation
in vivo. Therefore, ZA enhanced the antitumor effect of
gefitinib treatment in NSCLC cells with T790M mutations by
inhibiting STAT3 and p-STAT3 protein expression.
Another important mechanism of TKI resistance in
NSCLC is c-met amplification, accounting for ~22% of the acquired
resistance (8). The combining of
c-met and the ligand of HGF can activate downstream signaling
pathways PI3K/Akt, MAPK/ERK and STAT3 (42). The HCC827 GR cell line is a
gefitinib-acquired resistant cell line with c-met amplification
(8). Our study found that ZA
inhibited STAT3 protein phosphorylation in the HCC827 GR cells, and
the combined treatment inhibited EGFR and downstream molecules Akt
and STAT3 protein phosphorylation in vitro. We also
confirmed that combined treatment inhibited STAT3 protein
phosphorylation in vivo. Therefore, ZA enhanced the
antitumor effects of gefitinib treatment in NSCLC cells with c-met
amplification by inhibiting STAT3 protein phosphorylation.
As known, ZA induces osteoclast apoptosis by
inhibiting the mevalonate pathway. The mevalonate pathway plays an
important role in the activation of EGFR and downstream signaling
molecules. Drug targeting of the mevalonate pathway can increase
the antitumor effect of EGFR-TKI treatment in NSCLC with EGFR
mutations (43,44). Therefore, ZA enhanced the antitumor
effect of gefitinib treatment in NSCLC with EGFR mutations through
inhibition of the mevalonate pathway.
Our study has some limitations. i) We detected the
effects of ZA and gefitinib on the protein expression of EGFR and
downstream signaling molecules (ERK, STAT3 and Akt), but we did not
detect the expression of other upstream and downstream proteins.
Thus, we did not confirm whether the drug could affect expression
of other proteins. ii) A previous study confirmed that ZA can
enhance the antitumor effect of gefitinib treatment in HCC827 cells
in vivo (16). Therefore,
our study did not detect the antitumor effect of both drugs in
HCC827 nude mice and iii) we detected only the expression of STAT3
protein in tumor tissues, therefore expression of other proteins in
the tumor tissues were not determined.
In summary, we demonstrated that ZA enhanced the
antitumor effects of gefitinib in NSCLC with EGFR activating
mutation and TKI resistance by regulation of the tumor cell cycle,
induction of caspase-3 protein expression and inhibition of STAT3
protein expression. Our study provides a new treatment strategy for
NSCLC patients with EGFR mutations, particularly for TKI-resistant
NSCLC.
Acknowledgments
We thank Novartis Pharma Stein AG who kindly
provided the zoledronic acid for this research. The present study
was supported by the National Natural Science Foundation of China
(no. 81472197).
Abbreviations:
|
EGFR
|
epithelial growth factor receptor
|
|
TKI
|
tyrosine kinase inhibitor
|
|
NSCLC
|
non-small cell lung cancer
|
|
ZA
|
zoledronic acid
|
|
BPs
|
bisphosphonates
|
|
FPP
|
farnesyl pyrophosphate
|
|
PFS
|
progression-free survival
|
|
OS
|
overall survival
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
ERK
|
extracellular regulated protein
kinase
|
|
Akt
|
protein kinase B
|
|
MTT
|
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium
bromide
|
|
DMSO
|
dimethyl sulfoxide
|
|
IHC
|
immunohistochemistry
|
|
IOD
|
intensity optical density
|
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosell R, Moran T, Queralt C, Porta R,
Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M,
et al Spanish Lung Cancer Group: Screening for epidermal growth
factor receptor mutations in lung cancer. N Engl J Med.
361:958–967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu YL, Zhong WZ, Li LY, Zhang XT, Zhang L,
Zhou CC, Liu W, Jiang B, Mu XL, Lin JY, et al: Epidermal growth
factor receptor mutations and their correlation with gefitinib
therapy in patients with non-small cell lung cancer: A
meta-analysis based on updated individual patient data from six
medical centers in mainland China. J Thorac Oncol. 2:430–439. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gazdar AF: Activating and resistance
mutations of EGFR in non-small cell lung cancer: Role in clinical
response to EGFR tyrosine kinase inhibitors. Oncogene. 28(Suppl 1):
S24–S31. 2009. View Article : Google Scholar
|
|
5
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al North-East Japan Study Group: Gefitinib or chemotherapy for
non-small cell lung cancer with mutated EGFR. N Engl J Med.
362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin Y, Wang X and Jin H: EGFR-TKI
resistance in NSCLC patients: Mechanisms and strategies. Am J
Cancer Res. 4:411–435. 2014.PubMed/NCBI
|
|
7
|
Kosaka T, Yatabe Y, Endoh H, Yoshida K,
Hida T, Tsuboi M, Tada H, Kuwano H and Mitsudomi T: Analysis of
epidermal growth factor receptor gene mutation in patients with
non-small cell lung cancer and acquired resistance to gefitinib.
Clin Cancer Res. 12:5764–5769. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luckman SP, Coxon FP, Ebetino FH, Russell
RGG and Rogers MJ: Heterocycle-containing bisphosphonates cause
apoptosis and inhibit bone resorption by preventing protein
prenylation: Evidence from structure-activity relationships in J774
macrophages. J Bone Miner Res. 13:1668–1678. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dunford JE, Thompson K, Coxon FP, Luckman
SP, Hahn FM, Poulter CD, Ebetino FH and Rogers MJ:
Structure-activity relationships for inhibition of farnesyl
diphosphate synthase in vitro and inhibition of bone resorption in
vivo by nitrogen-containing bisphosphonates. J Pharmacol Exp Ther.
296:235–242. 2001.PubMed/NCBI
|
|
11
|
Di Salvatore M, Orlandi A, Bagalà C,
Quirino M, Cassano A, Astone A and Barone C: Anti-tumour and
anti-angiogenetic effects of zoledronic acid on human non-small
cell lung cancer cell line. Cell Prolif. 44:139–146. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Morgan G and Lipton A: Antitumor effects
and anticancer applications of bisphosphonates. Semin Oncol.
37(Suppl 2): S30–S40. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kijima T, Koga F, Fujii Y, Yoshida S,
Tatokoro M and Kihara K: Zoledronic acid sensitizes renal cell
carcinoma cells to radiation by downregulating STAT1. PLoS One.
8:e646152013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fukai J, Koizumi F and Nakao N: Enhanced
anti-tumor effect of zoledronic acid combined with temozolomide
against human malignant glioma cell expressing
O6-methylguanine DNA methyltransferase. PLoS One.
9:e1045382014. View Article : Google Scholar
|
|
15
|
Huang CY, Wang L, Feng CJ, Yu P and Wang
YS: Cooperation of bisphosphonates with tyrosine kinase inhibitors
in advanced non-small cell lung cancer with EGFR activation
mutation: A retrospective study. J Clin Oncol. 32:e190732014.
|
|
16
|
Chang JW, Hsieh JJ, Shen YC, Yeh KY, Wang
CH, Li YY and Hsu T: Bisphosphonate zoledronic acid enhances the
inhibitory effects of gefitinib on EGFR-mutated non-small cell lung
carcinoma cells. Cancer Lett. 278:17–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen W, Shen X, Xia X, Xu G, Ma T, Bai X
and Liang T: NSC 74859-mediated inhibition of STAT3 enhances the
anti-proliferative activity of cetuximab in hepatocellular
carcinoma. Liver Int. 32:70–77. 2012. View Article : Google Scholar
|
|
18
|
Wang M, Chen GY, Song HT, Hong X, Yang ZY
and Sui GJ: Significance of CXCR4, phosphorylated STAT3 and VEGF-A
expression in resected non-small cell lung cancer. Exp Ther Med.
2:517–522. 2011.PubMed/NCBI
|
|
19
|
Fromigue O, Lagneaux L and Body J-J:
Bisphosphonates induce breast cancer cell death in vitro. J Bone
Miner Res. 15:2211–2221. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Matsumoto S, Kimura S, Segawa H, Kuroda J,
Yuasa T, Sato K, Nogawa M, Tanaka F, Maekawa T and Wada H: Efficacy
of the third-generation bisphosphonate, zoledronic acid alone and
combined with anti-cancer agents against small cell lung cancer
cell lines. Lung Cancer. 47:31–39. 2005. View Article : Google Scholar
|
|
21
|
Li YY, Chang JW, Chou WC, Liaw CC, Wang
HM, Huang JS, Wang CH and Yeh KY: Zoledronic acid is unable to
induce apoptosis, but slows tumor growth and prolongs survival for
non-small-cell lung cancers. Lung Cancer. 59:180–191. 2008.
View Article : Google Scholar
|
|
22
|
Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang
PY, Xu HX, Kong LQ, Wang L, Wu WZ and Tang ZY: Depletion of
tumor-associated macrophages enhances the effect of sorafenib in
metastatic liver cancer models by antimetastatic and antiangiogenic
effects. Clin Cancer Res. 16:3420–3430. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Previdi S, Scolari F, Chilà R, Ricci F,
Abbadessa G and Broggini M: Combination of the c-Met inhibitor
tivantinib and zoledronic acid prevents tumor bone engraftment and
inhibits progression of established bone metastases in a breast
xenograft model. PLoS One. 8:e791012013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Beuselinck B, Wolter P, Karadimou A,
Elaidi R, Dumez H, Rogiers A, Van Cann T, Willems L, Body JJ,
Berkers J, et al: Concomitant oral tyrosine kinase inhibitors and
bisphosphonates in advanced renal cell carcinoma with bone
metastases. Br J Cancer. 107:1665–1671. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Keizman D, Ish-Shalom M, Pili R, Hammers
H, Eisenberger MA, Sinibaldi V, Boursi B, Maimon N, Gottfried M,
Hayat H, et al: Bisphosphonates combined with sunitinib may improve
the response rate, progression free survival and overall survival
of patients with bone metastases from renal cell carcinoma. Eur J
Cancer. 48:1031–1037. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ling YH, Lin R and Perez-Soler R:
Erlotinib induces mitochondrial-mediated apoptosis in human H3255
non-small-cell lung cancer cells with epidermal growth factor
receptorL858R mutation through mitochondrial oxidative
phosphorylation-dependent activation of BAX and BAK. Mol Pharmacol.
74:793–806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Okamoto K, Okamoto I, Okamoto W, Tanaka K,
Takezawa K, Kuwata K, Yamaguchi H, Nishio K and Nakagawa K: Role of
survivin in EGFR inhibitor-induced apoptosis in non-small cell lung
cancers positive for EGFR mutations. Cancer Res. 70:10402–10410.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gong YX, Somwar R, Politi K, Balak M,
Chmielecki J, Jiang X and Pao W: Induction of BIM is essential for
apoptosis triggered by EGFR kinase inhibitors in mutant
EGFR-dependent lung adenocarcinomas. PLoS Med. 4:e2942007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Senaratne SG, Pirianov G, Mansi JL, Arnett
TR and Colston KW: Bisphosphonates induce apoptosis in human breast
cancer cell lines. Br J Cancer. 82:1459–1468. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tassone P, Tagliaferri P, Viscomi C,
Palmieri C, Caraglia M, D'Alessandro A, Galea E, Goel A, Abbruzzese
A, Boland CR, et al: Zoledronic acid induces antiproliferative and
apoptotic effects in human pancreatic cancer cells in vitro. Br J
Cancer. 88:1971–1978. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu M, Yuan Y, Pan YY and Zhang Y:
Antitumor activity of combination treatment with gefitinib and
docetaxel in EGFR-TKI-sensitive, primary resistant and acquired
resistant human non-small cell lung cancer cells. Mol Med Rep.
9:2417–2422. 2014.PubMed/NCBI
|
|
32
|
Mu X, Zhang Y, Qu X, Hou K, Kang J, Hu X
and Liu Y: Ubiquitin ligase Cbl-b is involved in icotinib
(BPI-2009H)-induced apoptosis and G1 phase arrest of EGFR
mutation-positive non-small cell lung cancer. BioMed Res Int.
2013:7263752013. View Article : Google Scholar
|
|
33
|
Li T, Ling YH, Goldman ID and Perez-Soler
R: Schedule-dependent cytotoxic synergism of pemetrexed and
erlotinib in human non-small cell lung cancer cells. Clin Cancer
Res. 13:3413–3422. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li YY, Chang JW, Liu YC, Wang CH, Chang
HJ, Tsai MC, Su SP and Yeh KY: Zoledronic acid induces cell-cycle
prolongation in murine lung cancer cells by perturbing cyclin and
Ras expression. Anticancer Drugs. 22:89–98. 2011. View Article : Google Scholar
|
|
35
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sordella R, Bell DW, Haber DA and
Settleman J: Gefitinib-sensitizing EGFR mutations in lung cancer
activate anti-apoptotic pathways. Science. 305:1163–1167. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wen Z, Zhong Z and Darnell JE Jr: Maximal
activation of transcription by Stat1 and Stat3 requires both
tyrosine and serine phosphorylation. Cell. 82:241–250. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kiper HD, Tezcanli Kaymaz B, Gokbulut AA,
Selvi N, Avci CB, Kosova B, Iskender G, Yandim MK, Gunduz C, Sahin
F, et al: STAT pathway in the regulation of zoledronic acid-induced
apoptosis in chronic myeloid leukemia cells. Biomed Pharmacother.
67:527–532. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Reuben JS, Dinh L, Lee J, Stateson J,
Kamara H, Xiang L and Opperman LA: Bisphosphonates inhibit
phosphorylation of signal transducer and activator of transcription
3 and expression of suppressor of cytokine signaling 3:
Implications for their effects on innate immune function and
osteoclastogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol
Endod. 111:196–204. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Koto K, Murata H, Kimura S, Sawai Y, Horie
N, Matsui T, Ryu K, Ashihara E, Maekawa T, Kubo T, et al:
Zoledronic acid significantly enhances radiation induced apoptosis
against human fibrosarcoma cells by inhibiting radioadaptive
signaling. Int J Oncol. 42:525–534. 2013.
|
|
41
|
Yun CH, Mengwasser KE, Toms AV, Woo MS,
Greulich H, Wong KK, Meyerson M and Eck MJ: The T790M mutation in
EGFR kinase causes drug resistance by increasing the affinity for
ATP. Proc Natl Acad Sci USA. 105:2070–2075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Robinson KW and Sandler AB: The role of
MET receptor tyrosine kinase in non-small cell lung cancer and
clinical development of targeted anti-MET agents. Oncologist.
18:115–122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ringerike T, Blystad FD, Levy FO, Madshus
IH and Stang E: Cholesterol is important in control of EGF receptor
kinase activity but EGF receptors are not concentrated in caveolae.
J Cell Sci. 115:1331–1340. 2002.PubMed/NCBI
|
|
44
|
Mantha AJ, Hanson JEL, Goss G, Lagarde AE,
Lorimer IA and Dimitroulakos J: Targeting the mevalonate pathway
inhibits the function of the epidermal growth factor receptor. Clin
Cancer Res. 11:2398–2407. 2005. View Article : Google Scholar : PubMed/NCBI
|