Introduction
Esophageal carcinoma (EC) is the eighth most common
malignancy and the sixth leading cause of cancer-related death
worldwide (1). In China, EC is the
fourth leading cause of cancer-associated death (2). EC remains an aggressive disease with a
high mortality rate. Despite major progress in clinical diagnostics
and therapy, EC represents a tumor entity with a limited prognosis.
The overall survival of EC patients remains poor, and the 5-year
survival rate is 17% (3–5). In multimodal therapy concepts of
locally advanced EC, radiotherapy is one of the well-established
therapeutic modalities for cancer treatment (6). However, radioresistance is a major
challenge during the treatment of EC. A better understanding of the
mechanisms of radioresistance may identify strategies through which
to address this challenge.
Autophagy is an evolutionarily conserved process
designed for the degradation and turnover of long-lived proteins
and dysfunctional organelles. Activation of autophagic pathways can
serve as a survival and adaptive mechanism, providing metabolic
support in times of cellular stress, such as exposure to radiation,
limiting the efficacy of radiotherapy (6). However, few studies have shown that
autophagy is a protective mechanism against radiation damage for
tumor cells (7). Autophagy protects
the cells against radiation damage by supplying the products of
catabolism to repair cells and by eliminating damaged cells and
protein aggregates, but whether the response is actually
cytoprotective and the identity of the pathways involved remain
undetermined. Sapkota et al reported that the
tumor-suppressor protein LKB1 (serine-threonine kinase liver kinase
B1) is induced by activated ATM after radiation (8). Furthermore, AMPK and LKB1, which are
signaling molecules, stimulate autophagic activity via p27 or mTOR
signaling (9). Therefore, we
hypothesized that radiation promotes autophagy as a cytoprotective
adaptive mechanism via the LKB1 pathway.
Here, we report that autophagy was induced during
exposure to radiation and promoted esophageal squamous cell
carcinoma cell survival. We then investigated the role of the LKB1
pathway in activating autophagy. We demonstrated the effects of
this novel mechanism in vitro and in vivo and showed
that pharmacological or genetic autophagy inhibition contributed to
EC cell radiosensitization.
Materials and methods
Cells and reagents
The esophageal carcinoma cell line Eca-109 was
obtained from the China Center For Type Culture Collection. A total
of 1×105 cells/well were plated in 6-well plates in
RPMI-1640 medium containing 10% fetal calf serum (FCS) and were
incubated in a humidified atmosphere of 95% air and 5%
CO2 at 37°C. The primary antibodies were against LC3B,
phospho-AMPKα (Thr 172), LKB1 (all from Cell Signaling Technology),
and GAPDH (Abcam). The phospho(p)-LKB1 (Thr 366) antibody was
produced by Sangon Biotech (Shanghai, China) against the peptide
KIEDGIIYTQDFTVPGK, in which the underlined residue is a
phosphothreonine. 3-Methyladenine (3-MA) and chloroquine
diphosphate (CQ) were purchased from Sigma-Aldrich.
Western blotting
Cell lysates were centrifuged at 12,000 rpm for 5
min at 4°C. The supernatants were collected, and 30 µg of
protein was electrophoresed on 12% SDS-PAGE gels, transferred to
PVDF membranes, and incubated with the primary antibody at 4°C
overnight. Goat anti-rabbit IgG secondary antibody conjugated to
horseradish peroxidase and a chemiluminescence system (Thermo
Fisher Scientific) were used for detection. The band intensities
were determined by densitometry using ImageJ software.
GFP-LC3 redistribution evaluation
Eca-109 cells were transfected with PBABEpuro
GFP-LC3 plasmid (Addgene, Cambridge, MA, USA) using the MACSfectin™
reagent (Miltenyi, Bergisch Gladbach, Germany). Quantification of
green dots was performed using the particle analysis plugin of
ImageJ (NIH). A cell was considered to be GFP-LC3-positive when it
had 10 or more green dots per cell, and a total of 100 cells were
counted per treatment.
Viability assay
Cells were seeded at a density of
5×104/ml in 96-well plates. After 24 h, the cells were
treated with a single-dose of radiation (0, 2, 4, or 6 Gy), and CQ
was added 1 h before irradiation in the experimental study groups.
At 24 h after irradiation, 20 µl of a 5 mg/ml MTT solution
was added to each well, and the plates were incubated at 37°C for 4
h. The medium was removed, and 150 µl of DMSO per well was
added. The absorbance was measured at 490 nm with a Enspire
Multimode reader. The percentage of cell survival was calculated
relative to the control cells, which was set to 100%.
Hoechst 33342 staining
Eca-109 cells were plated in 6-well plates and fixed
with 4% paraformaldehyde for 10 min at 25°C. Then, the cells were
washed twice with PBS. Hoechst 33342 staining (1 mM; Sigma-Aldrich)
solution was added to the plate and incubated for 15 min in the
dark at 37°C. The staining solution was then discarded, and the
cells were washed twice with PBS. Stained cells were observed by
fluorescence microscopy (Olympus).
Flow cytometric analysis
Cells were collected 24 h after radiation. For cell
cycle distribution analysis, the cells were stained with propidium
iodide (PI) (50 µg/ml) solution. For apoptosis detection,
the cells were stained with the Annexin V-FITC apoptosis detection
kit (Abcam). Stained cells were detected by flow cytometry (BD
Biosystems, USA), and data were analyzed with the ModFit LT v2.0
and CellQuest software programs.
Clonogenic assay
Cells were treated with single-dose irradiation (0,
1, 2, 4, or 6 Gy), and CQ was added 1 h before irradiation in the
experimental study groups. Irradiation was performed at 37°C, and
the dose rate was 0.4 Gy/min. At 24 h after irradiation, varying
cell numbers were seeded in triplicate in 60-mm culture dishes with
5 ml of regular cell culture medium and incubated for 7–14 days.
The cells were fixed with ethanol and then stained with 0.5%
crystal violet. Colonies that contained ≥50 cells were counted.
siRNA transfection
A total of 1×106 cells/well in 12-well
plates were transfected overnight using MACSfection reagent
(Miltenyi) with LKB1 siRNA, AMPKα siRNA (both from Santa Cruz
Biotechnology) or a negative control. After culture with fresh
medium for 24 h, knockdown was confirmed by western blotting.
RNA isolation and conventional reverse
transcription-PCR
Total cellular RNA from 5×105 cells was
prepared with TRIzol reagent (Invitrogen). A total of 1 µg
of RNA per sample was converted to cDNA with the Qiagen OneStep
RT-PCR kit (Qiagen) using a C1000 Touch Thermal Cycler (Bio-Rad).
The cDNA synthesis reactions were pre-incubated at 94°C and then
amplified over 30 cycles consisting of a 30-sec melting step
(94°C), a 15-sec annealing step at 58°C, and a 25-sec extension
step (72°C).
Real-time RT-PCR
We reverse-transcribed 1 µg of RNA with
Reverse Transcriptase M-MLV (Takara). PCR reactions were performed
in triplicate with the SYBR Green Master Mix (Toyobo). Reactions
were pre-incubated at 95°C and then amplified over 40 cycles
consisting of a 30-sec melting step (94°C), a 15-sec annealing step
at 58°C, and a 25-sec extension step (72°C). Amplification was
measured using a CFX96 Real-Time PCR detection system (Bio-Rad).
The primers included LC3B forward, GGTAAACGGGCTGTGTGAGA and
reverse, AAGGCAGAAGGGAGTGTGTC; and LKB1 forward,
ACTGAGGAGGTTACGGCAVA and reverse, TGGTGATGTTGTAGAGGGTGA.
Xenografts
Animal procedures were approved by the University
Committee on the Use and Care of Animals of the Huazhong University
of Science and Technology. A total of 1×106 Eca-109
cells were injected subcutaneously into the right posterior limb of
6- to 8-week old male Balb/c nu/nu mice. After the tumors were
established (mean subcutaneous volume, 50 mm3), the mice
were randomly divided into four groups. Groups 1 and 2 were
injected intraperitoneally with PBS or CQ (50 mg/kg), respectively.
Group 3 was administered a single dose of localized irradiation (6
Gy), and group 4 was irradiated and then administered an
intraperitoneal injection of CQ. All of the experiments used five
mice per group. Tumors were measured with calipers twice weekly.
The tumor volume was the length × (width)2/2, and
fold-growth was relative to treatment day one. Tumor tissues were
removed on day 27.
Immunohistochemistry
Slides were stained with primary antibody overnight
at 4°C followed by HRP-conjugated secondary antibodies (1:250) at
room temperature for 30 min. TUNEL staining was performed according
to the manufacturer's instructions. The One Step TUNEL apoptosis
assay kit was obtained from the Beyotime Institute of Biotechnology
(China). Afterwards, the slides were observed under an Olympus BX51
research microscope and images were obtained. The cells with green
fluorescence were defined as apoptotic cells.
Staining quantification
Images were imported into ImageJ and either
converted to binomy with the quantification of the percent of the
area positively stained or the determination of the relative
intensity of the color staining of interest using the measure RGB
feature. Data are expressed in arbitrary units per high powered
field.
Results
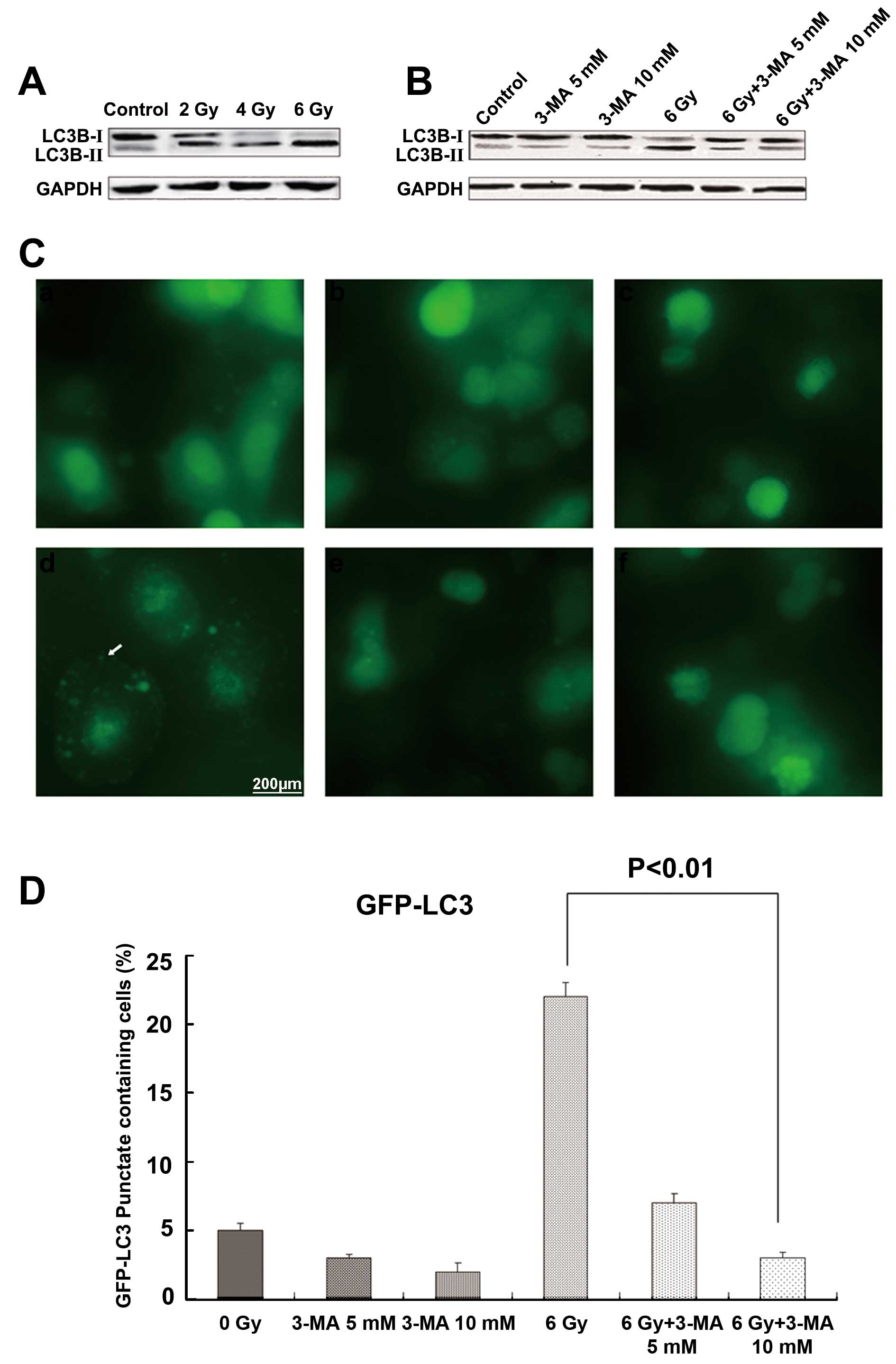
Autophagy inhibitors disrupt autophagy in
radiation-induced Eca-109 cells
3-MA is typically used as an autophagy inhibitor.
The extent of conversion from LC3B-I to LC3B-II reflects the level
of autophagosome formation. Western blotting demonstrated that
after radiation, the ratio of LC3B-II/LC3B-I was significantly
increased in the Eca-109 cells, whereas the levels of
LC3B-II/LC3B-I were decreased following 3-MA treatment in a
dose-dependent manner (Fig. 1A and
B). These results indicated that autophagy inhibitors disrupted
the radiation-induced Eca-109 cell autophagy. Similar effects were
noted in the Eca-109 cells transduced to express a GFP-LC3 fusion
protein, in which radiation increased autophagy, as determined by
the number of Eca-109 cells with punctuate green staining, the
marker reduced by 3-MA. After radiation exposure, 3-MA decreased
the number of cells with punctate green staining (P<0.01;
Fig. 1C and D).
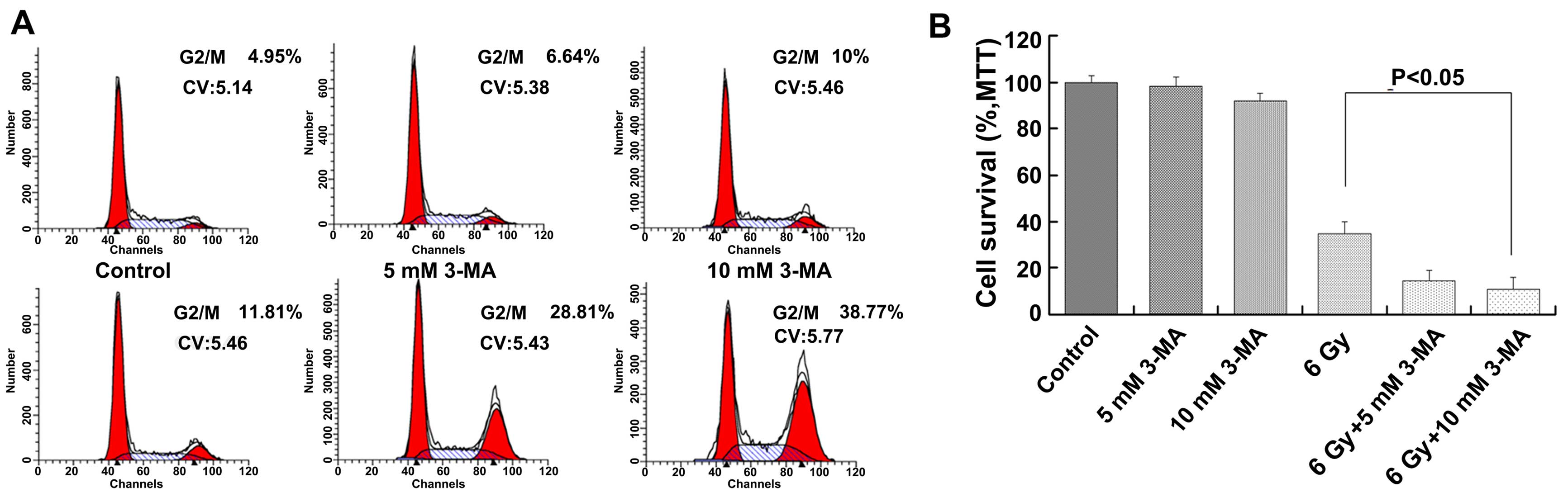
Inhibition of radiation-induced autophagy
increases cell cycle arrest and cell death
Flow cytometry showed that when Eca-109 cells were
treated with 3-MA, irradiation or combined therapy, the cell cycle
distribution was different. When Eca-109 cells were treated with
irradiation and 3-MA, we observed G2/M phase arrest of the Eca-109
cells; however, the Eca-109 cells in the G2/M phase were increased
only briefly. Notably, the Eca-109 cells in the G2/M phase were
significantly increased when the Eca-109 cells were treated with
combined therapy for 24 h (Fig.
2A). We measured the cell survival after radiation exposure
combined with 3-MA. 3-MA significantly decreased the number of
viable Eca-109 cells after radiation exposure (Fig. 2B). Based on the survival-promoting
effect of radiation-induced autophagy, we characterized the type of
cell death occurring after radiation exposure when autophagy was
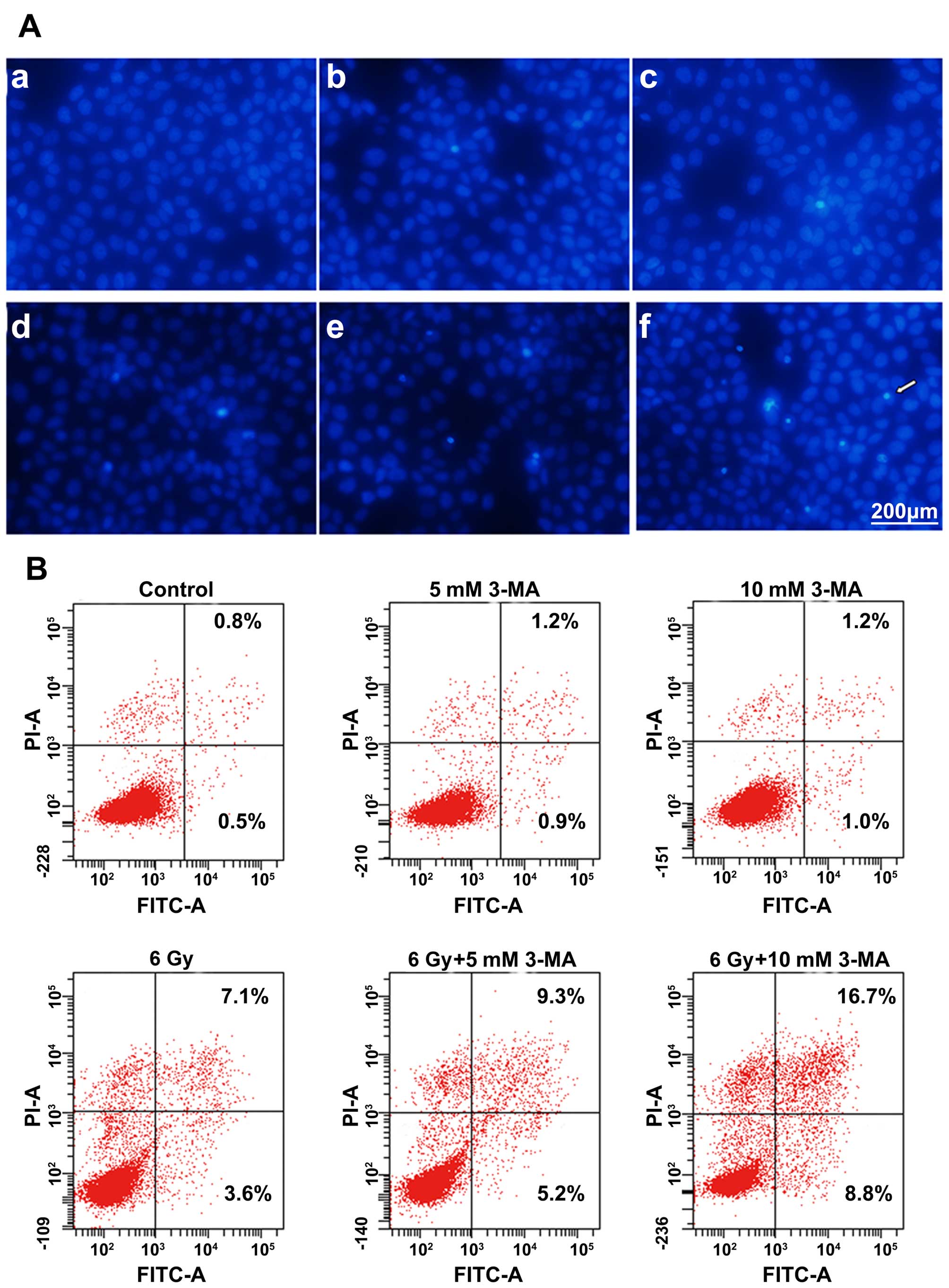
inhibited. We stained Eca-109 cells with Hoechst 33342, a
fluorescent dye that accumulates in apoptotic cells, and showed
that the dye increased after the Eca-109 cells were treated with
irradiation, 3-MA or a combination of both. In particular, Hoechst
33342 clearly accumulated following the combined therapy with
irradiation and 3-MA. The percentages of early and late apoptotic
cells were significantly increased after combined therapy with
irradiation and 3-MA (Fig. 3).
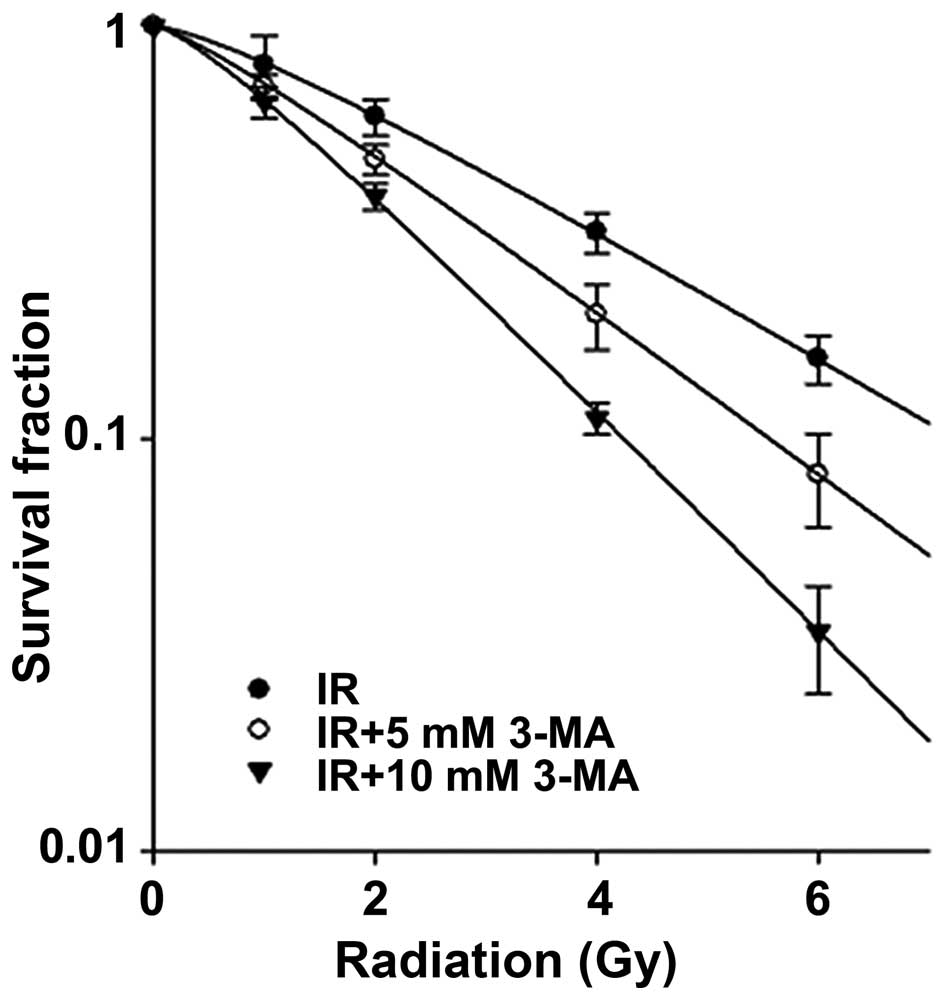
Clonogenic survival of Eca-109 cells
after inhibition of radiation-induced autophagy
To determine whether inhibition of autophagy
sensitizes Eca-109 cells to radiation, we performed colony forming
assays. As shown in Fig. 4, the
number of surviving Eca-109 cells was significantly decreased with
increasing concentrations of 3-MA. The SF2 (survival fraction at 2
Gy) and D0 values (dose of radiation producing a 37% survival rate)
were 61±5% and 2.75±0.26 Gy when irradiation was used alone.
However, for the combined therapy with irradiation and 3-MA, the
SF2 and D0 values were decreased in a concentration-dependent
manner and were 48±4% and 2.19±0.18 Gy and 38±5% and 1.6±0.05 Gy
for 5 and 10 mM 3-MA, respectively.
Dependence of radiation-induced autophagy
on LKB1
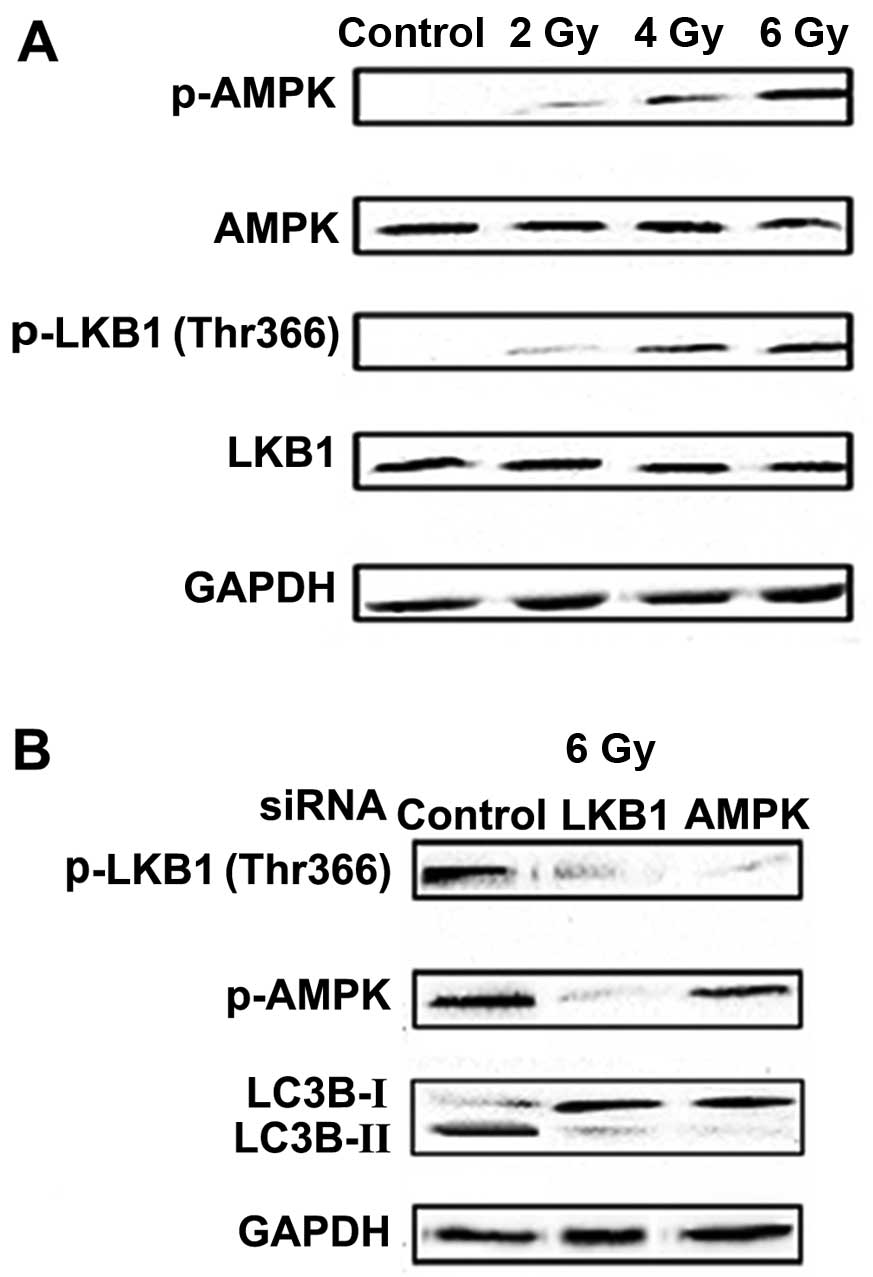
Sapkota et al observed radiation-induced
phosphorylation of LKB1 at Thr-366 (8). After radiation exposure, both LKB1 and
AMPK were activated in the Eca-109 cells. In particular, the
expression of p-LKB1 (Thr-366) and p-AMPK was increased in a
dose-dependent manner (Fig. 5A).
Furthermore, siRNA-mediated knockdown of LKB1 or AMPK in the
Eca-109 cells blocked the radiation-mediated LC3B-I to LC3B-II
conversion, and LKB1 siRNA reduced LKB1 and AMPK phosphorylation
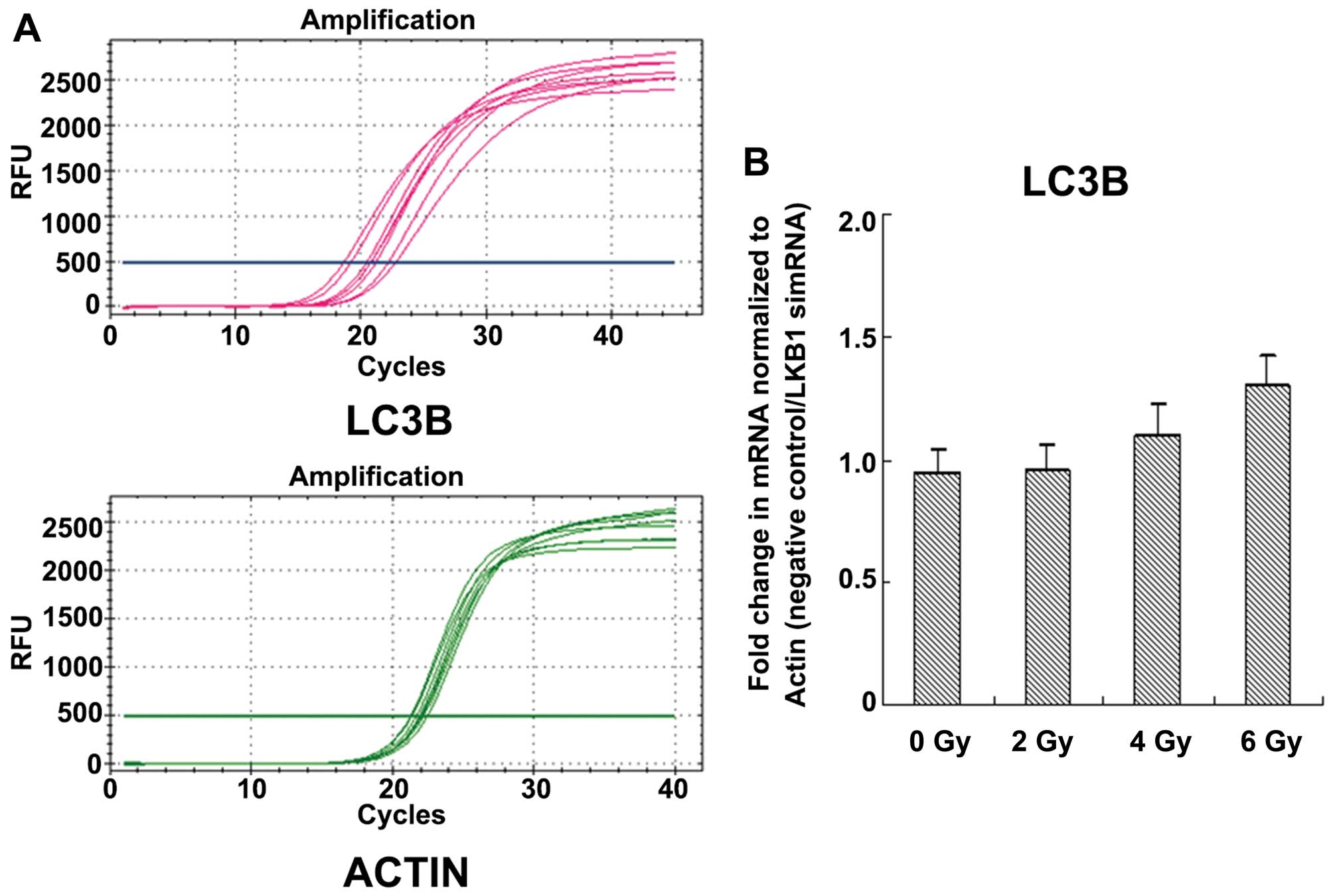
(Fig. 5B). We then investigated the
transcriptional changes to radiation-induced autophagy. The changes
in LC3B transcripts in the negative control relative to the LKB1
siRNA after radiation exposure were significant (P<0.05;
Fig. 6).
Combined inhibition of autophagy and
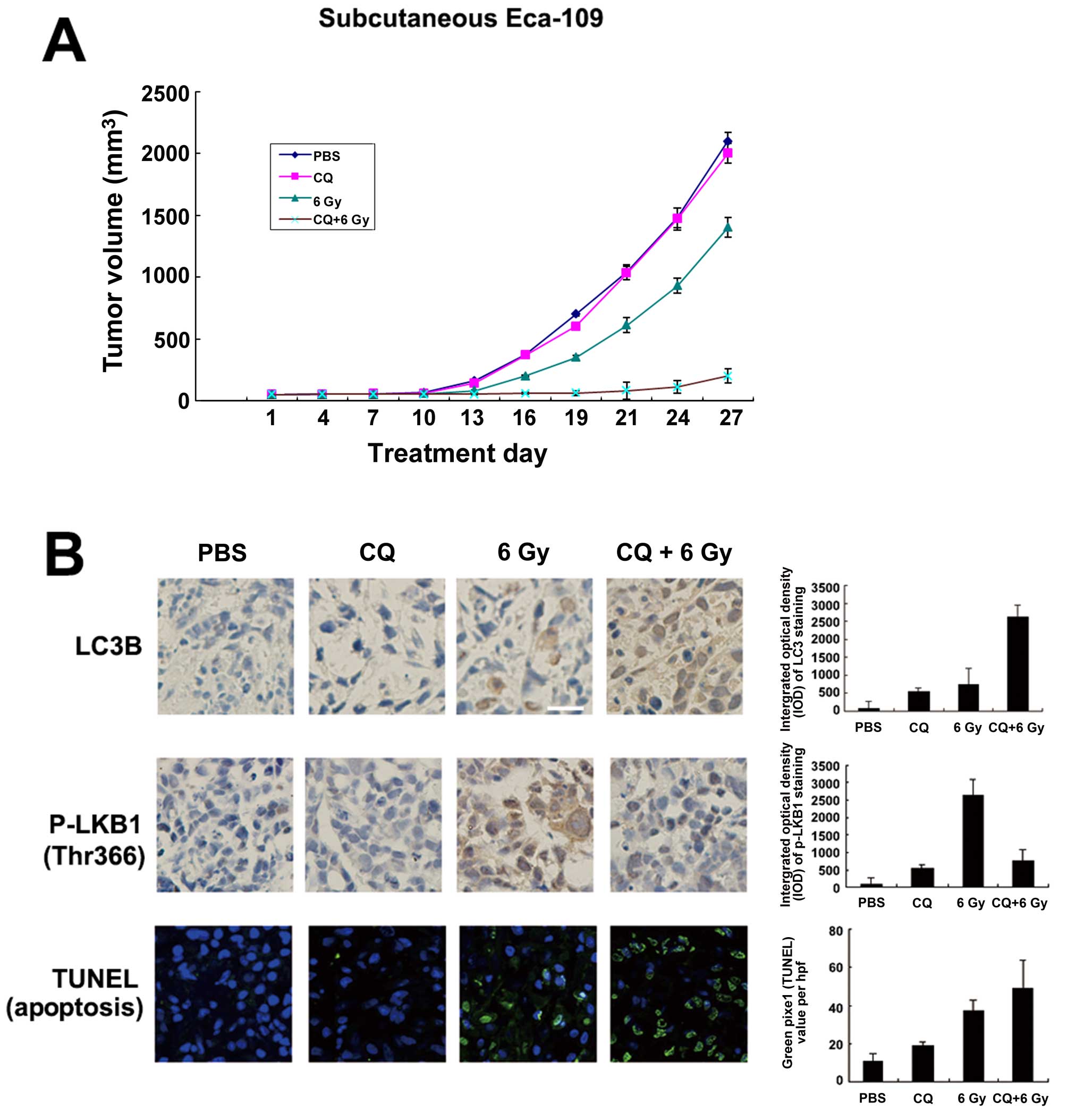
ionizing radiation exerts a potent antitumor effect in vivo
Chloroquine (CQ) is used to inhibit autophagy in
vivo, partly because it is the only FDA-approved autophagy
inhibitor. We then determined whether CQ counteracted the
survival-promoting effects of radiation-induced autophagy by
treating subcutaneous tumors derived from Eca-109 cells with the
autophagy inhibitor CQ and/or ionizing radiation of 6 Gy. After 27
days, the tumor volumes differed between the 4 treatment groups
(P<0.05), and combined therapy (CQ+6 Gy) inhibited tumor growth
significantly vs. ionizing radiation of 6 Gy alone (P<0.05)
(Fig. 7A). Furthermore, ionizing
radiation of 6 Gy significantly increased LC3B expression and
decreased p-LKB1 (Thr366) simultaneously. Compared to PBS or CQ
treatment alone (P<0.05), combined therapy (CQ+6 Gy) increased
LC3B expression and decreased p-LKB1 (Thr366) (P<0.05; Fig. 7B). Cell death in these xenografts
was characterized using TUNEL staining to detect cells in late
apoptosis. Staining was increased in the CQ+6 Gy groups compared to
the 6 Gy groups (P<0.05; Fig.
7B).
Discussion
Autophagy is a fundamental and phylogenetically
conserved self-degradation process that is characterized by the
formation of double-layered vesicles (autophagosomes) around
intracellular cargo for delivery to lysosomes and proteolytic
degradation (10). Autophagy has
two functions in cancer. It can be tumor suppressive through the
elimination of oncogenic protein substrates, toxic unfolded
proteins and damaged organelles or, alternatively, it can be tumor
promoting in established cancers via autophagy-mediated
intracellular recycling that provides substrates for metabolism and
maintains the functional pool of mitochondria (11). Autophagy, an alternative form of
programmed cell death, contributes significantly to the
anti-neoplastic effects of radiation therapy.
The ability of autophagy to capture, degrade,
eliminate and recycle intracellular components affects metabolism,
enables host defenses, remodels the proteome, regulates
trafficking, alters signaling and influences cellular interactions
(10). In this study, we
demonstrate that Eca-109 cells treated with autophagy inhibitors
after radiation exhibited decreased survival and upregulated
apoptosis. Our in vivo data showed increased TUNEL staining
in CQ plus radiation-treated xenografts, suggesting an increased
number of apoptotic cells during combined treatment. We also found
that the combined treatment induced G2/M cell cycle arrest, which
may contribute to cell death. In addition, we also demonstrated
that 3-MA, an inhibitor of autophagy, significantly decreased the
clonogenic survival of the Eca-109 cells. These findings indicate
that autophagy functions as a tumor-protective effect after
exposure to stressors, such as radiation. A few similar reports
have demonstrated that cell apoptosis can delay or evade death via
the autophagosome-lysosome pathway in response to various stressors
(12).
There is ample evidence that radiation-induced cell
death is affected by various related biochemical processes in the
autophagic pathways. Moretti et al reported that radiation
may activate autophagy via the adaptive endoplasmic reticulum
stress response (13). Nam et
al reported the activation of autophagy via inhibition of the
mTOR pathway, leading radioresistant cancer cells to senescence
(6). We found that LKB1 was
phosphorylated at Thr-366 after radiation exposure in a
dose-dependent manner, and AMPK was also activated. Both LKB1 and
AMPK could contribute to autophagy, with P27(kip1) activation as a
possible mechanism (14). These
findings suggest an internal relationship between autophagy, LKB1,
AMPK and radiation. We found that radiation-mediated LC3B-I to
LC3B-II conversion and overall LC3B degradation was dependent upon
both LKB1 and AMPK.
Interestingly, CQ decreased p-LKB1 expression in
radiotherapy xenografts. CQ is a late autophagy inhibitor, acting
on cells downstream of LKB1 (15).
Theoretically, CQ would not affect P-LKB1 expression based on the
regulatory mechanism of autophagy. The differences may reflect a
potential negative feedback system of autophagy when autophagosomes
rapidly increase in vivo. Future studies are necessary to
clarify this contradiction.
In conclusion, our findings reveal an important
relationship between radiation and autophagy signaling via LKB1 and
show that autophagy promotes esophageal squamous carcinoma cell
survival through pharmacological or genetic means. Targeting
autophagy is a novel method to enhance the effect of radiation
therapy as an anticancer modality.
Acknowledgments
This study was supported by Grant WX13B02 from the
Foundation of Health and Family Planning Commission of Wuhan
Municipality, China.
References
|
1
|
Enzinger PC and Mayer RJ: Esophageal
cancer. N Engl J Med. 349:2241–2252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sudo T, Iwaya T, Nishida N, Sawada G,
Takahashi Y, Ishibashi M, Shibata K, Fujita H, Shirouzu K, Mori M,
et al: Expression of mesenchymal markers vimentin and fibronectin:
The clinical significance in esophageal squamous cell carcinoma.
Ann Surg Oncol. 20(Suppl 3): S324–S335. 2013. View Article : Google Scholar
|
|
4
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Graf D, Vallböhmer D, Knoefel WT, Budach W
and Häussinger D: Multimodal treatment of esophageal carcinoma.
Dtsch Med Wochenschr. 139:2141–2147. 2014.In German. PubMed/NCBI
|
|
6
|
Nam HY, Han MW, Chang HW, Kim SY and Kim
SW: Prolonged autophagy by mTOR inhibitor leads radioresistant
cancer cells into senescence. Autophagy. 9:1631–1632. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zeng X and Kinsella TJ: Impact of
autophagy on chemotherapy and radiotherapy mediated tumor
cytotoxicity: 'To live or not to live'. Front Oncol. 1:302011.
View Article : Google Scholar
|
|
8
|
Sapkota GP, Deak M, Kieloch A, Morrice N,
Goodarzi AA, Smythe C, Shiloh Y, Lees-Miller SP and Alessi DR:
Ionizing radiation induces ataxia telangiectasia mutated kinase
(ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem J.
368:507–516. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu WK, Coffelt SB, Cho CH, Wang XJ, Lee
CW, Chan FK, Yu J and Sung JJ: The autophagic paradox in cancer
therapy. Oncogene. 31:939–953. 2012. View Article : Google Scholar
|
|
10
|
Barth S, Glick D and Macleod KF:
Autophagy: Assays and artifacts. J Pathol. 221:117–124. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Carew JS, Kelly KR and Nawrocki ST:
Autophagy as a target for cancer therapy: New developments. Cancer
Manag Res. 4:357–365. 2012.PubMed/NCBI
|
|
13
|
Moretti L, Cha YI, Niermann KJ and Lu B:
Switch between apoptosis and autophagy: Radiation-induced
endoplasmic reticulum stress? Cell Cycle. 6:793–798. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sanli T, Steinberg GR, Singh G and
Tsakiridis T: AMP-activated protein kinase (AMPK) beyond
metabolism: A novel genomic stress sensor participating in the DNA
damage response pathway. Cancer Biol Ther. 15:156–169. 2014.
View Article : Google Scholar :
|
|
15
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|