Introduction
Colon cancer is one of the most common cancers and
its prevalence is increasing gradually due to changes in lifestyle
(1). Epidemiological studies have
attempted to elucidate an association between a high-fiber diet and
decreased incidence and progression of colon cancer. Butyrate is a
short-chain fatty acid that is naturally produced by the intestinal
microflora during dietary fiber fermentation and has been suggested
as a target for colon cancer therapy because it inhibits cell
proliferation and causes apoptosis in colon cancer cells (2). Butyrate suppresses cell proliferation
by causing cell cycle arrest at the G1 and G2
phases, which involves cyclin D1 and retinoblastoma (Rb) signaling
(3). In addition, butyrate is a
histone deacetylase (HDAC) inhibitor, similar to trichostatin
A(TSA), and results in apoptosis by increasing the expression of
pro-apoptotic proteins, such as Bak, Bad and Bim, and decreasing
that of anti-apoptotic proteins, such as Bcl-2 and Bcl-xL (4).
Butyrate plays an important role in normal
colonocytes as a major energy source and promotes their
proliferation (5). However, most
cancer cells prefer to utilize glucose as an energy source even
under abundant oxygen conditions, so colon cancer cells cannot use
butyrate efficiently, which enhances its anticancer effects
(6).
Recently, it has been reported that butyrate may
trigger resistance in colon cancer cells (7). As mentioned above, butyrate is a
natural product of dietary fiber fermentation to which colonocytes
are consistently exposed. Acquisition of resistance to butyrate can
affect the proliferation, cell cycle and invasiveness of colon
cancer cells. However, the mechanism of resistance to butyrate is
unclear. Drug efflux pumps, such as P-glycoprotein (P-gp), breast
cancer-resistant protein (BCRP) and the multidrug resistance
associated protein 1 (MRP1), may be related to the acquisition of
resistance to butyrate. Such proteins are considered to influence
drug absorption, bioavailability and resistance (8).
In this study, we established a butyrate-resistant
colon cancer cell line and investigated the effect of butyrate
resistance in colon cancer cells, focusing on sensitivity to
chemotherapeutic agents, the cell cycle and invasiveness.
Materials and methods
Chemicals
Sodium butyrate, TSA, mitomycin C,
3-(4,5-dimeth-ylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT)
and protease inhibitor cocktail were purchased from Sigma-Aldrich
(St. Louis, MO, USA). Fetal bovine serum (FBS), RPMI-1640 medium,
and Dulbecco's phosphate-buffered saline (DPBS) were from
Invitrogen (Carlsbad, CA, USA). Antibodies against Rb, Bax, Bim,
BclXL, cyclin E, cyclin D1, cyclin-dependent kinase
(CDK) 2, CDK4, p21Waf1/Cip1, extracellular
signal-regulated kinases (Erk) 1/2 and β-actin were obtained from
Cell Signaling Technology (Beverly, MA, USA). MRP1, BCRP and P-gp
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA) and anti-glycer-aldehyde-3-phosphate
dehydrogenase (GAPDH) antibody was obtained from Calbiochem (EMD
Biosciences Inc., San Diego, CA, USA). Secondary antibodies were
purchased from Bio-Rad (Hercules, CA, USA). Enhanced
chemiluminescence solution (ECL) was kindly donated by Detroit
R&D, Inc. (Detroit, MI, USA). Propidium iodide and RNase A were
from Abcam (Cambridge, UK).
Establishment of a butyrate-resistant
cell line
The HCT116 human colon cancer cell line was obtained
from the American Type Culture Collection (ATCC, Manassas, VA, USA)
and cultured in RPMI-1640 medium supplemented with 10% FBS and 1%
penicillin-streptomycin at 37°C in a humidified 5% CO2
incubator. To establish the butyrate-resistant HCT116 colon cancer
cell line (HCT116/BR), HCT116 parental cells (HCT116/PT) were
initially incubated with serum-containing media supplemented with
0.2 mM sodium butyrate. Butyrate treatment induced the death of
most cells while some cells survived, but they proliferated slower
compared to HCT116/PT cells. When cells were 80% confluent, the
media was changed to a fresh media containing the same
concentration of butyrate. By continuously growing cells in the
presence of the same concentration of butyrate, no more detectable
death of cells was observed and then the treating concentration of
butyrate increased 2-fold. After culturing cells with the
increasing concentration of sodium butyrate to a maximum of 1.6 mM
for 3 months, butyrate-resistant cells were established. Butyrate
stock was prepared by dissolving sodium butyrate in DPBS. The
treatment was carried out after overnight incubation to allow cell
attachment.
Cell proliferation assay
Cell proliferation was determined in both cell lines
according to a method described previously with slight
modifications (9). HCT116/PT and
HCT116/BR cells were seeded onto 96-well plates. Cells were treated
with various concentrations of butyrate, paclitaxel, 5-fluorouracil
(5-FU), doxorubicin and TSA. After 72-h incubation, medium was
removed, 0.5% MTT stock was diluted 1:10 with medium and 100
µl added to each well, followed by incubation at 37°C for a
further 2 h. Subsequently, dimethylsulfoxide (DMSO) was added to
solubilize the purple formazan crystals and then absorbance at 540
nm was measured using an ELISA reader (Bio-Tek Instruments Inc.,
Winooski, VT, USA).
Flow cytometry
To assess cell cycle progression, HCT116/PT and
HCT116/BR cells were plated onto 60-mm dishes. After overnight
incubation, HCT116/PT cells were treated with 6.4 mM butyrate for
24 h. Cells were then trypsinized and centrifuged for 5 min at 500
× g at 4°C, and fixed with 70% ethanol at 4°C, followed by addition
of 1 ml of propidium iodide solution (final concentration, 50
µg/ml), which contained 200 µg/ml RNase A, for 30 min
in the dark (10). Flow cytometry
was then performed using a FACSCalibur flow cytometer and
identified using Cell Quest software (Becton-Dickinson, San Jose,
CA, USA). Red fluorescence, measured at 585/542 nm, indicative of
propidium iodide uptake by damaged cells, was measured by
logarithmic amplification and electronic compensation for spectral
overlap (11).
Protein extraction and
immunoblotting
Cells were lysed with cell lysis buffer containing
protease inhibitor cocktail, and protein concentration was
determined using the bicinchoninic acid (BCA) assay. Protein
samples (20–30 µg per lane) were resolved by 7.5–12.5%
sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred to a nitrocellulose membrane. For
immunodetection, blots were incubated with 1:1,000 diluted primary
antibodies in 5% bovine serum albumin (BSA) Tris-buffered saline
(TBS) containing 0.1% Tween-20 (TBS-T) at 4°C with gentle shaking
overnight, followed by incubation with a secondary antibody
conjugated to horseradish peroxidase at room temperature. The
antigen-antibody complexes were detected using ECL reagents with an
ImageQuant LAS-4000 mini (GE Healthcare Life Sciences, Piscataway,
NJ, USA). GAPDH or β-actin was used as the loading control
(12).
Wound healing assay
Cells were seeded onto 6-well plates to create a
confluent monolayer after overnight incubation. Mitomycin C was
treated (final concentration, 25 µg/ml) for 30 min and
removed. A straight line was scratched in the middle of the cell
monolayer. After washing with DPBS, cells were treated with
chemotherapeutic drugs and incubated up to 24 h. The widths of the
scratched line were measured at appropriate time intervals and
compared to that at 0 h.
Gelatin zymography
Activity of matrix metalloproteinases (MMPs) was
detected using gelatin zymography according to a method described
previously with slight modifications (13). Briefly, cells were incubated in
60-mm dishes and medium was replaced with serum-free medium to
promote MMP secretion when cells reached 80% confluence. After 48-h
incubation, medium was harvested and concentrated using a Speed-vac
(Savant/E-C Instruments, Niantic, CT, USA). Electrophoresis was
performed in 10% SDS-PAGE gels containing 0.1% gelatin (Wako Pure
Chemical Industries, Ltd., Osaka, Japan) without a reducing agent
or boiling. After electrophoresis, the gel was soaked with
renaturing buffer (2.5% Triton X-100, v/v) for 1 h, rinsed with
distilled water three times, incubated with developing buffer (50
mM Tris-base, 150 mM NaCl, 1 µM ZnCl2 and 50 mM
CaCl2) for 72 h at 37°C, stained with 0.5% (w/v)
Coomassie Blue, and destained in destaining buffer (methanol:acetic
acid:water, 10:5:85, v/v). Gel images were scanned using a Gel-Doc
Imaging System (Bio-Rad) and band density was measured using the
ImageJ software.
Statistical analysis
Data are presented as means ± standard deviation
(SD). Statistical analysis was performed using the GraphPad Prism 5
software. Data were compared using an unpaired two-tailed t-test or
one-way analysis of variance (ANOVA), followed by Dunnett's post
hoc test. P-values <0.05 were considered to indicate statistical
significance (14).
Results
Establishment of butyrate-resistant
HCT116 cells
HCT116/BR cells were derived from the HCT116 human
colon cancer cell line by chronic exposure to butyrate for 3
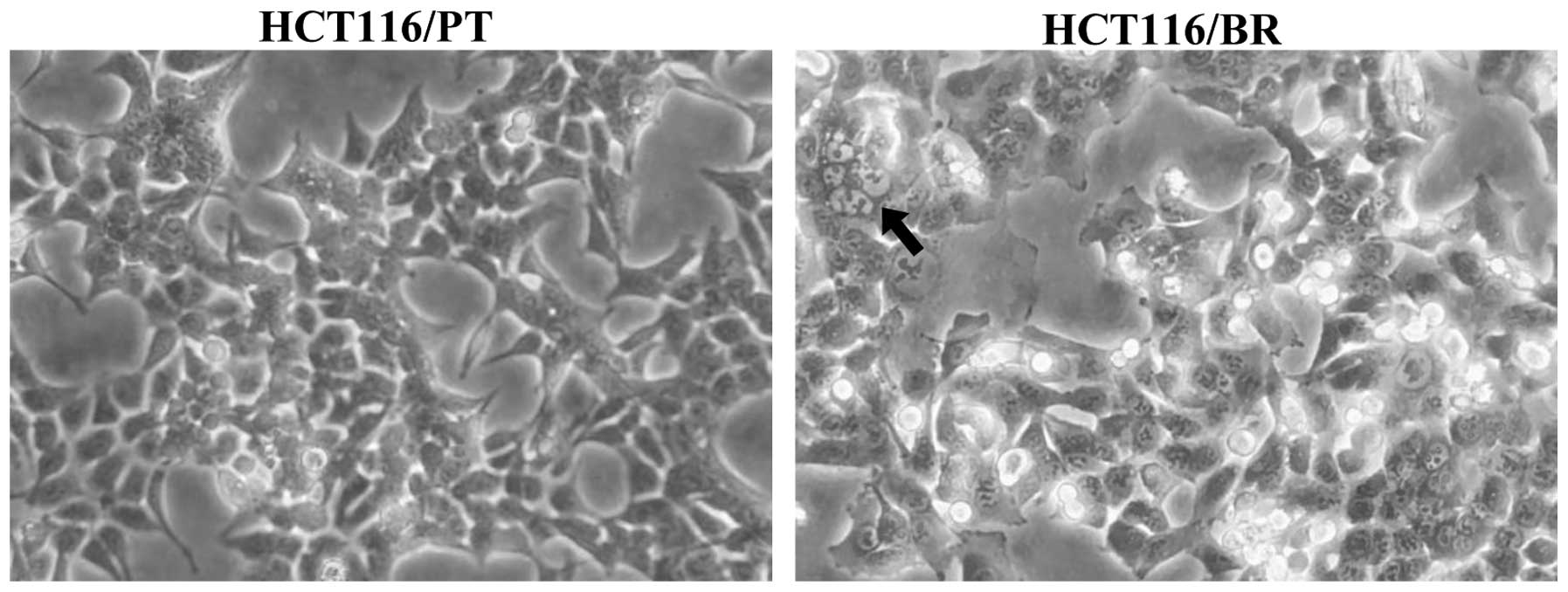
months. Cell morphology was altered slightly (Fig. 1). HCT116/PT cancer cells had
cuboidal and sharp-pointed shapes, but HCT116/BR cells were more
rounded and expanded. In addition, vacuolization was observed in
HCT116/BR cells. The growth rate of the cells was lower than that
of HCT116/PT cells.
Chemoresistance of HCT116/BR cells
Whether resistance to other chemotherapeutic agents
accompanied resistance to butyrate was assessed by determining cell
viability by MTT assay. HCT116/PT and HCT116/BR cells were treated
with butyrate, paclitaxel, 5-FU, doxorubicin and TSA, and the
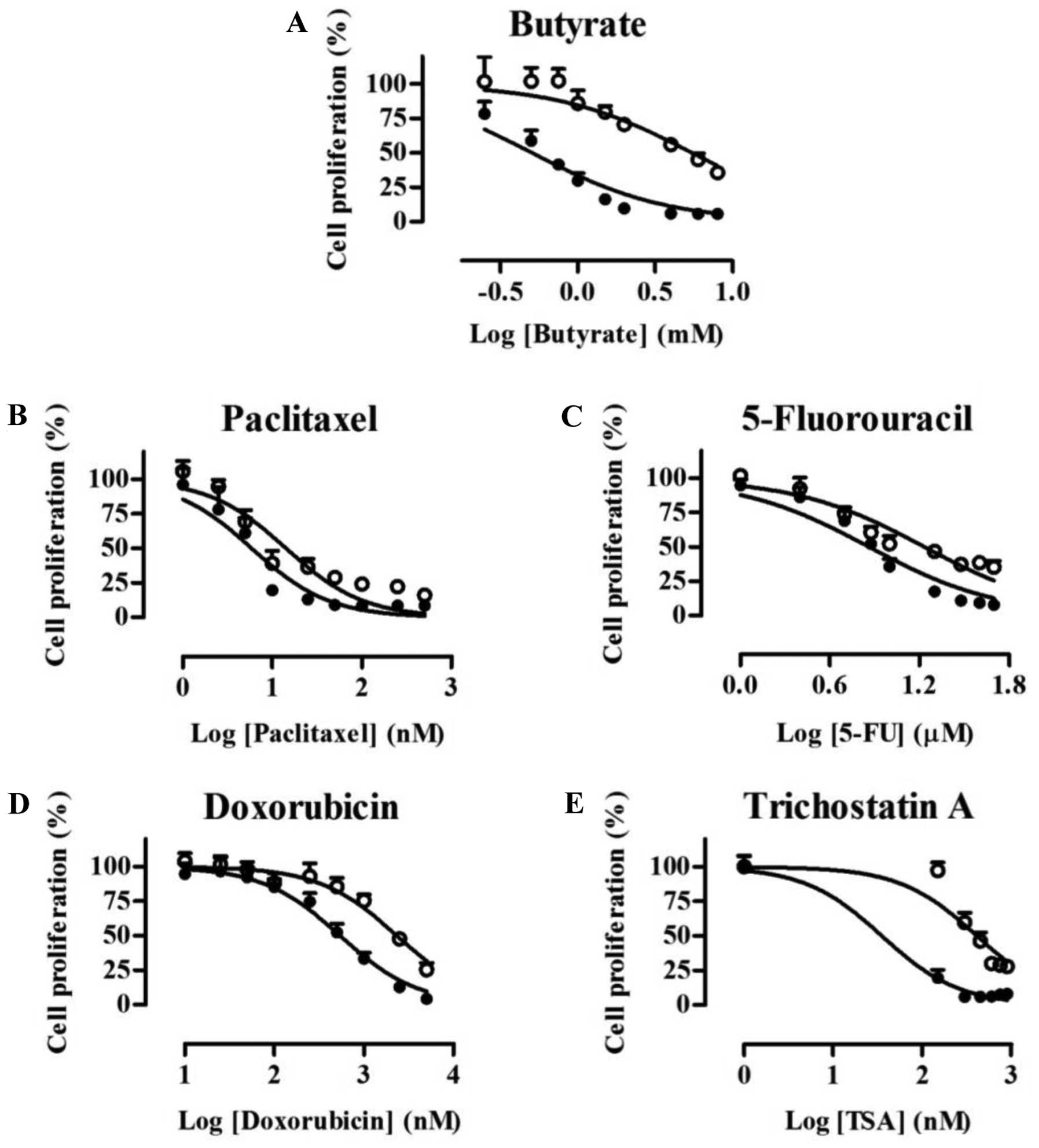
dose-response curve and the concentrations that inhibited cell
growth by 50% (IC50) values are shown in Fig. 2 and Table I, respectively. The inhibition of
cell proliferation was increased in a concentration-dependent
manner in both cell lines, but HCT116/BR cells exhibited greater
chemoresistance. The IC50 values of HCT116/BR cells for
butyrate, paclitaxel, 5-FU, doxorubicin and TSA were 10.8, 2.42,
2.36, 4.31 and 11.3-fold higher, respectively, than those of
HCT116/PT cells.
 | Table IIC50 values of
chemotherapeutic drugs in HCT116/PT and HCT116/BR cells. |
Table I
IC50 values of
chemotherapeutic drugs in HCT116/PT and HCT116/BR cells.
| Chemotherapeutic
drugs | HCT116/PT | HCT116/BR |
|---|
| Butyrate (mM) | 0.508 | 5.50 |
| Paclitaxel (nM) | 5.89 | 14.3 |
| 5-FU (µM) | 7.28 | 17.1 |
| Doxorubicin (nM) | 541 | 2,330 |
| Trichostatin
A(nM) | 36.3 | 412 |
Drug efflux pump expression
Drug efflux pumps are related to drug resistance
(8). One possible mechanism of
chemoresistance to butyrate is regulation of the expression of drug
efflux pumps. Therefore, we determined P-gp, BCRP and MRP1 protein
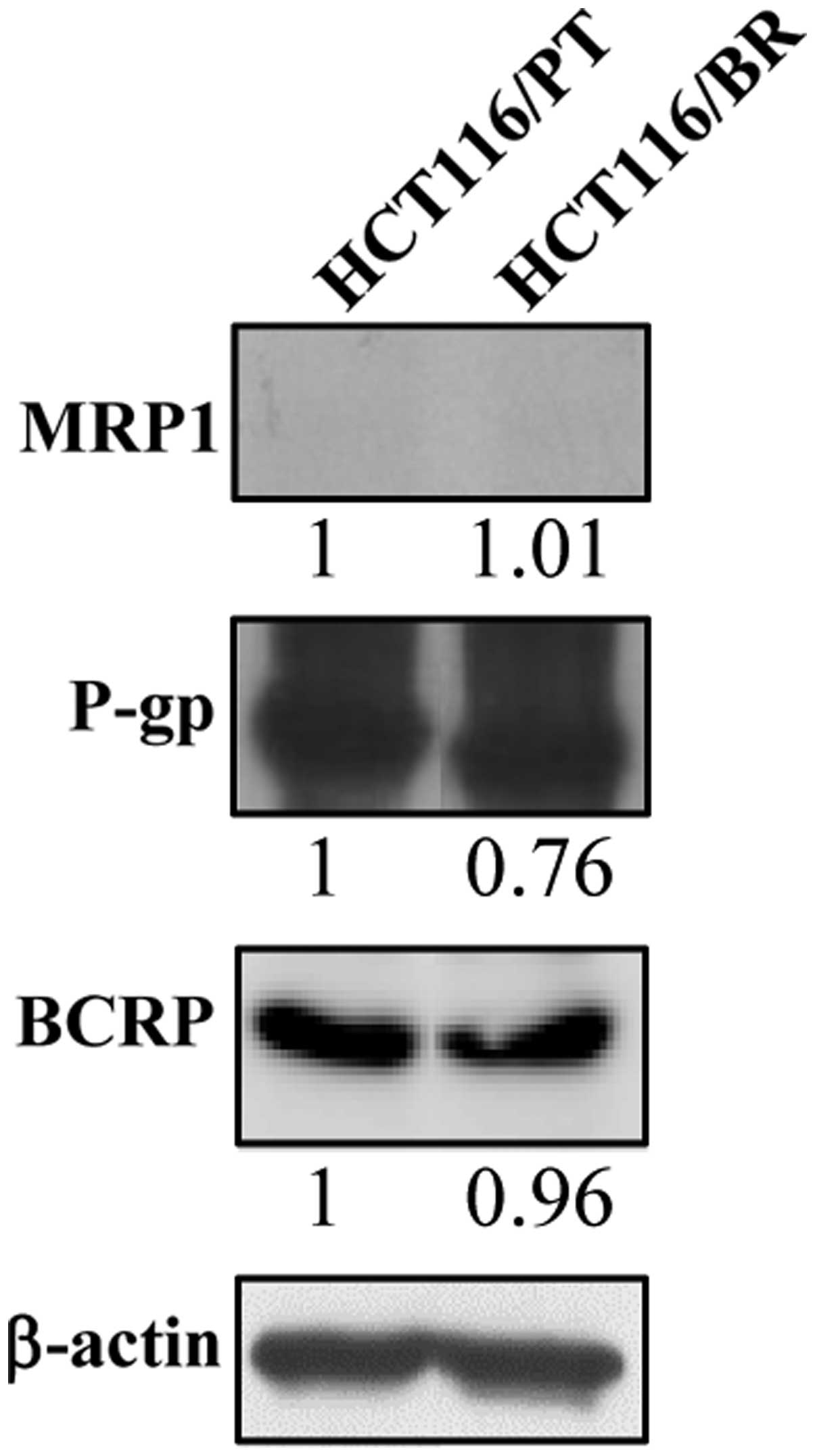
levels by immunoblotting (Fig. 3).
There was no significant difference in P-gp and BCRP expression
between the cell lines, and MRP1 was not detected in HCT116/PT or
HCT116/BR cells. Thus, drug efflux pumps did not affect the
chemoresistance of HCT116/BR cells.
Induction of cell cycle arrest by
butyrate
Butyrate induces cell cycle arrest and apoptosis in
cancer cells (15), but our
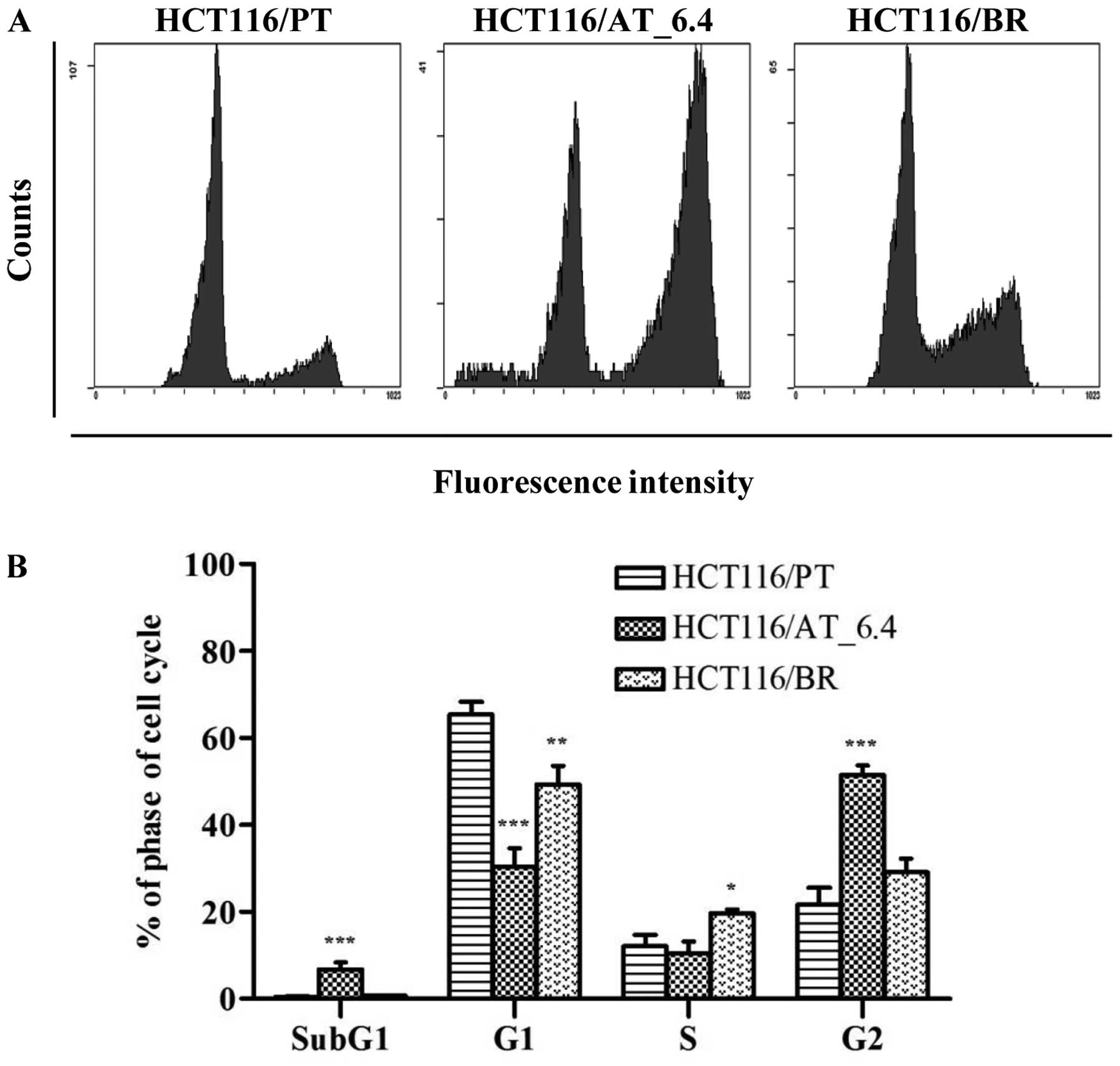
findings indicated no such effects in HCT116/BR cells. We performed
flow cytometry to assess the effect of butyrate on cell cycle
progression in HCT116/BR cells (Fig.
4). We also treated HCT116/PT cells with 6.4 mM butyrate
(4-fold of 1.6 mM) for 24 h (HCT116/AT_6.4) and compared the
results with those of HCT116/PT and HCT116/BR cells. The proportion
of subG1-phase cells was increased by treatment with 6.4
mM butyrate compared to untreated HCT116/PT cells (6.69 vs. 0.47%)
suggesting that apoptosis was triggered. The proportion of
subG1-phase HCT116/BR cells was similar (0.68%). The
proportion of cells in S phase, which is a DNA-replication phase of
the cell cycle, was elevated by 62.0% in HCT116/BR compared to
HCT116/PT cells. In addition, the proportion of HCT116/AT_6.4 cells
in G2 phase was >2-fold greater than that of
HCT116/PT cells (51.4 vs. 21.6%), but was restored to the control
level in HCT116/BR cells (29.1%).
Protein expression on cell cycle and
apoptosis
We conducted an immunoblotting analysis to examine
the expression of proteins involved in the cell cycle and apoptosis
in HCT116/PT and HCT116/BR cells. We treated HCT116 cells with 1.6
and 6.4 mM butyrate for 24 h (HCT116/AT_1.6 and HCT116/AT_6.4,
respectively) to determine the effects of acute butyrate
treatment.
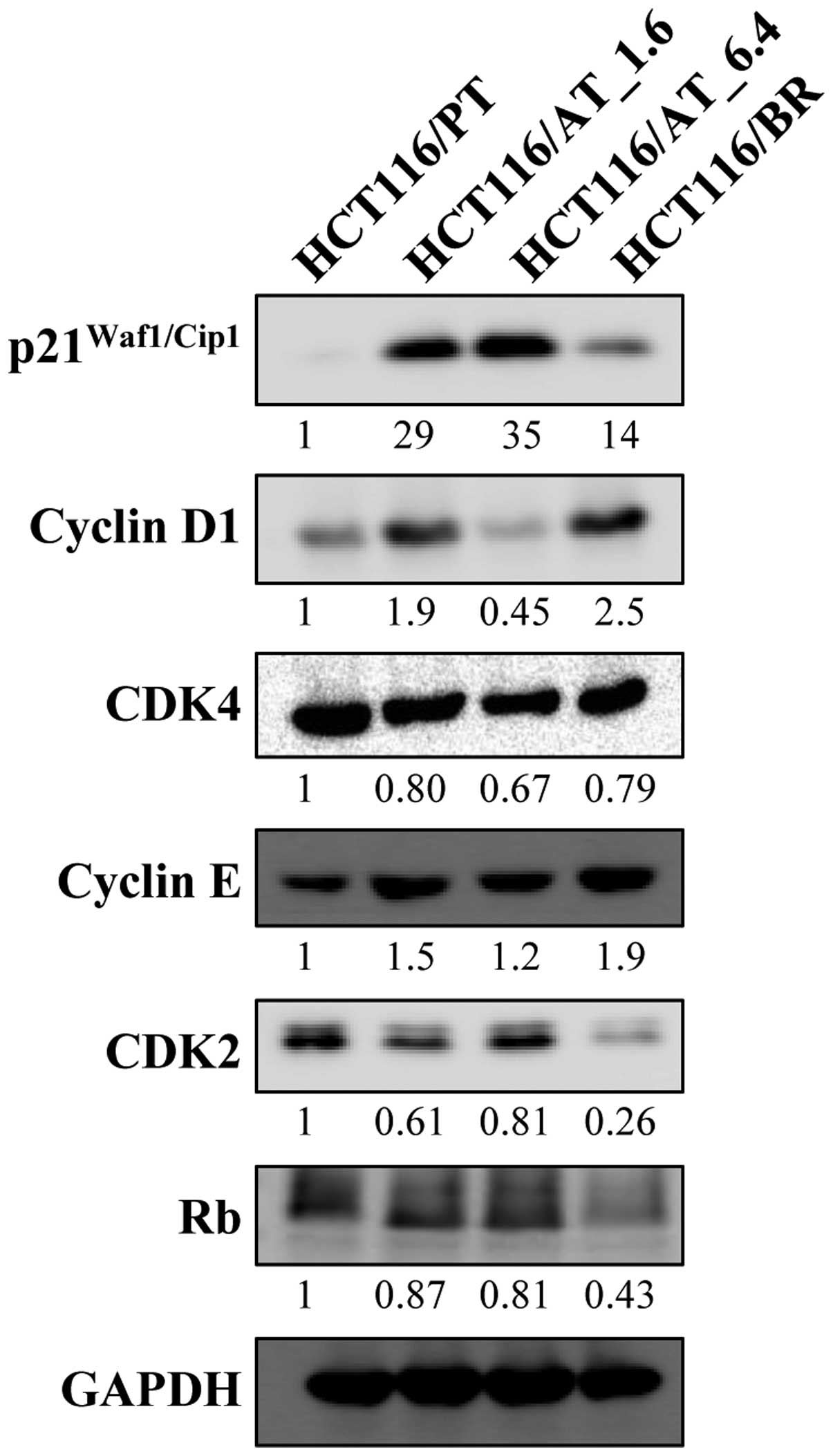
Expression of p21Waf1/Cip1 was greatly
increased in HCT116/AT_1.6 and HCT116/AT_6.4 cells, but the
increase was less marked in HCT116/BR cells compared to HCT116/PT
cells (Fig. 5). HCT116/BR cells
showed greater cyclin D1 expression compared to HCT116/PT cells.
CDK4 protein expression was minimally changed. Rb expression was
decreased in HCT116/BR cells. Cyclin E expression in HCT116/BR
cells was slightly increased by butyrate; however, CDK2 expression
was greatly reduced.
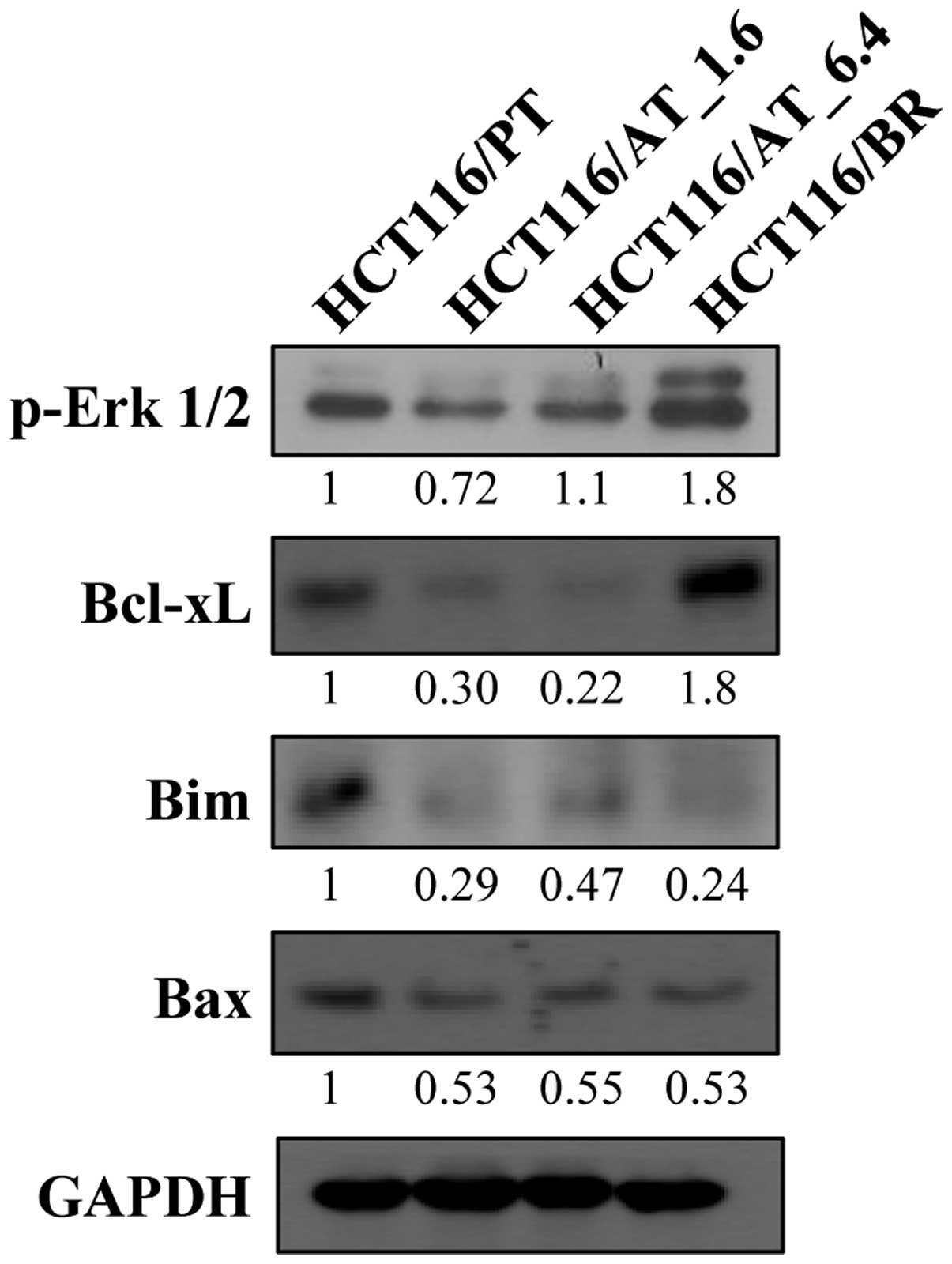
The Bcl-2 family, which includes Bcl-xL, Bim and
Bax, is involved in the regulation of apoptosis, and their
expression is shown in Fig. 6. The
expression of Bcl-xL, which is an important anti-apoptotic protein,
was decreased in HCT116/AT_1.6 and HCT116/AT_6.4 cells by the acute
butyrate treatment, but was significantly increased in HCT116/BR
cells. Bim and Bax are pro-apoptotic proteins, the expression of
which was decreased in HCT116/AT_1.6, HCT116/AT_6.4 and HCT116/BR
cells by both chronic and acute butyrate treatment. Moreover,
activation of Erk reduces the expression of Bim, and phospho-Erk1/2
expression was increased in HCT116/BR cells.
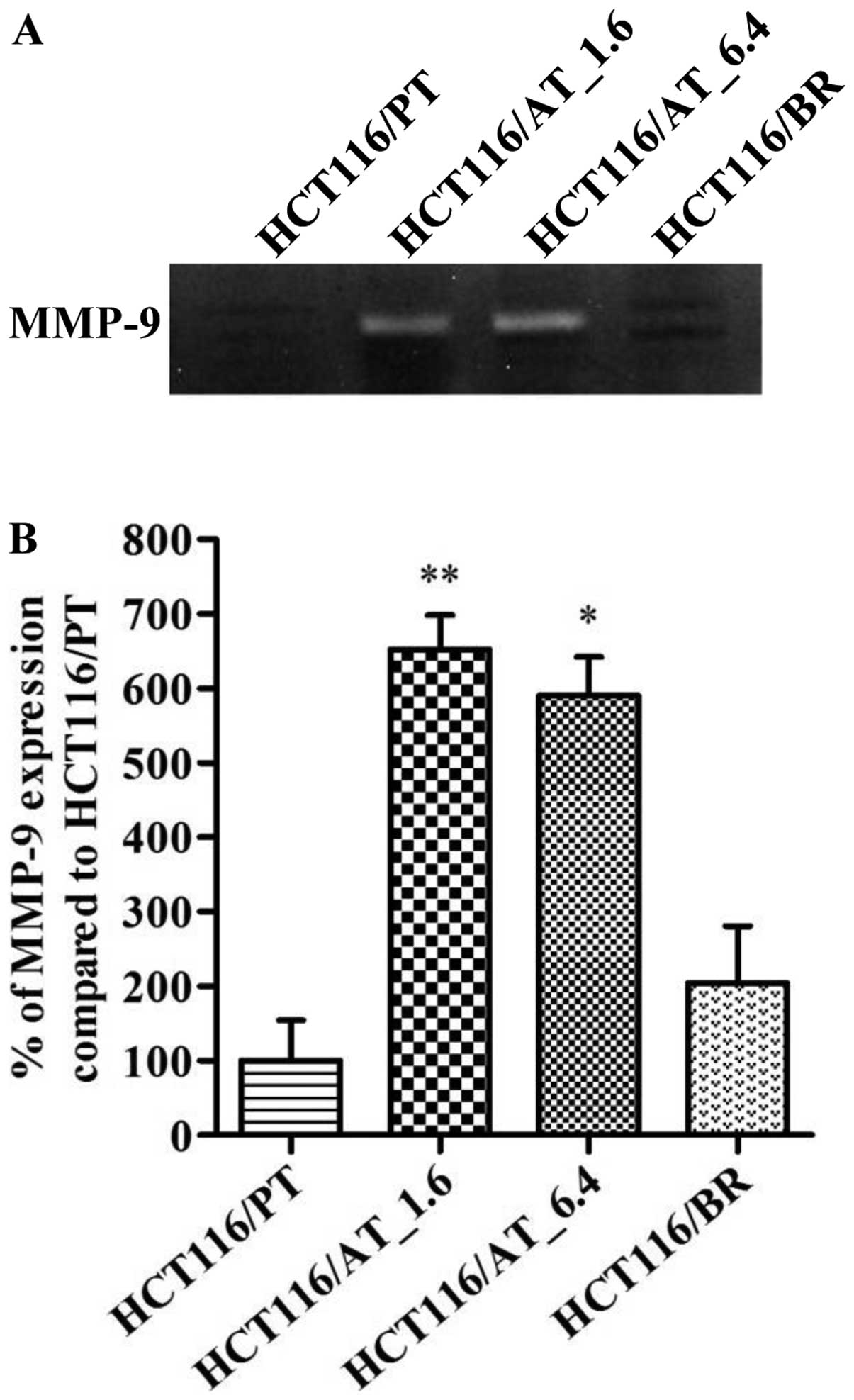
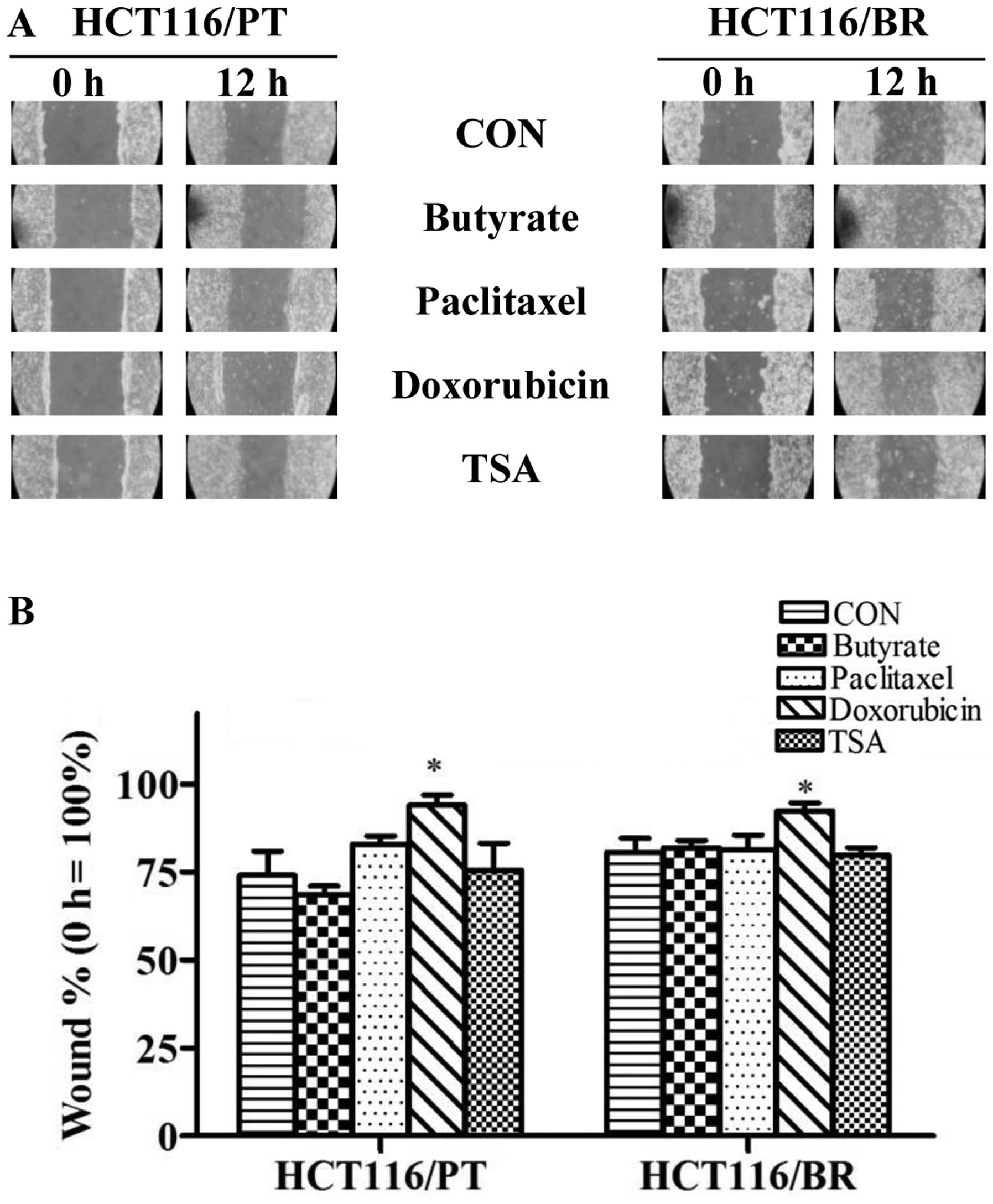
Effect of butyrate on invasiveness
We performed gelatin zymography and a wound healing
assay to evaluate cell invasiveness (Fig. 7) and migratory activity (Fig. 8). The wound healing rate was
generally slower in HCT116/BR than HCT116/PT cells. MMP-9 plays a
role in cancer cell metastasis, and there was no significant
difference in its expression between HCT116/PT and HCT116/BR cells
(Fig. 7). However, following
short-term butyrate treatment, MMP-9 expression was significantly
increased in both HCT116/AT_1.6 and HCT116/AT_6.4 cells. MMP-2 was
not detected in HCT116/PT and HCT116/BR cells. There was no
significant difference in wound healing rate between HCT116/PT and
HCT116/BR cells following treatment with butyrate, paclitaxel and
TSA; however, doxorubicin significantly inhibited wound healing in
both cell types.
Discussion
Butyrate inhibits proliferation and induces
apoptosis of cancer cells, although it promotes the growth of
normal cells. Recently, however, butyrate has been noted in colon
cancer cells but the mechanism is unclear. Investigating the
mechanism of resistance to butyrate in colon cancer cells is
important for establishing new therapeutic strategies (16). In this study, we assessed the effect
of resistance induced by butyrate in HCT116 human colon cancer
cells.
We established butyrate-resistant HCT116 cells by
chronic exposure to butyrate, which altered several of the cellular
characteristics. First, chronic exposure of HCT116/BR cells to
butyrate resulted in chemoresistance. Butyrate-resistant
BCS-TC2.BR2 human colon adenocarcinoma cells are more tolerant to
apoptosis induced by exogenous stimulation (7). Using a cell proliferation assay, we
found that HCT116/BR cells had enhanced survivability compared to
HCT116/PT cells following treatment with chemotherapeutic agents,
such as paclitaxel, doxorubicin, TSA and 5-FU, as well as butyrate.
This suggests that resistance to butyrate is accompanied by
tolerance to chemotherapeutic agents. We hypothesized drug efflux
pumps to be a possible resistance mechanism. However, there were no
significant differences in the protein levels of drug efflux pumps,
such as P-gp, BCRP and MRP1. Consequently, multidrug resistance in
butyrate-resistant HCT116/BR colon cancer cells is not associated
with resistance to chemotherapeutic agents.
We next examined the effect of butyrate on cell
cycle progression. The proportion of HCT116/PT cells in
G1 phase was higher, and that in G2 phase
lower compared to HCT116/AT_6.4 cells. Following short-term
treatment with butyrate, the proportion in G2 phase was
significantly increased in HCT116/AT_6.4 cells; however, HCT116/BR
cells exhibited a pattern similar to the parental cells, suggesting
that chronic exposure to butyrate acquired the resistance to
butyrate and thus resulted in restoration of a normal cell cycle
progression.
Protein expression data suggested that escape of
cell cycle arrest induced by butyrate in HCT116/BR cells was
dependent on p21Waf1/Cip1, cyclin D1 and Rb signaling. A
previous study reported that HeLa human cervical cancer cells
showed resistance to butyrate, which was associated with
upregulation of cyclin D1 (17).
Butyrate induced G1 arrest by disturbing Rb signaling
was mediated by inhibition of cyclin D1 and p53-independent
induction of p21Waf1/Cip1 (3). In contrast, CDK2 expression was
significantly decreased in HCT116/BR cells. This was supported by
the flow cytometry results, which indicated an increased proportion
of HCT116/BR cells in S phase, likely because the cyclin E/CDK2
complex is required for progression from G1 to S phase
(18). Furthermore, inhibition of
apoptosis was confirmed in HCT116/BR cells by upregulation of
Bcl-xL, and downregulation of Bim and Bax, as well as upregulation
of phospho-Erk, which is a regulator of Bim (4,16).
This is a unique mechanism that activation of Erk1/2 induces the
phosphorylation of Bim, leads Bim to degradation via the proteasome
and results in the protection against cell death (19,20).
We also examined MMP-9 expression to determine the
effect of butyrate on invasiveness. MMP-9 expression was increased
by acute butyrate treatment for 72 h. In contrast, MMP-9 expression
in HCT116/BR cells was negligible and comparable to that of
HCT116/PT cells. The mechanism underlying the increased MMP-9
expression in HCT116/AT cells was unclear; however, MMP-9
expression was reported to be increased in the HCT15 colon cancer
cell line by treatment with 1 mM butyrate for 5 days (21). Although short-term butyrate
treatment results in upregulation of MMP-9 expression, it was
gradually decreased to the level of parental cells by continuous
exposure to butyrate. This was confirmed by a wound-healing assay,
in which no significant differences were observed between HCT116/PT
and HCT116/BR cells. Therefore, chronic butyrate treatment did not
have a marked effect on cell invasiveness and migration, despite
the fact that acute butyrate treatment influenced MMP-9
expression.
Collectively, we assessed butyrate resistance and
noted several similarities between HCT116/PT and HCT116/BR cells.
Following acute butyrate treatment, the proportion of cells in
G2 phase was highly increased, but recovered to the
level of the control in HCT116/BR cells. However, there were
several differences between HCT116/BR compared to HCT116/PT cells.
HCT116/BR cells exhibited considerable chemoresistance, which was
not mediated by drug efflux pumps. Resistance to cell cycle arrest
and apoptosis induced by butyrate in HCT116/BR cells was related to
the regulation of Bcl-2 family proteins and cyclin D1 as well as
p21Waf1/Cip1.
Acknowledgments
This study was supported by Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Science, ICT and future Planning
(NRF-2015R1A2A2A01007546).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gaschott T and Stein J: Short-chain fatty
acids and colon cancer cells: The vitamin D receptor - butyrate
connection. Recent Results Cancer Res. 164:247–257. 2003.
View Article : Google Scholar
|
|
3
|
Vaziri C, Stice L and Faller DV:
Butyrate-induced G1 arrest results from p21-independent disruption
of retinoblastoma protein-mediated signals. Cell Growth Differ.
9:465–474. 1998.PubMed/NCBI
|
|
4
|
Ruemmele FM, Schwartz S, Seidman EG,
Dionne S, Levy E and Lentze MJ: Butyrate induced Caco-2 cell
apoptosis is mediated via the mitochondrial pathway. Gut.
52:94–100. 2003. View Article : Google Scholar
|
|
5
|
Guilloteau P, Martin L, Eeckhaut V,
Ducatelle R, Zabielski R and Van Immerseel F: From the gut to the
peripheral tissues: The multiple effects of butyrate. Nutr Res Rev.
23:366–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Donohoe DR, Collins LB, Wali A, Bigler R,
Sun W and Bultman SJ: The Warburg effect dictates the mechanism of
butyrate-mediated histone acetylation and cell proliferation. Mol
Cell. 48:612–626. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
López de Silanes I, Olmo N, Turnay J,
González de Buitrago G, Pérez-Ramos P, Guzmán-Aránguez A,
García-Díez M, Lecona E, Gorospe M and Lizarbe MA: Acquisition of
resistance to butyrate enhances survival after stress and induces
malignancy of human colon carcinoma cells. Cancer Res.
64:4593–4600. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leslie EM, Deeley RG and Cole SP:
Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2,
and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol.
204:216–237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lepiarczyk M, Kałuża Z, Bielawska A,
Czarnomysy R, Gornowicz A and Bielawski K: Cytotoxic activity of
octahydropyrazin[2,1-a:5,4-a′]diisoquinoline derivatives in human
breast cancer cells. Arch Pharm Res. 38:628–641. 2015. View Article : Google Scholar
|
|
10
|
Han YS, Lee JH and Lee SH: Antitumor
effects of fucoidan on human colon cancer cells via activation of
Akt signaling. Biomol Ther (Seoul). 23:225–232. 2015. View Article : Google Scholar
|
|
11
|
Lim SJ, Choi HG, Jeon CK and Kim SH:
Increased chemoresistance to paclitaxel in the MCF10AT series of
human breast epithelial cancer cells. Oncol Rep. 33:2023–2030.
2015.PubMed/NCBI
|
|
12
|
Song HM, Park GH, Eo HJ, Lee JW, Kim MK,
Lee JR, Lee MH, Koo JS and Jeong JB: Anti-proliferative effect of
naringenin through p38-dependent downregulation of cyclin D1 in
human colorectal cancer cells. Biomol Ther (Seoul). 23:339–344.
2015. View Article : Google Scholar
|
|
13
|
Zhang R, Pan X, Huang Z, Weber GF and
Zhang G: Osteopontin enhances the expression and activity of MMP-2
via the SDF-1/CXCR4 axis in hepatocellular carcinoma cell lines.
PLoS One. 6:e238312011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cook KL, Shajahan AN, Wärri A, Jin L,
Hilakivi-Clarke LA and Clarke R: Glucose-regulated protein 78
controls cross-talk between apoptosis and autophagy to determine
antiestrogen responsiveness. Cancer Res. 72:3337–3349. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gonçalves P and Martel F: Butyrate and
colorectal cancer: The role of butyrate transport. Curr Drug Metab.
14:994–1008. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barrasa JI, Santiago-Gómez A, Olmo N,
Lizarbe MA and Turnay J: Resistance to butyrate impairs bile
acid-induced apoptosis in human colon adenocarcinoma cells via
up-regulation of Bcl-2 and inactivation of Bax. Biochim Biophys
Acta. 1823:2201–2209. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Derjuga A, Richard C, Crosato M, Wright
PS, Chalifour L, Valdez J, Barraso A, Crissman HA, Nishioka W,
Bradbury EM, et al: Expression of p21Waf1/Cip1 and cyclin D1 is
increased in butyrate-resistant HeLa cells. J Biol Chem.
276:37815–37820. 2001.PubMed/NCBI
|
|
18
|
Zhang Y, Wang Z, Ahmed F, Banerjee S, Li Y
and Sarkar FH: Down-regulation of Jagged-1 induces cell growth
inhibition and S phase arrest in prostate cancer cells. Int J
Cancer. 119:2071–2077. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ley R, Balmanno K, Hadfield K, Weston C
and Cook SJ: Activation of the ERK1/2 signaling pathway promotes
phosphor-ylation and proteasome-dependent degradation of the
BH3-only protein, Bim. J Biol Chem. 278:18811–18816. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luciano F, Jacquel A, Colosetti P, Herrant
M, Cagnol S, Pages G and Auberger P: Phosphorylation of Bim-EL by
Erk1/2 on serine 69 promotes its degradation via the proteasome
pathway and regulates its proapoptotic function. Oncogene.
22:6785–6793. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Serpa J, Caiado F, Carvalho T, Torre C,
Gonçalves LG, Casalou C, Lamosa P, Rodrigues M, Zhu Z, Lam EWF, et
al: Butyrate-rich colonic microenvironment is a relevant selection
factor for metabolically adapted tumor cells. J Biol Chem.
285:39211–39223. 2010. View Article : Google Scholar : PubMed/NCBI
|