Introduction
Breast cancer is the most frequently diagnosed
cancer in women around the world (1). Although the mortality has declined
over the two decades mainly due to the deeper understanding of its
biology and advances in management approaches, it is still the most
life-threatening cancer in women, and, even worse, the incidence
rate is increasing gradually (2).
Through gene expression profiling, several intrinsic breast cancer
subtypes have been identified. Triple-negative breast cancer
(TNBC), one of the subtypes, accounting for 10–15% of invasive
breast cancers, is characterized by the absence of estrogen
receptor and progesterone receptor and no overexpression of human
epidermal growth factor receptor 2 (HER2) (3). Therefore, patients with TNBC cannot be
treated with endocrine therapy or therapies targeted to HER2. As a
group, they have a worse prognosis and tend to relapse early
compared with other subtypes of breast cancers (4). Hence, there is a compelling need to
find more effective treatments. It has been reported that the
overexpression of epidermal growth factor receptor (EGFR) was seen
in ~80% of TNBC (5,6). This discovery led to the investigation
of the EGFR inhibitors (EGFRI). However, both the anti-EGFR
monoclonal antibody cetuximab and the small molecular tyrosine
kinase inhibitors (TKIs) gefitinib and erlotinib seem to be
ineffective according to phase II studies (7-9). Thus,
more studies are needed to answer the question of why EGFR
inhibitors failed in treatment of those EGFR overexpressed breast
cancers.
TAZ (transcriptional coactivator with PDZ-binding
domain; also known as WWTR1) and its paralog YAP (YES associated
protein) are the two main downstream effectors of the Hippo
signaling pathway, which plays a major role in organ size control,
cell differentiation, and tumorigenesis across species (10). TAZ is preferentially overexpressed
in highly invasive breast cancer cells, most of which belong to
TNBC cell lines (11). In addition,
it has been reported that overexpression of TAZ induced the
activation of EGFR signaling, and one of the EGFR ligands,
amphiregulin (AREG), is a target of TAZ. AREG functions in a
non-cell-autonomous manner to mediate EGF-independent growth and
malignant behavior of mammary epithelial cells (12). These studies suggest that the high
expression of TAZ may be one of the reasons that TNBC was not
sensitive to EGFR inhibitors.
In this study, we successfully established two types
of TNBC cell lines with TAZ stably silenced. For the first time, we
observed TAZ gene silencing modified the drug sensitivity of breast
cancer cells to EGFRI in a cell context-depended manner. In
addition, also for the first time, we found YAP expression could be
upregulated both at mRNA and protein levels by TAZ inhibition in
certain breast cancer cell line, which leads to the EGFRI
resistance. These findings indicate that TAZ/YAP inhibition can
significantly improve EGFRI efficacy, which may pave the way for
TNBC therapy.
Materials and methods
Cell culture and antibodies
Human breast cancer cell lines (MCF-7, MDA-MB-231,
MDA-MB-468, and BT-549) were obtained from American Type Culture
Collection. These original cells were routinely cultured at 37°C in
the presence of 5% CO2 in RPMI-1640 supplemented with
10% fetal bovine serum (FBS, Hyclone). Cells in the exponential
growth phase were used for all experiments. The primary antibodies,
anti-YAP, anti-TAZ/YAP, and anti-GAPDH, were purchased from Cell
Signaling Technology Inc., and HRP-conjugated secondary antibodies
against rabbit from GE Amersham.
Cell lysates preparation and western blot
analysis
Briefly, cells were washed twice with
phosphate-buffered saline (PBS) and then lysed on ice in RIPA
buffer (50 mM Tris-HCl pH 7.4, 1% Nonidet P-40, 0.5% sodium
deoxycholate, 150 mM NaCl, 0.02% sodium azide, and 0.1% SDS)
containing protease and phosphatase inhibitors (Sigma-Aldrich, St.
Louis, MO, USA) for 15 min and cleared of debris by centrifugation
at 12,000 rpm for 15 min at 4°C. After boiling with an equal volume
of 2X SDS loading buffer for 5 min, cell lysates were
electrophoresed with 10% SDS-PAGE and blotted to PVDF membranes
(Millipore). The membranes were blotted with 5% non-fat milk in
TBS-T (10 mmol/l Tris-HCl pH 7.5, 0.5 mol/l NaCl, and 0.05% w/v
Tween-20) buffer at room temperature for 1 h, and then incubated
with primary antibodies overnight at 4°C. The membranes were washed
and then incubated with suitable peroxidase conjugated secondary
antibodies for 1 h at room temperature. After washing three times
with TBS-T, antibody binding was visualized using chemiluminescence
detection system as described by the manufacturer (Millipore).
Molecular weights of the immunoreactive proteins were estimated
based on PageRuler Prestained Protein ladder (MBI, Fermentas).
Experiments were repeated at least three times.
RNA purification and quantitative reverse
transcriptase-PCR (RT-qPCR)
Total RNA was extracted using TRIzol reagent
according to the protocol provided by the manufacturer
(Invitrogen). RNA concentrations were quantified by NanoDrop 2000
(Nanodrop). Reverse transcription reaction was performed using 2
µg of total RNA with Reverse Transcription System (Promega).
The mRNA levels of TAZ and YAP were analyzed using SYBR-Green qPCR
Master Mix kit (Promega) in ABI PRISM 7500 fast Sequence Detection
System (Applied Biosystems). The real-time qPCR reaction was
carried out in triplicate for each sample. The GAPDH gene was used
as an endogenous control for normalization and the mRNA levels of
TAZ and YAP were determined using the 2−ΔΔCt methods
(13). Specific primer pairs are
listed in Table I.
 | Table ISequences of primers for real-time
qPCR. |
Table I
Sequences of primers for real-time
qPCR.
| Name | Primer sequences |
|---|
| GAPDH | F:
TGATGACATCAAGAAGGTGGTGAAG |
| R:
TCCTTGGAGGCCATGTGGGCCAT |
| TAZ | F:
CAGCAATGTGGATGAGATGG |
| R:
AAGGAGGGAGCACGAGTCA |
| YAP | F:
GGAACACTGGAAGGAGATGG |
| R:
AGCAATGGACAAGGAAGAGC |
siRNA transfection and lentivirus
infection
The small interfering RNAs (siRNAs) against TAZ and
YAP were designed and synthesized by GenePharma Inc. (GenePharma)
and transfection was done with Lipofectamine™ RNAiMAX (Life
Technologies) regent according to the manufacturer's protocol. The
regions of the TAZ mRNA (GenBank accession no. NM_001168278) and
YAP mRNA (GenBank accession no. NM_006106) were selected as the
RNAi target sites. The RNA interfering sequences were
5′-CCGUUUCCCUGAUUUCCUUTT-3′ (sense) for si-TAZ and
5′-GGUGAUACUAUCAACCAAATT-3′ (sense) for si-YAP. BLAST analysis
shows no homology of the siRNA sequences to any other sequences in
the Human Genome Database. Scrambled siRNA as negative control was
also obtained from GenePharma.
To establish stable TAZ knockdown cells
(BT-549/sh-TAZ; MDA-MB-231/sh-TAZ), biologically active short
hairpin RNA (shRNA) was subcloned into lentiviral vector
hU6-MCS-Ubiquitin-EGFP-IRES-puromycin (GV248, Genechem), which
carried the transgene for green fluorescent protein (GFP), and used
to infect BT-549 and MDA-MB-231 cells. The shRNA target sequence
was the same target sequence of the si-TAZ mentioned above. Breast
cancer cells were incubated with viral supernatants for 24 h and
then returned to normal growth medium. For confirmation of
downregulation of TAZ gene, after 72 h cells were harvested and
analyzed for reduction of TAZ expression by real-time RT-qPCR and
western blot analysis.
Flow cytometry assay
Apoptosis induction in siRNA treated cells was
assayed by the detection of membrane externalization of
phosphatidylserine using an Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) apoptosis assay kit (KeyGen Biotech).
At 72 h after transfection, cells were harvested and washed with
ice-cold PBS twice and resuspended in 500 µl of binding
buffer, 5 µl Annexin v-FITC and 5 µl PI were added,
and then cells were incubated for 15 min in the dark. Finally, the
cells were analyzed within 1 h by flow cytometry.
Cell proliferation assay
Cell proliferation was investigated by colorimetric
assay using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium
bromide (MTT). In brief, breast cancer cells were seeded in a
96-well flat-bottomed plate at 5×103 cells/well in
triplicate. At each experimental point, 0.5 mg/ml MTT
(Sigma-Aldrich) was added into the medium and cells were cultured
for additional 4 h. Afterwards, the supernatant was removed and the
formazan crystals were dissolved in 150 ml dimethyl sulfoxide
(DMSO) at room temperature for 15 min. Absorbance of the solution
was then measured at 490 nm wavelength using an ELx800 Absorbance
Microplate Reader (Biotek). The experiments were performed
independently in triplicate.
Chemosensitivity assay
EGFR inhibitor, gefitinib or AG-1478 (Selleckchem)
was initially dissolved in DMSO, which was diluted with fresh
medium immediately before each experiment. Briefly, BT-549/sh-TAZ
or MDA-MB-231/sh-TAZ cells were seeded in a 96-well plate at
5×103 cells/well in triplicate and incubated for 24 h.
Then the medium was moved and replaced with the fresh medium
containing the drugs with different concentrations. After
incubation for another 48 h, the cell viability was examined by the
MTT assay.
Colony formation assay
Log-phase cells were seeded in triplicate onto
6-well plates with 2 ml of complete media (500 cells/well) and
incubated at 37°C in a humidified incubator. Every week the medium
was replaced with fresh medium. After 3 weeks, the colonies were
fixed with 100% methanol, stained with 0.1% crystal violet, and
washed with PBS. The numbers of colonies were counted using a light
microscope.
Statistical analysis
The results were expressed as mean ± standard
deviation. Statistical significance was assessed by Student's
t-test or one-way ANOVA followed by Bonferroni multiple comparison
post-tests. Statistical analyzes were performed using GraphPad
Prism v.5.0 package. Differences were considered statistically
significant at a level of P<0.05 (shown as
*P<0.05; **P<0.01 in the figures).
Results
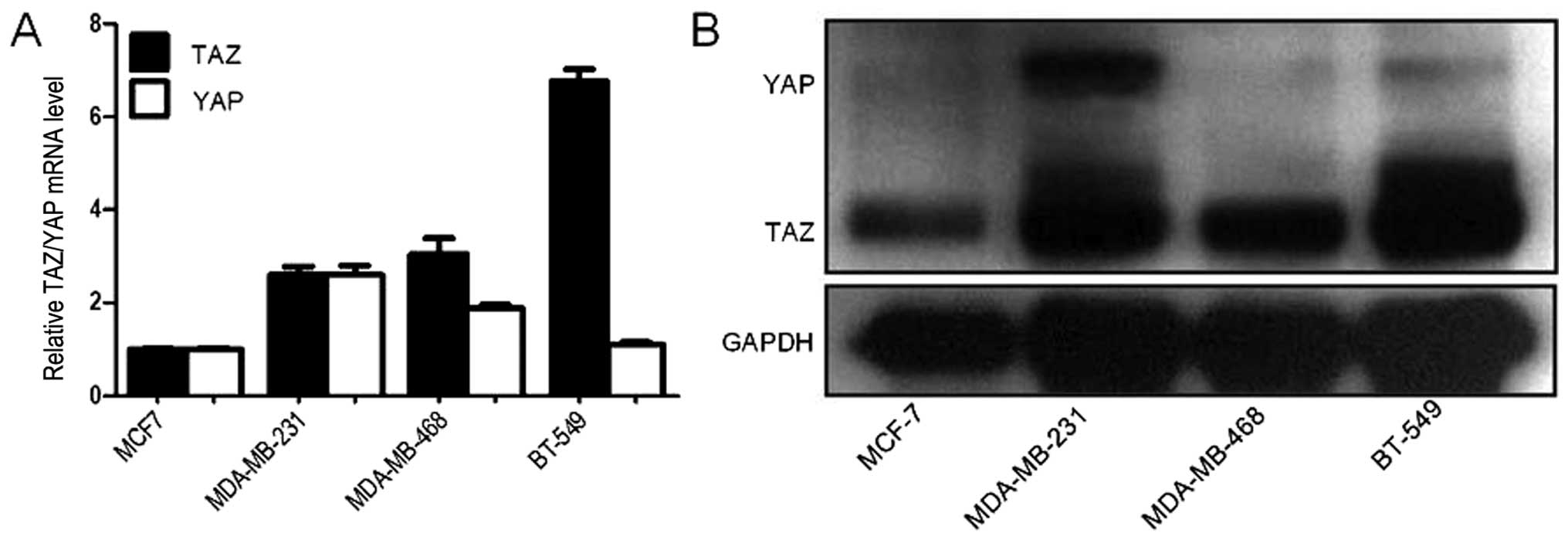
TAZ expression was upregulated in
triple-negative breast cancer cells
The expression of TAZ and YAP was examined by
western blotting and RT-qPCR in 4 human breast cancer cell lines.
TAZ is preferentially overexpressed in TNBC cells (BT-549,
MDA-MB-468, and MDA-MB-231); however, high expression level of YAP
was only seen in MDA-MB-231 cells (Fig.
1). These results are consistent with a previous study
(14). TAZ is comparably highly
expressed in TNBC cells suggesting that it may be correlated with
certain characteristics of TNBC (15).
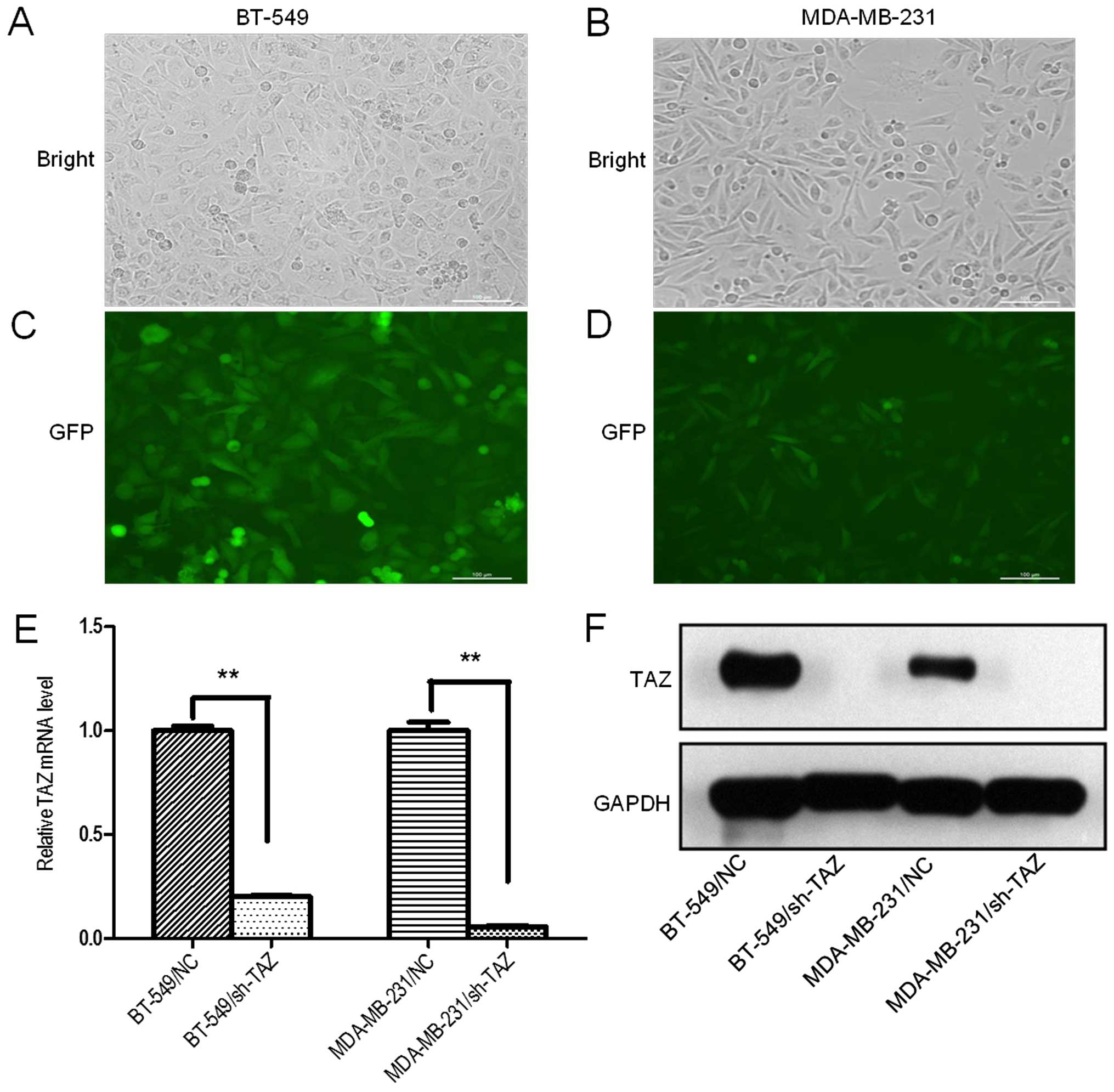
sh-TAZ regulates breast cancer cell
sensitivity to EGFRI gefitinib and AG-1478
In order to establish breast cancer cells with TAZ
stably silenced, we successfully constructed a lentivirus vector
harboring shRNA against TAZ. We chose TAZ overexpressing cells,
BT-549 and MDA-MB-231, for TAZ knockdown. The knockdown efficiency
was evaluated using western blotting and RT-qPCR. The results
disclosed that the best knockdown effect was with shRNA at
multiplicity of infection (MOI) 30 both in BT-549 and MDA-MB-231
cells. After 72 h post-transfection, >90% of the survived cells
were GFP-positive (Fig. 2A–D).
RT-qPCR analyses showed that TAZ mRNA levels were significantly
reduced when compared with corresponding negative control
transfection (Fig. 2E). To
correlate the decreases in TAZ mRNA expression with TAZ protein
levels, western blot analysis was performed at 72 h after shRNA
silencing and showed that TAZ protein levels were also reduced,
thereby confirming efficient knockdown (Fig. 2F).
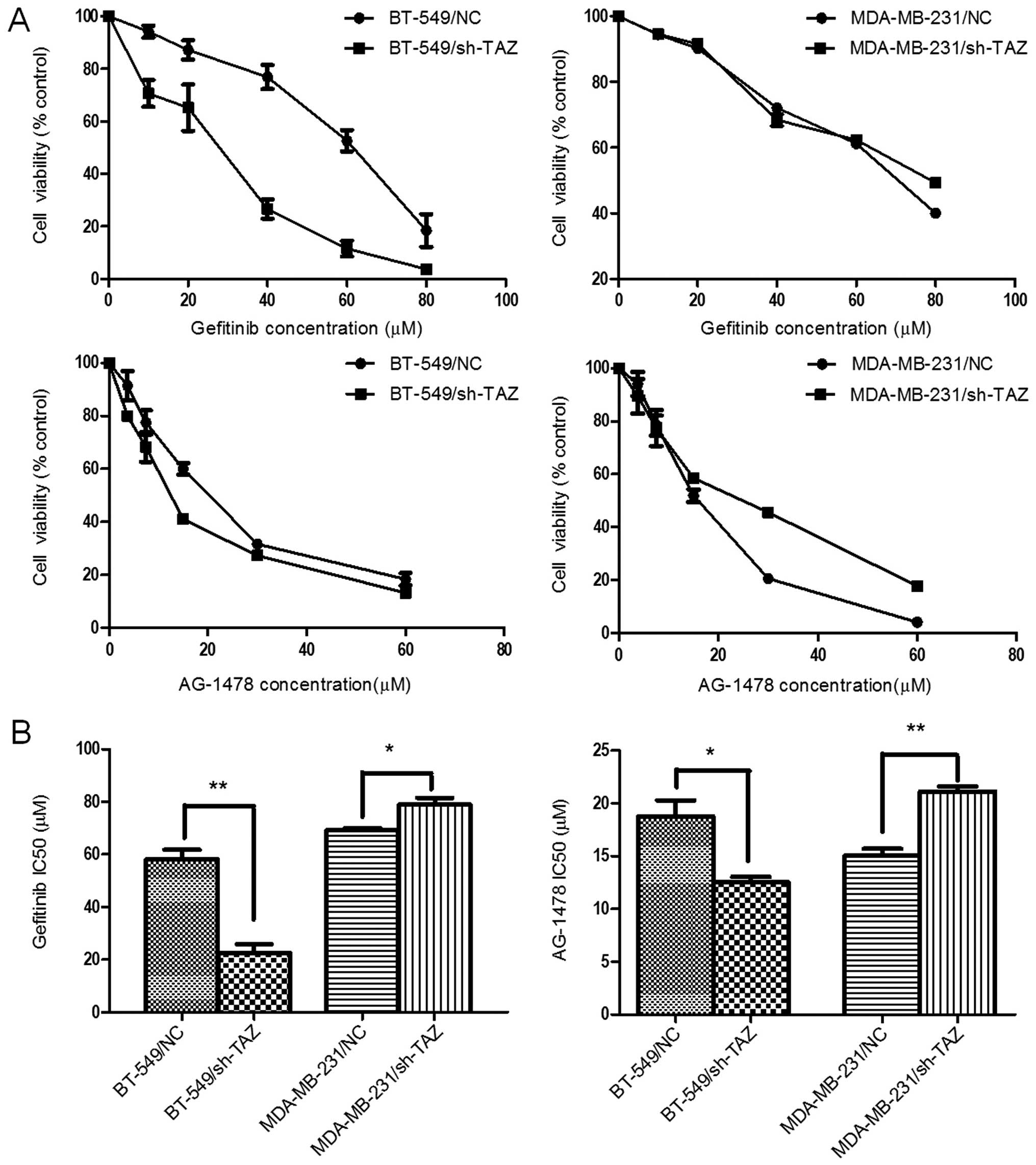
To investigate the effect of sh-TAZ on EGFRI
sensitivity in the breast cancer cells, we treated TAZ shRNA or
negative control (NC) shRNA transfected cells with gefitinib or
AG-1478 separately and the cell viability curves are shown in
Fig. 3A. The silencing of TAZ
expression in BT-549 cells resulted in strikingly higher cell
growth inhibition at different drug concentrations, with the
IC50 of gefitinib being 22.65±3.28 µM in
BT-549/sh-TAZ cells, significantly lower than 58.19±3.58 µM
in BT-549/NC cells (P=0.002). The same trend was found in AG-1478
treatment, with the IC50 reduced from 18.76±1.52 to
12.52±0.53 µM (P=0.018). In contrast, shRNA-TAZ in
MDA-MB-231 cells led to EGFRI resistance, with the IC50
values of gefitinib and AG-1478 in MDA-MB-231/NC cells being
69.27±0.86 and 15.02±0.68 µM, respectively, significantly
lower than 79.01±2.54 (P=0.022) and 21.14±0.49 µM (P=0.002)
in MDA-MB-231/sh-TAZ cells (Fig.
3B). These results suggest that TAZ inhibition by lentiviral
shRNA resulted in enhancement in chemosensitivity of EGFRI in
BT-549 cells, but was diminished in MDA-MB-231 cells.
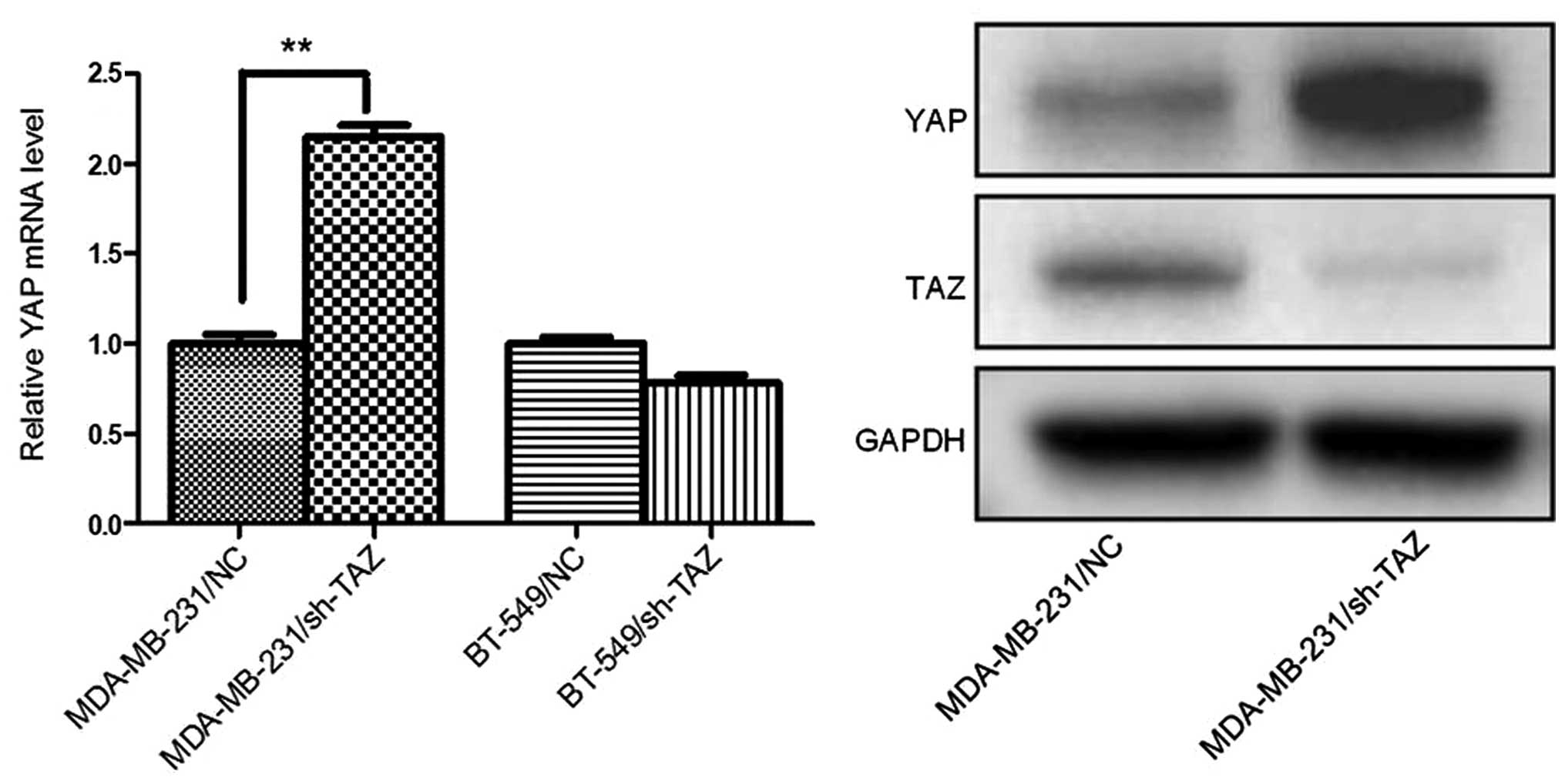
The sh-TAZ led EGFRI resistance in
MDA-MB-231 cells was mediated by upregulation of YAP
expression
To understand the underlying mechanism of the effect
of sh-TAZ on EGFRI sensitivity in the breast cancer cells, we
further analyzed the expression change of YAP before and after TAZ
knockdown. As shown in Fig. 1, YAP
expression in MDA-MB-231 cells was higher than that in MCF-7, but
was slight or absent in BT-549 cells. In addition, after TAZ
knockdown, the expression of YAP in MDA-MB-231/sh-TAZ cells was
markedly elevated both in mRNA and protein levels, which was not
seen in BT-549/sh-TAZ cells (Fig.
4).
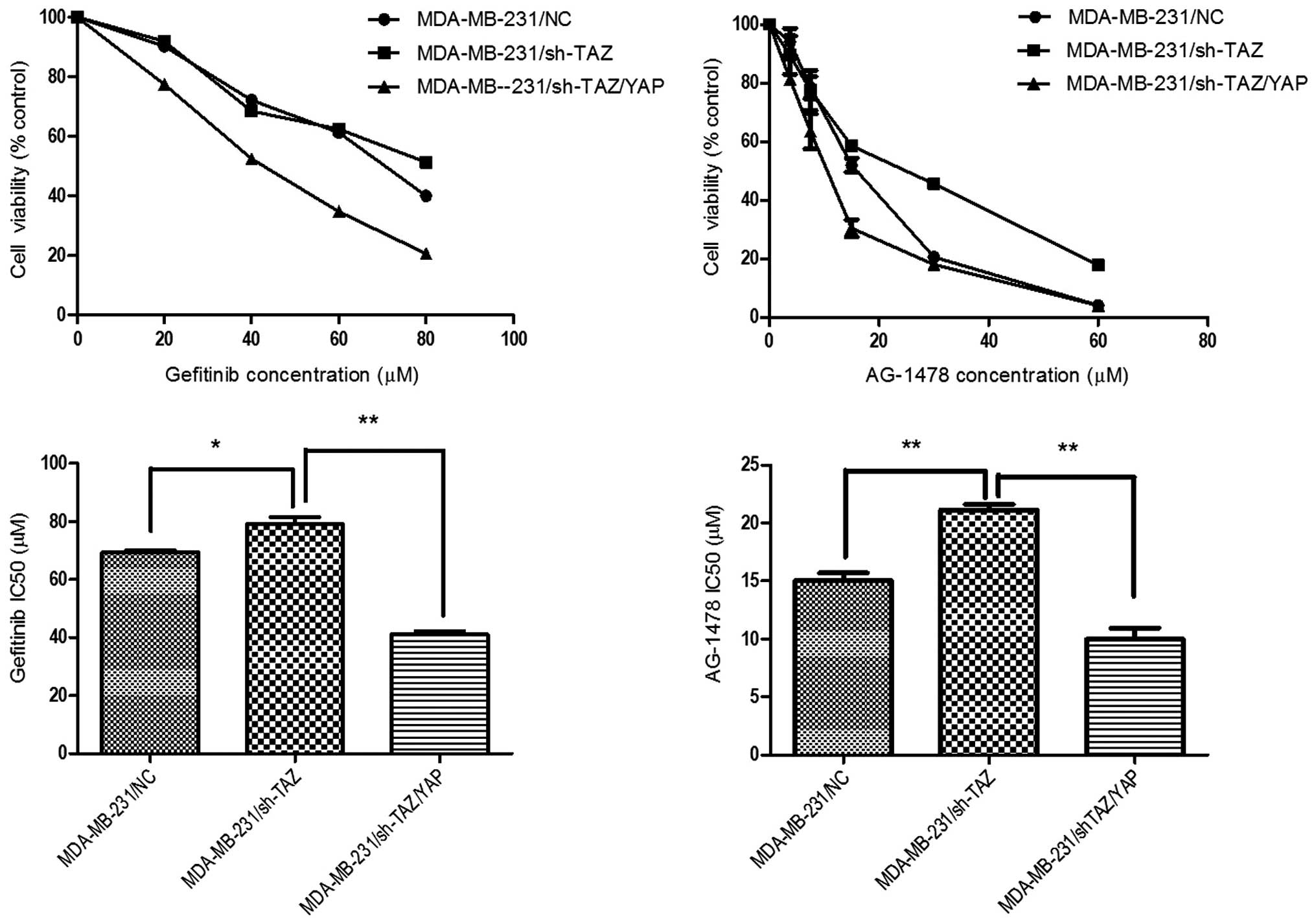
In order to study the relationship between YAP
expression and EGFRI resistance, we further knocked down YAP in
MDA-MB-231/sh-TAZ cells by YAP specific siRNA, and investigated the
changes of IC50. The results show that IC50
values of gefitinib and AG-1478 were 79.01±2.54 and 21.14±0.49
µM for MDA-MB-231/sh-TAZ cells, respectively, but
IC50 values in TAZ/YAP co-knockdown MDA-MB-231 cells
declined to 41.02±1.26 (P<0.01) and 9.98±0.96 µM
(P<0.01), respectively (Fig. 5).
These results suggest that compared with the MDA-MB-231/sh-TAZ
cells, the TAZ/YAP co-knockdown MDA-MB-231 cells restored the EGFRI
sensitivity.
TAZ inhibition affects apoptosis and
proliferation of breast cancer cells
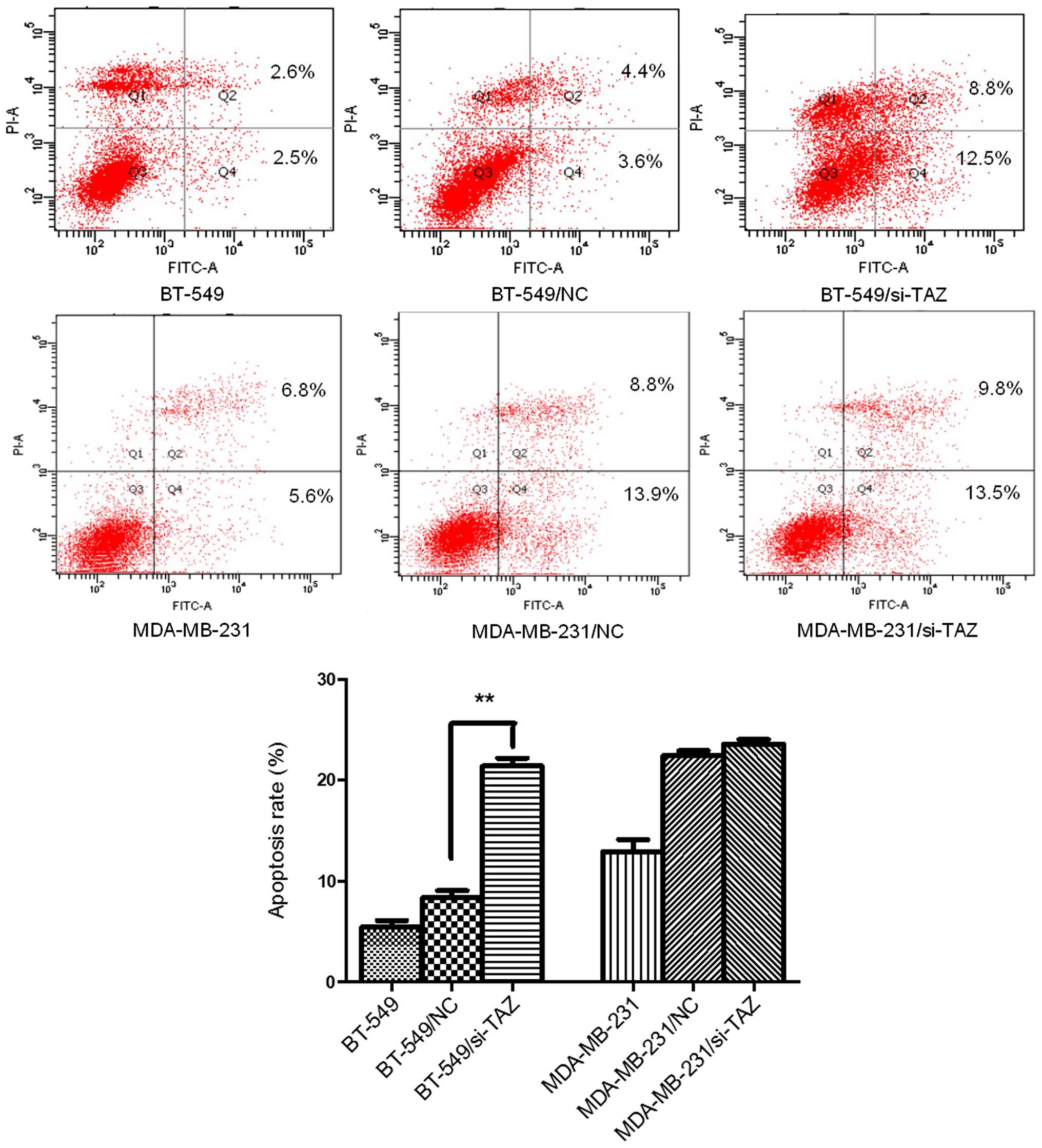
To determine whether inhibition of TAZ affected
apoptosis and proliferation in breast cancer cells, we performed
flow cytometry, cell proliferation curve, and colony forming
assays. Flow cytometry showed the cellular apoptosis was
significantly increased in the BT-549 cells transfected with TAZ
specific siRNA (BT-549/si-TAZ) compared with control, while no
significant change was seen in MDA-MB-231/si-TAZ cells, suggesting
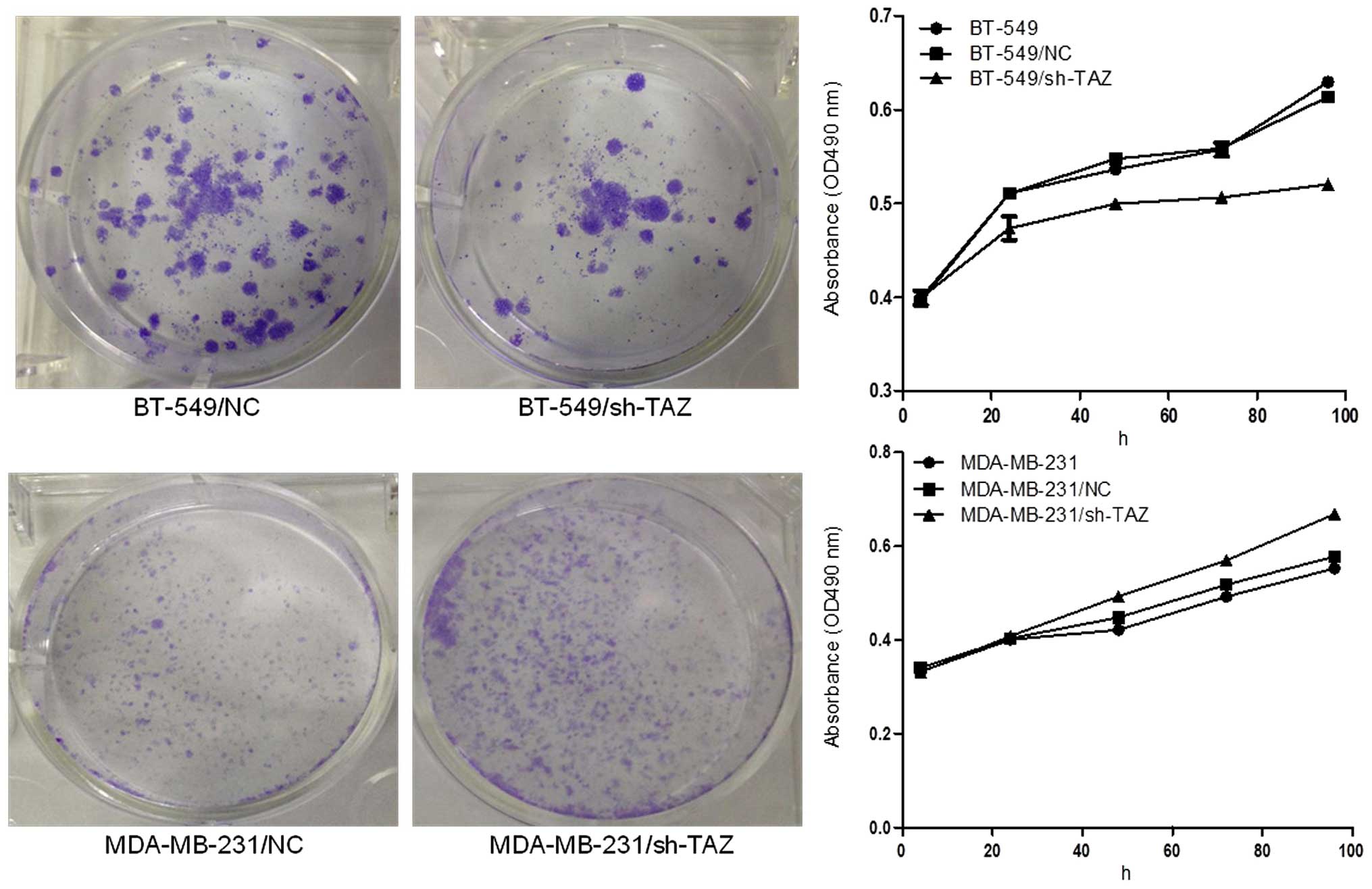
that si-TAZ induced spontaneous apoptosis in BT-549 cells (Fig. 6). Similarly, cell proliferation
curves by MTT assay show that silencing of TAZ gene substantially
affected BT-549 cells on proliferation compared with control;
however, silencing of TAZ gene slightly promoted cell proliferation
in MDA-MB-231 cells (Fig. 7). The
colony forming efficiency of the transfected cells was investigated
by colony forming assay, and the results show that TAZ-shRNA
transfected BT-549 cells had significantly fewer colonies than
control, while, on the contrary, MDA-MB-231/sh-TAZ cells showed
more colonies than control (Fig.
7).
Discussion
The Hippo signaling pathway is a newly discovered
and evolutionally conserved signal cascade, which plays a pivotal
role in regulating organ size, stem cell pluripotency, and
tumorigenesis from Drosophila to mammals (15). Mechanically, when the main
downstream effectors TAZ or YAP translocated into the nucleus, they
will act as transcription coactivators to promote
proliferation-associated gene expression (16). Despite highly conserved sequence and
domain organization, TAZ and YAP have their own specific
transcription factor partners, and some researchers believe they
can hardly compensate each other (17).
Indeed, it has been reported that TAZ expression
level was increased in a broad range of different human cancers,
such as colorectal, breast, and lung cancers. Moreover, the higher
TAZ protein level is always associated with poorly differentiated
tumors and shorter patient overall survival (18). In vitro experiments
demonstrate that downregulation of TAZ expression not only reduced
cancer cell migration and invasion, but also inhibited
tumorigenesis in nude mice, while upregulation was able to induce
cell malignant transformation (11). Therefore, TAZ is proposed as an
oncogene and a potentially attractive therapeutic target for cancer
treatment (19).
We have noted that TAZ can induce growth factors to
promote independent proliferation of breast cancer cells through
activation of its transcription target EGFR ligand AREG, and
expression of TAZ and EGFR is positively correlated with the
invasiveness of breast cancer cell lines (12). These observations implicate the
potential benefit of TAZ knockdown on EGFR targeted therapy.
However, no published studies exist focused on this issue in the
PubMed, although TAZ mediated Taxol resistance in breast cancer
cells have been reported (20).
In this study, we investigated the therapeutic
effects of EGFRI on human breast cancer cells overexpressing TAZ.
Gefitinib, an EGFRI, has been shown to be highly effective in
clinical treatment of certain pathological types of lung cancer,
and AG-1478, which is a highly selective EGFRI, has almost no
activity on HER2, platelet-derived growth factor receptor (PDGFR),
tyrosine kinase receptor (Trk), Bcr-abl or insulin receptor (InsR).
Interestingly, BT-549 and MDA-MB-231 cell lines showed different
responses to EGFRI treatment when TAZ was silenced, with an
increase in chemosensitivity of BT-549/sh-TAZ cells to EGFRI and a
decrease in MDA-MB-231/sh-TAZ cells.
Further research found that BT-549 cells expressed a
high level of TAZ, but a low level of YAP, and the level of YAP did
not increase after TAZ knockdown. By contrast, MDA-MB-231 cells
expressed both TAZ and YAP, and the level of YAP significantly
increased after TAZ knockdown. In order to identify whether the
EGFRI resistance of MDA-MB-231/sh-TAZ cells was caused by increased
expression of YAP, we further knocked down the YAP in
MDA-MB-231/sh-TAZ cells, and found that the resistance to EGFRI in
MDA-MB-231/sh-TAZ cells was reversed.
Previous studies have shown that TAZ may compensate
for the loss of YAP functions. Huang et al reported that
knockdown of YAP significantly increased EGFRI erlotinib
sensitivity in ovarian cancer cell lines that express little or no
TAZ (21). In addition, knockdown
of YAP in ovarian cancer cell lines that express both YAP and TAZ
only led to a very moderate effect on cancer cell growth or drug
sensitivity (22,23). Here, we have shown that knockdown of
TAZ in breast cancer cell lines that express little or no YAP, such
as BT-549 cells, increased EGFRI sensitivity; however, for breast
cancer cell lines that express both YAP and TAZ, such as MDA-MB-231
cells, knockdown of TAZ may not help improve sensitivity of EGFRI
treatment. These findings indicate YAP may also compensate for the
loss of TAZ functions. Therefore, simultaneous inhibition of the
functions of TAZ and YAP is needed in some cases.
In conclusion, this study highlights the potential
for TAZ to be a therapeutic target in breast cancers, as reducing
TAZ levels can partially revert resistance to EGFR inhibitors. In
addition, for the first time, we found upregulation of YAP could be
induced by TAZ inhibition in a certain breast cancer cell line,
which leads to EGFRI resistance. For patients with high expression
of both TAZ and YAP, anti-YAP drugs need to be added. Therefore, we
propose to develop new therapeutic agents that can simultaneously
target TAZ and YAP. We believe that a specific inhibitor to TAZ/YAP
combined with anti-EGFR therapy may improve the therapeutic
efficacy in TNBC treatment.
Acknowledgments
This study was supported in part by the National
Natural Science Foundation of China for the youth (no. 81301809),
and the Cultivation of High-level Innovation Health Talents of
Zhejiang (grant no. 2012-241).
References
|
1
|
Boyle P and Howell A: The globalisation of
breast cancer. Breast Cancer Res. 12(Suppl 4): S72010. View Article : Google Scholar :
|
|
2
|
Youlden DR, Cramb SM, Yip CH and Baade PD:
Incidence and mortality of female breast cancer in the Asia-Pacific
region. Cancer Biol Med. 11:101–115. 2014.PubMed/NCBI
|
|
3
|
Brenton JD, Carey LA, Ahmed AA and Caldas
C: Molecular classification and molecular forecasting of breast
cancer: Ready for clinical application? J Clin Oncol. 23:7350–7360.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Criscitiello C, Azim HA Jr, Schouten PC,
Linn SC and Sotiriou C: Understanding the biology of
triple-negative breast cancer. Ann Oncol. 23(Suppl 6): vi13–vi18.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De Laurentiis M, Cianniello D, Caputo R,
Stanzione B, Arpino G, Cinieri S, Lorusso V and De Placido S:
Treatment of triple negative breast cancer (TNBC): Current options
and future perspectives. Cancer Treat Rev. 36(Suppl 3): S80–S86.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Corkery B, Crown J, Clynes M and O'Donovan
N: Epidermal growth factor receptor as a potential therapeutic
target in triple-negative breast cancer. Ann Oncol. 20:862–867.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bernsdorf M, Ingvar C, Jörgensen L, Tuxen
MK, Jakobsen EH, Saetersdal A, Kimper-Karl ML, Kroman N, Balslev E
and Ejlertsen B: Effect of adding gefitinib to neoadjuvant
chemotherapy in estrogen receptor negative early breast cancer in a
randomized phase II trial. Breast Cancer Res Treat. 126:463–470.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carey LA, Rugo HS, Marcom PK, Mayer EL,
Esteva FJ, Ma CX, Liu MC, Storniolo AM, Rimawi MF, Forero-Torres A,
et al: TBCRC 001: Randomized phase II study of cetuximab in
combination with carboplatin in stage IV triple-negative breast
cancer. J Clin Oncol. 30:2615–2623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dickler MN, Cobleigh MA, Miller KD, Klein
PM and Winer EP: Efficacy and safety of erlotinib in patients with
locally advanced or metastatic breast cancer. Breast Cancer Res
Treat. 115:115–121. 2009. View Article : Google Scholar
|
|
10
|
Hong W and Guan KL: The YAP and TAZ
transcription co-activators: Key downstream effectors of the
mammalian Hippo pathway. Semin Cell Dev Biol. 23:785–793. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chan SW, Lim CJ, Guo K, Ng CP, Lee I,
Hunziker W, Zeng Q and Hong W: A role for TAZ in migration,
invasion, and tumorigenesis of breast cancer cells. Cancer Res.
68:2592–2598. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang N, Morrison CD, Liu P, Miecznikowski
J, Bshara W, Han S, Zhu Q, Omilian AR, Li X and Zhang J: TAZ
induces growth factor-independent proliferation through activation
of EGFR ligand amphiregulin. Cell Cycle. 11:2922–2930. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
14
|
Cordenonsi M, Zanconato F, Azzolin L,
Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR,
Poletti A, et al: The Hippo transducer TAZ confers cancer stem
cell-related traits on breast cancer cells. Cell. 147:759–772.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pan D: The hippo signaling pathway in
development and cancer. Dev Cell. 19:491–505. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Piccolo S, Cordenonsi M and Dupont S:
Molecular pathways: YAP and TAZ take center stage in organ growth
and tumorigenesis. Clin Cancer Res. 19:4925–4930. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang K, Degerny C, Xu M and Yang XJ: YAP,
TAZ, and Yorkie: A conserved family of signal-responsive
transcriptional coregulators in animal development and human
disease. Biochem Cell Biol. 87:77–91. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harvey KF, Zhang X and Thomas DM: The
Hippo pathway and human cancer. Nat Rev Cancer. 13:246–257. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Edgar BA: From cell structure to
transcription: Hippo forges a new path. Cell. 124:267–273. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lai D, Ho KC, Hao Y and Yang X: Taxol
resistance in breast cancer cells is mediated by the hippo pathway
component TAZ and its downstream transcriptional targets Cyr61 and
CTGF. Cancer Res. 71:2728–2738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang JM, Nagatomo I, Suzuki E, Mizuno T,
Kumagai T, Berezov A, Zhang H, Karlan B, Greene MI and Wang Q: YAP
modifies cancer cell sensitivity to EGFR and survivin inhibitors
and is negatively regulated by the non-receptor type protein
tyrosine phosphatase 14. Oncogene. 32:2220–2229. 2013. View Article : Google Scholar
|
|
22
|
Zhang X, George J, Deb S, Degoutin JL,
Takano EA, Fox SB, Bowtell DD and Harvey KF; AOCS Study group: The
Hippo pathway transcriptional co-activator, YAP, is an ovarian
cancer oncogene. Oncogene. 30:2810–2822. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hall CA, Wang R, Miao J, Oliva E, Shen X,
Wheeler T, Hilsenbeck SG, Orsulic S and Goode S: Hippo pathway
effector Yap is an ovarian cancer oncogene. Cancer Res.
70:8517–8525. 2010. View Article : Google Scholar : PubMed/NCBI
|