1. Introduction
Endocrine disruptors (EDs) are exogenous chemicals
that can interfere with any aspect of hormone action; therefore,
they can disrupt normal mammary and female genitalia development,
function and carcinogenesis, especially when exposure occurs during
early life (1).
The mechanisms by which EDs can also modify the risk
of hormone-sensitive female neoplasms, such as breast or
endometrial cancers, are multiple, interacting and complex. They
can act at very low doses, with non-linear dose response curves
(2). They can affect the synthesis
of natural hormones, their release and/or transport. In target
tissues, EDs can reduce or increase the effects of natural hormones
on their receptors and/or they can change signaling cascades.
Hormone metabolism and changes in elimination can be the final part
of their indirect endocrine interference.
Carcinogens induce cancer or promote tumor growth by
mechanisms such as increased expression of oncogenes, decreased
expression of tumor suppressors, changes in expression of cell
cycle or apoptosis regulator genes (3). With the exception of
diethylstilboestrol (DES), EDs cannot be considered carcinogens
per se, but they can indirectly interfere with the endocrine
and immune systems favoring true carcinogenic effects (4).
Breast cancer is the most common cancer in women.
Mammary gland growth and function are influenced by multiple
endocrine-mediated mechanisms/pathways that may be altered by
thousands of EDs, mostly when they act during vulnerable periods,
such as embryogenesis or during breast maturation, from puberty to
the first full term pregnancy (5).
Currently available epidemiologic human data are
inadequate to support a conclusion concerning the association of
EDs and endometrial, tubal or ovarian cancer, mostly because they
are based on adult data, on professional or accidental high
dose/short term exposure while there are no data on newly
introduced environmental pollutants (6).
2. Literature search methods
We performed a review mainly based on Medline
search. Other databases were used to retrieve literature including
Scopus and Trip Database.
3. Endocrine disruptors and their effects on
the development of female tumors
The supposed carcinogenic mechanisms of the most
known EDs (pesticides, DDT, dioxins, phthalates, bisphenol A,
diethylstilbestrol, heavy metals) are here reviewed and summarized
in Figs. 1Figure 2Figure 3Figure 4Figure 5–6.
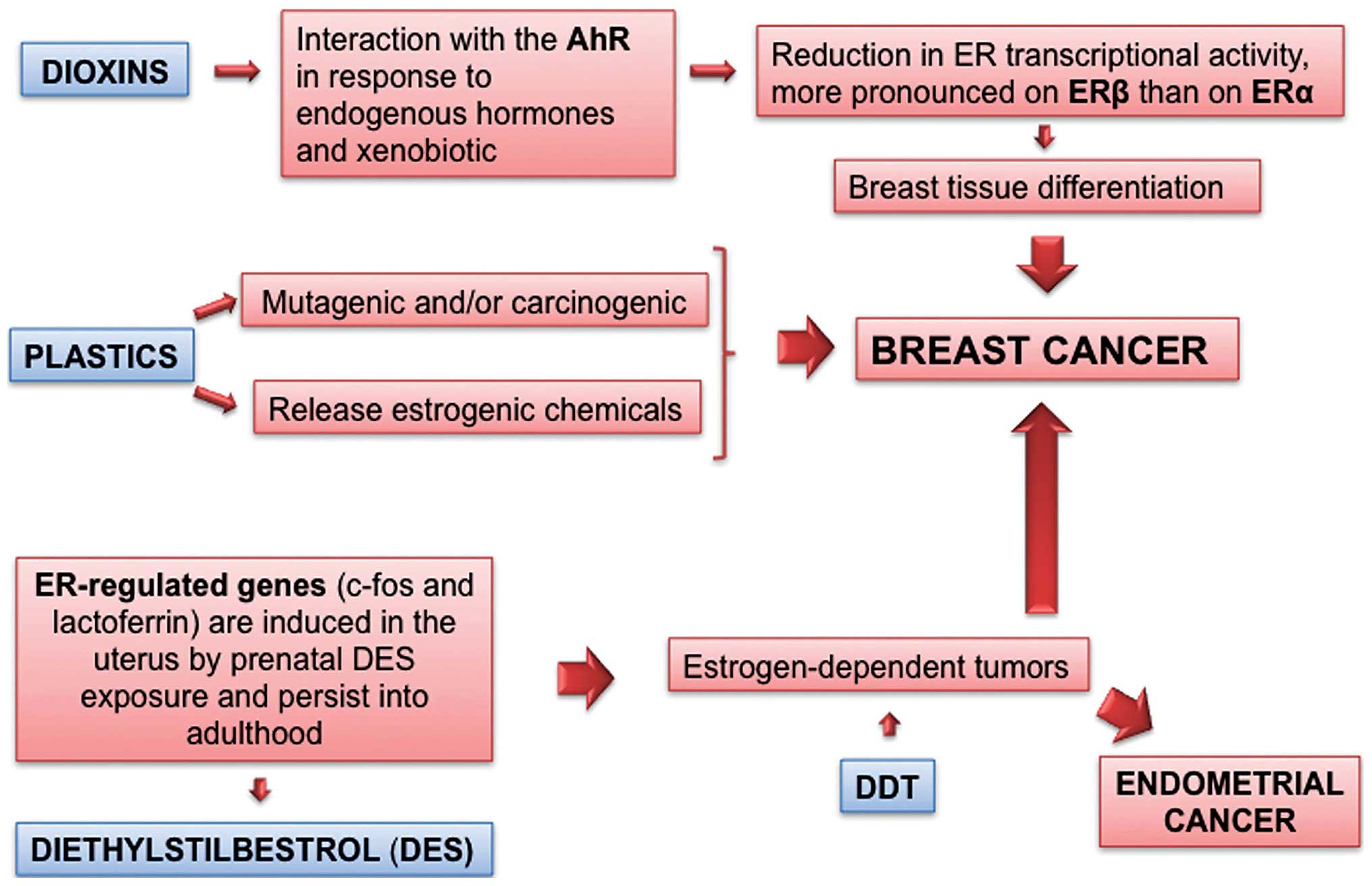
Dioxins
To date dioxins are the EDs most convincingly
associated with breast cancer in exposed humans. Dioxins are a
class of environmental chemicals exemplified by
2,3,7,8-tetra-chlorodibenzo-p-dioxin (TCDD). Dioxins and
dioxin-like compounds interact with specific polymorphisms to
enhance breast cancer risk (7)
(Fig. 1).
Dioxins act through the aryl hydrocarbon receptor
(AhR), a ligand-activated nuclear transcription factor,
intracellular mediator of xenobiotic signaling pathways. This
ligand-dependent transcription factor mediates a range of
biological and toxicological effects, all pivoting around the cell
responses to endogenous (e.g., endogenous hormones) and exogenous
(e.g., xenobiotic) challenges. Due to the common mechanisms and
occurrence in the same matrices, the intake and body burden of
dioxins are evaluated in a cumulative way, each compound
contributing to the toxic equivalent (TEQ) of a given mixture in
biological samples, its concentration and potency (expressed as AhR
activation); the dioxin TEQ is a rare example of a straightforward
use of in vitro cell biology data (AhR activation potency)
into risk assessment. Dioxins in females act as anti-estrogenic EDs
as AhR activation leads to a reduction in ER transcriptional
activity, which is more pronounced on ERβ than on ERα.
Interestingly, several studies have found an increased risk of
breast cancer associated with high body burdens of dioxin-like PCB
in conjunction with certain genetic polymorphisms involved in
carcinogen activation and steroid hormone metabolism (7).
A single oral dose of the most potent dioxin
compound (TCDD) to pregnant rats at term organogenesis and just
prior migration of the mammary bud into the fat pad (gestational
day 15) is sufficient to impair differentiation and increase
expression levels of ERα of terminal ductal structures in adult
rats, although the tissue retained the ability to differentiate in
response to estrogen. Mammary epithelial transplantation between
control and TCDD-exposed females suggests that the stroma plays a
major role in the TCDD-induced retarded development of the mammary
gland. In mice exposed in utero, the TCDD effect on breast
histogenesis was much more pronounced than the slight effects on
hormone levels, suggesting that, rather than affecting steroid
balance, dioxins act directly on breast tissue differentiation. Due
to the persistence and long half-life of TCDDs, in these studies,
the exposure of rodent conceptuses and pups likely continued beyond
the initial dosing and throughout lactation. This is supported by
in vitro findings on SCp2 mammary epithelial cells, where
TCDD reduced the expression of genes involved in cell adhesion and
milk secretion (8). Interestingly,
downregulation of superoxide dismutase was the main long-term
change in protein expression observed in mammary glands from rats
exposed in utero; thus, TCDD may render the mammary tissue
ill-equipped to deal with subsequent free radical exposure.
The Seveso Women's Health Study was comprised of 981
women, who were infants to 40-years of age in 1976, resided in the
most contaminated areas and had archived data that were collected
soon after the infamous chemical accident in 1976. In breast cancer
patients, individual TCDD body burden was significantly related
with tumor risk (9). The
pleiotropic effects of such a potent hormone trigger as TCDD was
also associated with an increased morbidity and mortality from
lymphohematopoietic and other neoplasms as well as markers of
altered endocrine-immune function and they were often
gender-related. In the Seveso industrial accident, TCDD exposure
appeared to reduce the risk of uterine cancer, but the number of
cases was too small for a comprehensive evaluation (10). Finally, one potentially relevant
effect was the clear link between exposure levels in men and a
lowered male/female gender ratio in their offspring (11).
DDT
DDT is a prototypal representative of persistent,
bioaccumulating contaminants of the food chain. Notwithstanding the
severe use restrictions since the 1970's and the banning in Europe
as a pesticide in 1986, residues are still found in feeds, foods
and in the fat deposits of living organisms, including humans.
Actually DDT is a mixture of DDT and DDE isomers, all able to
bioaccumulate and make up the body burden of DDT and related
compounds. The isomers p,p′-DDT and o,p′-DDE show
estrogenic activity both in vitro and in vivo,
whereas other isomers, such as the very persistent p,p′-DDE
(also a metabolite) are mainly antiandrogenic.
Long-term exposure to DDT through food could be
hypothesized to increase the risk for developing estrogen-dependent
tumors such as breast cancer. However literature is discordant
(12). In 1993 Wolff et al
(13) observed that the risk of
breast cancer was higher among women with high serum concentrations
of DDE, the major metabolite of DDT, compared with women with low
levels. Since then, a substantial number of epidemiologic studies
have investigated this hypothesis. In humans DDT/DDE body burden
has been associated with breast cancer (early, peripubertal
exposure) and with endometrial cancer [ongoing exposure (14)] (Fig.
1). The antiandrogenic p,p′-DDE may accelerate tumor
onset in mice and there is a trend for positive correlation of
breast cancer with DDE, which was not significant, due to low
statistical power (15).
A review article by Calle et al (16) and a meta-analysis (17) of the epidemiologic evidence for
tissue DDE concentrations and breast cancer, have not found an
association between breast cancer risk and ED body burden in
different scenarios. However, an important caveat to these studies
remains largely unexplored: the importance that age at exposure may
have in breast cancer development. Indeed there is a paucity of
evidence regarding exposure at critical time periods (17). Some studies (18–20)
have observed positive associations between insecticides and breast
cancer. The reasons for these discrepancies are difficult to
identify, but it may be plausible that these studies provide a
different focus on the role of contributing factors impinging on
breast cancer risk together, and in synergy, with organochlorines.
A finding worth mentioning has been observed in the US Agricultural
Health Study: breast cancer risk was elevated among women whose
husbands had used dieldrin (RR=2.0), a chlorinated insecticide
closely related to DDT, but not when used by the women themselves.
There was no clear association of breast cancer risk with farm size
or washing of clothes worn during pesticide application, but the
risk was modestly elevated among women whose homes were closest to
areas of pesticide application (21).
However, conclusions are somewhat different when
examining the risk related to the body burden resulting from the
dietary exposure in the general population.
Evidence from a prospective study of young women in
California who had their blood samples drawn in 1959–1967 found a
significantly increased risk of breast cancer with increasing
levels of serum p,p′-DDT. Women in the highest exposure
category had a 5-fold significant increase in risk (22). This was a nested case-control study
among a cohort of female members of the Kaiser Permanente Health
Plan in Oakland, CA and used stored blood samples collected to
assay for serum p,p′-DDT. The unique circumstances
surrounding the study have permitted the investigation of
early-life exposure to DDT and future breast cancer risk during a
time when DDT was actively being used in the United States. A
recent meta-analysis has not confirmed that current exposure levels
to DDT/DDE increases the risk of breast cancer in humans (23).
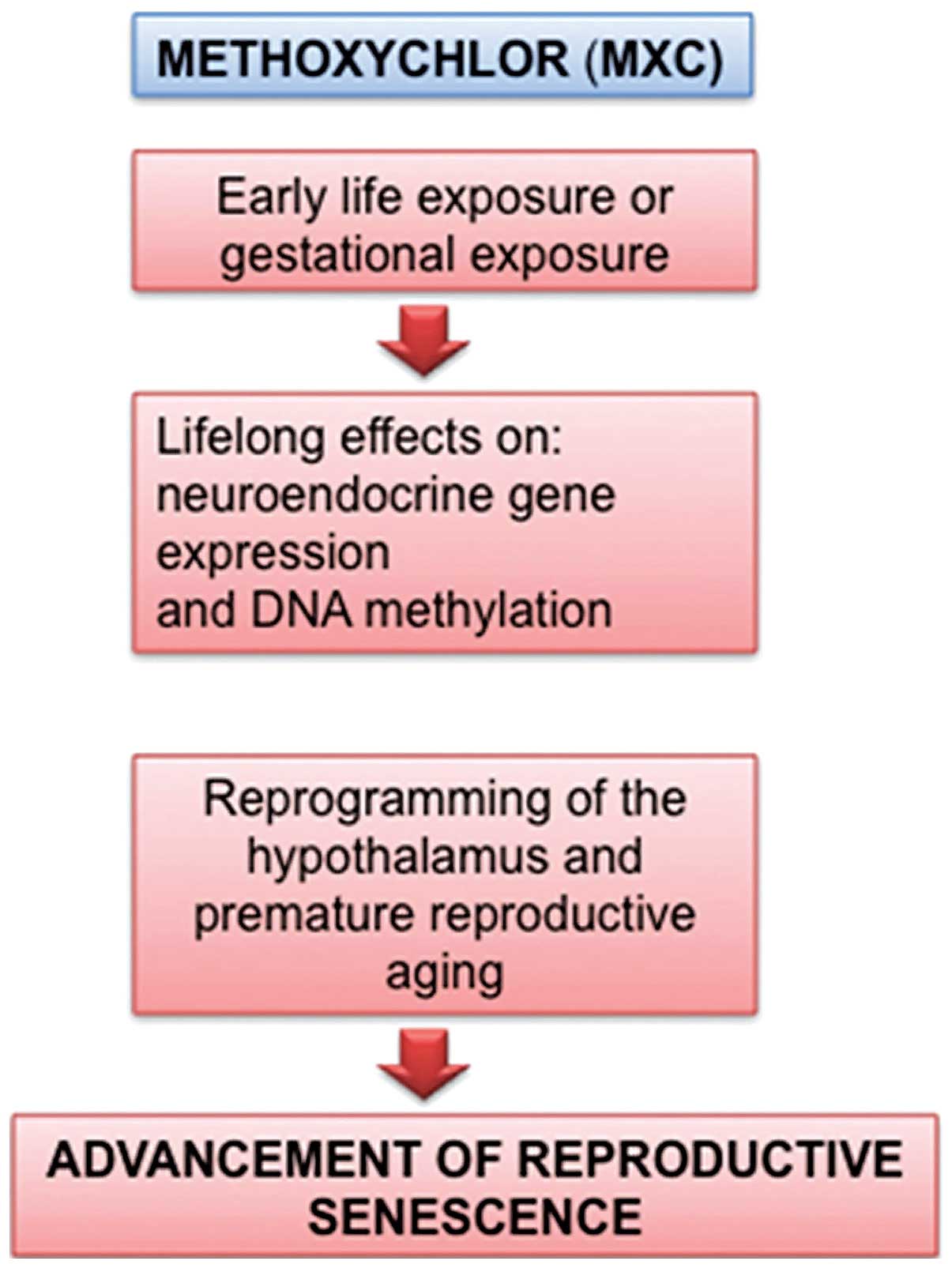
Methoxychlor
Methoxychlor (MXC) was introduced as a less
persistent alternative to DDT but, unfortunately, many studies have
demonstrated that MXC is also an estrogenic ED (24). Gestational exposure to MXC disrupts
the female reproductive system with lifelong effects on
neuroendocrine gene expression and DNA methylation, as well as
faster reproductive senescence. Animal and recent epidemiological
studies showed that reproductive aging was accelerated by ED
developmental exposures: besides MXC, also bisphenol A, dioxins and
perfluorocarbons were highlighted (Fig.
2). By hastening senescence and/or increasing cancer risk, EDs
may eliminate the possibility of biological children for women who
may postpone childbirth for personal or professional reasons.
Considering the important roles of estrogens on targets in the body
and brain, early reproductive senescence may also accelerate
various disease-related states associated with menopause, and
affect the quality of life in the aging population of women
(25).
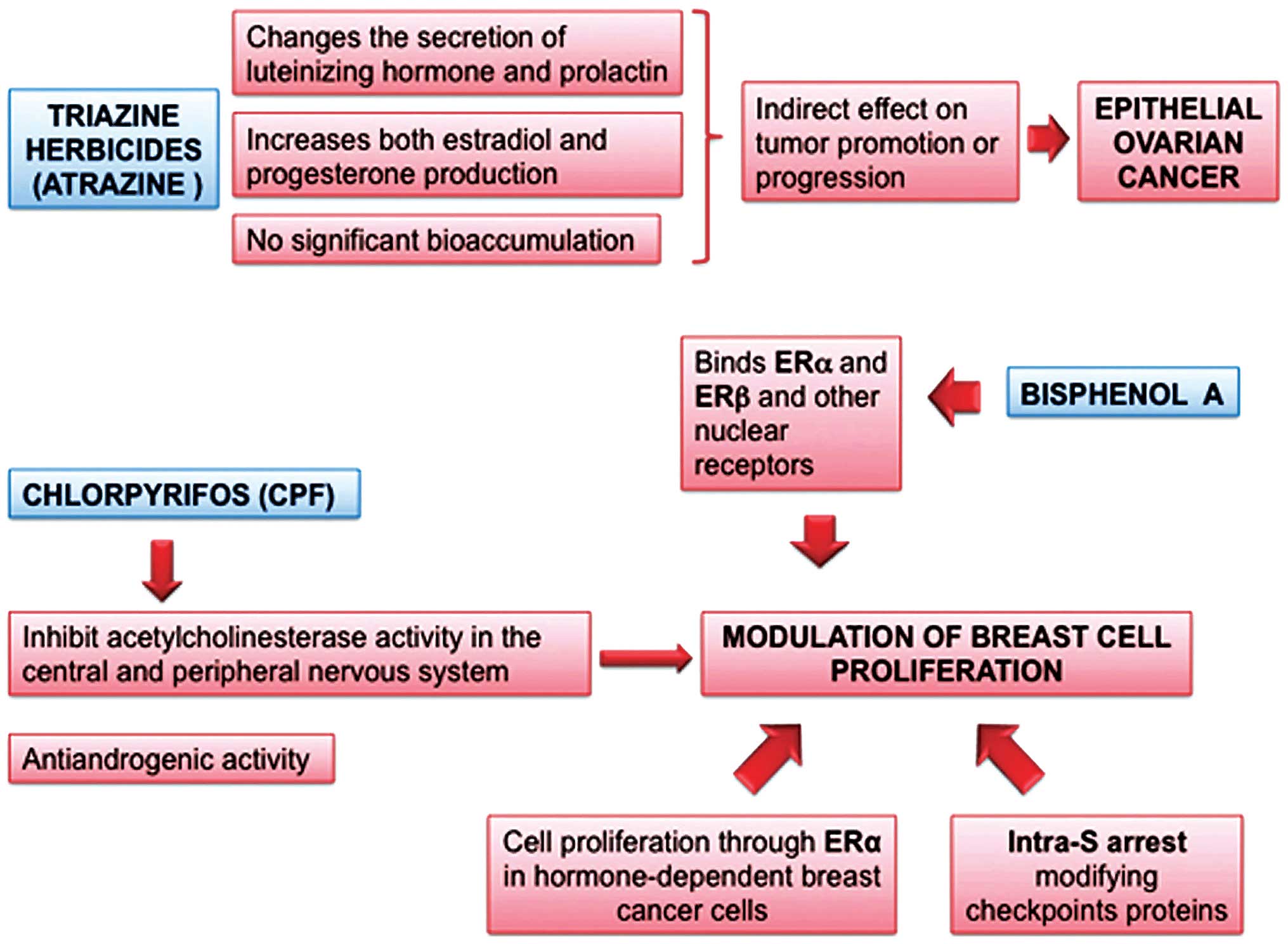
Atrazine and triazine herbicides
Atrazine is the prototypal representative of this
group of herbicides. These compounds act as EDs in female rats by
altering the secretion of luteinizing hormone (LH) and prolactin at
the hypothalamic-pituitary level. Triazine herbicides have no
significant bioaccumulation potential, but they can be significant
pollutants of water bodies (Fig.
3). Atrazine is an example of species and strain-specific
induction of mammary tumors. Adult female Sprague-Dawley rats fed
with diet-supplemented atrazine developed mammary tumors earlier
and with increased incidence, when compared to controls, while this
findings was not observed in other rats (26). In Sprague-Dawley rats, atrazine
caused a persistent estrus with a prolonged and sustained estrogen
secretion from ovarian follicles, which failed to ovulate due to
the herbicidal action on gonadotropin balance. This mechanism of
ovulation failure is fundamentally different from menopause in
women.
Previous studies have demonstrated an increased risk
of ovarian cancer among women exposed to triazine herbicides.
Notably, triazine herbicides are among the very few EDs for which
an epidemiological association with epithelial ovarian cancer might
be hypothesized (27).
Chlorpyrifos and other pesticides
The 2,4,5-trichloro-phenoxypropionic acid and the
fungicide captan were found to significantly increase the risk of
post-menopausal breast cancer among women whose husbands used such
pesticides (RR=2.0 and 2.7, respectively). The risk was moderately
higher when the residence was close to the pesticide application
area. Some indications, not statistically significant, of an
increased risk for breast cancer were found in premenopausal women
using specific organophosphorus insecticides, in particular
chlorpyrifos, dichlorvos, and terbufos (Fig. 3) (28). A significantly increased risk of
breast cancer was also associated with self-reported residential
pesticide use; however, due to the study design, no dose response
trend was observed and it was, indeed, difficult to detect
(29).
The Agricultural Health Study also found increased
ovarian cancer risk among women employed as private pesticide
applicators (SIR=2.97) (30).
Low and environmentally relevant concentrations of
glyphosate possess estrogenic activity and alter both ERα and ERβ
expression. Glyphosate-based herbicides are widely used for soybean
cultivation, and our results also found that there is an additive
estrogenic effect between glyphosate and genistein, a phytoestrogen
in soybeans (31).
Summing up the overall data, there is no conclusive
evidence on non-occupational insecticide exposure and breast
cancer. These compounds can modulate several mechanisms of
carcinogenesis, in particular in the breast, but the cell
type-specific responses are at nanomolar range concentrations.
While some cancer cell lines, such as MCF-7, exhibit a significant
increase in all the cancer mechanisms, other cell lines, such as
MDA-MB-231, exhibit a markedly reduced invasion potential following
exposure to pesticides (32). Thus
it is difficult to predict the overall net cancer effect, without
specifying the ED, dose, exposure time and the particular type of
cancer cell.
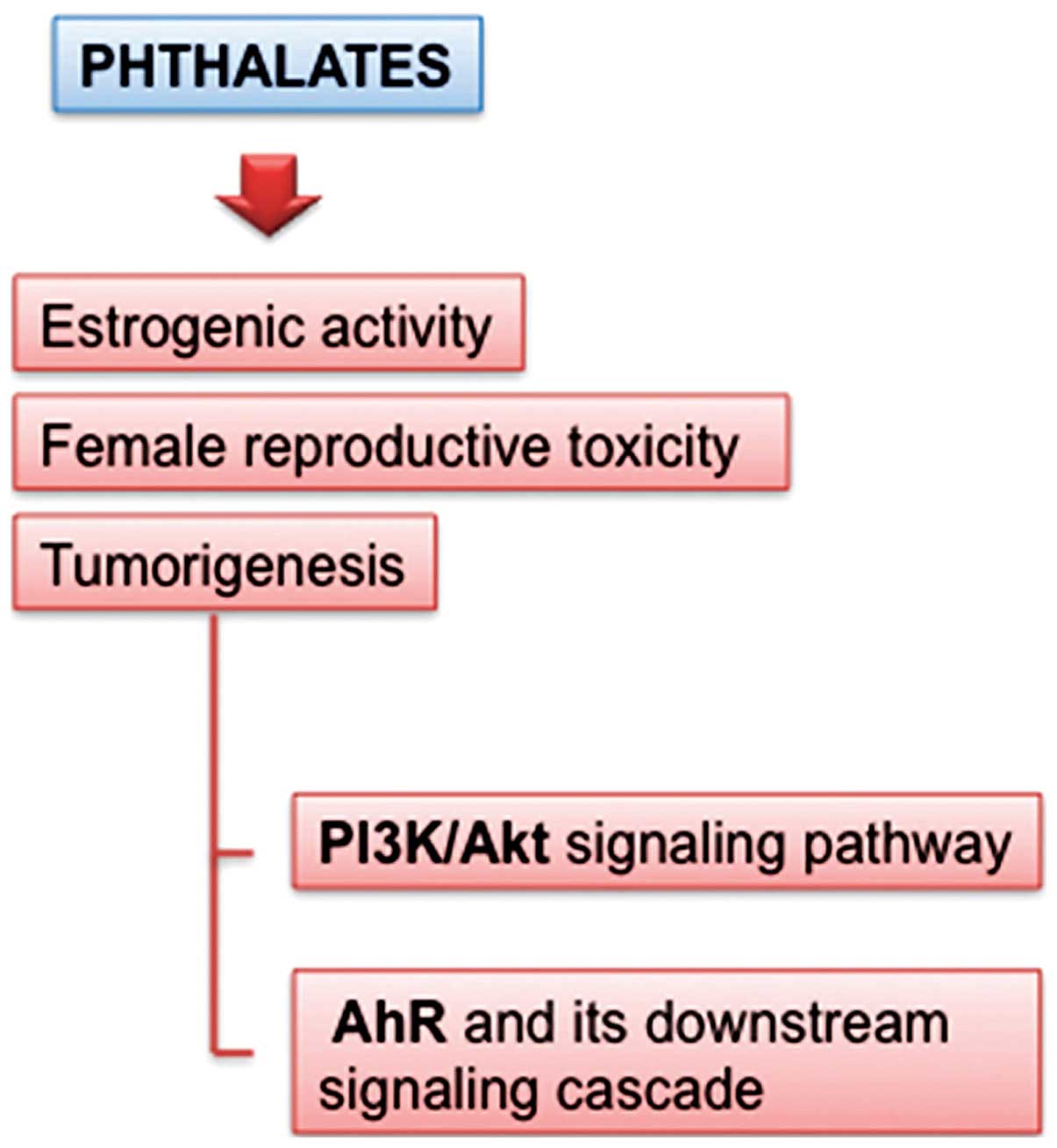
Phthalates
Phthalates are a large family of synthetic
high-production chemicals used in polyvinyl chloride (PVC)
plastics, beauty and infant products, medical devices, as well as
in enteric coating of some medications. In the general population,
exposure accounts for approximately 2 mg/day, while occupational
and medical exposures can reach much higher levels. Phtalates are
believed to act on steroid biosynthesis, affecting also the early
steps in liver.
Several groups have documented the ability of
phthalates to cause female reproductive toxicity and the subject
has been previously reviewed (Fig.
4) (33). Phtalates such as
butyl phthalate (BBP), di-n-butyl phthalate (DBP) and
di-2-ethylhexyl phthalate (DEHP) are not only capable of inducing a
proliferative effect through the PI3K/AKT signaling pathway but
also of displaying estrogenic activity even at a very low
concentration (34).
Notwithstanding the widespread diffusion and the
relevant effects and mechanisms, no studies have been performed on
the possible association between phthalate exposure and female
cancer risk. Nevertheless a growing body of evidence suggests that
EDs contribute to female reproductive disorders, mostly phthalates.
Phthalate attributable endometriosis cases across the European
Union have been estimated at 56,700 and 145,000 women,
respectively, with total combined economic and health care costs
potentially reaching €163 million and €1.25 billion (35). These public health costs should be
considered as the EU contemplates regulatory action on EDs. When
compared with population-based controls, the risk of endometrioid
clear cell ovarian cancer for women with endometriosis is 3-times
greater (36). As the life-time
risk of ovarian cancer is significantly increased in endometriosis
patients from approximately 1 to 2% (37) and phthalates can contribute to
endometriosis with a probability of causation of 20–39%, further
studies on phthalates and ovarian cancer are warranted. Breast
cancer and endometriosis share some common environmental and
molecular risk factors; thus this is a further area of research
(38).
Bisphenol A
Bisphenol A [BPA, 2,2,-bis(hydroxyphenyl) propane]
is one of the highest-volume chemicals produced worldwide. BPA is a
plasticizer found in reusable plastic containers, food and beverage
can liners, baby bottles and dental sealants, among others.
Originally synthesized as an estrogenic compound, it is currently
utilized to manufacture food and beverage containers resulting in
uptake with food and drinks. According to an opinion issued by EFSA
in 2015 (39), adolescents are the
population group with the highest aggregate (oral plus dermal)
exposure to BPA (1.449 µg/kg BW), taking into account that
the substance has been forbidden in baby bottles in Europe since
2011.
Humans are widely exposed to BPA through plastic
goods, food and drink packaging, and thermal paper receipts. BPA is
the best-studied ED and it is the only one for which effects of
exposure have been described at multiple time points spanning fetal
development and postnatal life. Bisphenol A is such a widespread
industrial chemical, that, albeit a non-persistent chemical, it is
consistently found in body fluids, due to continuous environmental
exposure. Internal levels appear to be higher in infertile compared
to fertile women (40). There is
concern that exposure to low doses of BPA, defined as less than or
equal to 5 mg/kg body weight/day, may have developmental effects on
various hormone-responsive organs as it is detected in body fluids
of more than 90% of the human population.
BPA exposure has become an important health concern
based on its ability to 'leach' from these products and enter the
materials contained within them. Elevated temperature and extreme
pH further increase the leaching of BPA from food containers. Human
exposure has been confirmed in various tissues including ovarian
follicular fluid, and various exposures have been linked to
reproductive effects in animal models (41).
In the long-term, BPA exposure was found to result
in an increase in the number of epithelial structures and in the
development of pre-cancerous and cancerous lesions in the mammary
glands of rodents that are manifested in adulthood. The effects of
BPA on mammary development and tumorigenesis in rodents are used as
a paradigmatic example of how altered prenatal mammary development
may lead to breast cancer in humans (Fig. 3). Changes in the stroma and its
extracellular matrix led to altered ductal morphogenesis.
Additionally, gestational and lactational exposure to BPA increased
the sensitivity of rats and mice to mammotropic hormones during
puberty and beyond, thus suggesting a plausible explanation for the
increased incidence of breast cancer (41).
Perinatal exposure to environmentally relevant doses
of BPA alters long-term hormone response that may increase the
propensity to develop breast cancer (42). BPA may also alter epigenetic
regulation of relevant gene panels, possibly supporting breast
cancer promotion.
BPA is considered an 'estrogenic' ED, binding to
both ERα and ERβ, but it may interact with other nuclear receptors
as well, e.g., antagonism with androgen receptor and agonism with
pregnane X receptor. As for other EDs, experimental studies on BPA
essentially addressed the possible link with breast cancer, rather
than with ovarian or endometrial cancer.
BPA-induced tissue changes in the mammary gland were
found to include enhanced estradiol sensitivity, increased presence
of progesterone receptor-positive ductal cells and enhanced ductal
terminal end branching in the epithelium, while the stroma
associated with hyperplastic ducts had more fibrous tissue and mast
cells (43). Ductal hyperplasias
and carcinoma in situ with increased number of estrogen
receptor-α positive cells were present in adult rats (44). This gland phenotype was not be
unique to rats as it was observed also in mice exposed from
organogenesis through to lactation (45). The mode of action could involve a
stromal effect: BPA accelerates the maturation of the adipose
tissue pad of the mammary gland and alters collagen localization,
thereby possibly altering the fat-epithelium interactions. Oral BPA
exposure of rat dams during lactation increased the susceptibility
to a later challenge with DMBA as evidenced by the increased
numbers of tumors per rat and the shortened latency period
(46); BPA exposure caused
increased cell proliferation and progesterone receptor expression
and decreased apoptosis. Since BPA (contrary to, e.g., DDT or PCB)
is still a widely used chemical, there is (understandably) a lively
debate about the relevance of these findings to risk assessment.
The BPA effects on mammary programming are observed at dose levels
(down to 0.25 mg/kg BW) far below the NOAEL (5 mg/kg BW) derived
from standard toxicological studies; contrary to fat-soluble
chemicals, the bioaccumulation potential and excretion in milk are
low for BPA, thus there are uncertainties about the actual neonatal
and peripubertal exposure. Most important, several experiments used
injection, thus using a route not relevant for the general
population and by-passing liver-mediated conjugation, an important
detoxification step in BPA metabolism. On the other hand, the
effects observed are consistent across experiments and across
species (rats and mice); the internal exposure achieved may be
relevant to actual human exposures (47).
Continuous and/or repeated exposure to BPA may also
play a role in tumor progression and poorer patient outcome
(49). For instance, BPA exposure
in vitro elicited a pattern of gene expression related with
higher tumor aggressiveness in epithelial-stromal co-cultures from
breast cancer patients (48).
Diethylstilboestrol
Diethylstilboestrol (DES) is a potent estrogen
agonist. It is the most well-known example of an ED eliciting
transplacental carcinogenesis. Young adult offsprings exposed in
utero to this potent drug had a higher rate of reproductive
tract abnormalities in both genders as well as a higher rate of the
rare clear-cell vaginal adenocarcinoma in female ones (50). Neonatal treatment of mice on
post-natal days 1–5 with DES has been shown to cause uterine
adenocarcinoma by 18 months, similar to the use of the
phytoestrogen genistein (51).
The timing of exposure is critical to the potential
development of uterine cancer. In fact, treatment of adult mice
with comparable levels of DES did not induce uterine neoplasms.
Direct evidence of a link between prenatal estrogen
exposure and breast cancer risk has been gathered from a cohort of
women born to mothers treated with the potent synthetic estrogen
DES during pregnancy (Fig. 1).
Breast cancer risk at 40 years of age and older was 2.5-fold higher
in the DES-exposed women.
This potent estrogenic agent, known for its
transplacental carcinogenesis when used as a drug in the 1960's,
has been used as an experimental model to explore the developmental
anomalies that increase the susceptibility to mammary gland
neoplasia later in life, due to the proliferative estrogen-like
effects.
DES has been investigated also as an experimental
model for endometrial cancer. Since the endometrium in primates is
shed and regenerates monthly, the key target cells for initiating
endometrial cancer are most likely a lineage of non-shedding cells,
possibly of a stem cell-type. This scenario is in accordance with
the hypothesized tumorigenic action of ED by altering the
developmental programming of target tissues. ER-regulated genes,
such as c-fos and lactoferrin, are induced in the uterus by
prenatal DES exposure and persist into adulthood (Fig. 1). Also neonatal exposure of mice to
DES was found to lead to ERα-dependent tumorigenesis in the uterus.
The Eker rat strain is specifically vulnerable to another uterine
alteration induced by neonatal DES, leiomyomas (52). Leiomyomas developed in rats exposed
in utero, displayed an enhanced proliferative response to
steroid hormones compared to tumors in unexposed animals. The
induction of leiomyomas was highly dependent on the developmental
window. In adult myometrium, the expression of the
estrogen-responsive gene calbindinD(9)K and the progesterone receptor were
reprogrammed in females exposed to DES at days 3 to 5
(pre-implantation) and days 10 to 12 (organogenesis) but not in
those exposed at days 17 to 19 (late fetal period). Reprogramming
in response to DES exposure resulted in a hyper-responsiveness to
ovarian hormones. Since the resistant period coincided with the
time at which reproductive tract tissues were exposed to endogenous
estrogen, the data suggested that the programming was most
vulnerable to estrogenic ED when the uterine tissue was in an
estrogen-naïve state (52). The
experimental studies performed with such a potent endocrine-active
agent as DES, often performed by parenteral administration, have
been difficult to extrapolate to environmental EDs, also because
these are usually far less potent. However, the DES studies
provided a detailed insight on the mechanisms by how early ED
exposure may predispose reproductive tissues to cancer.
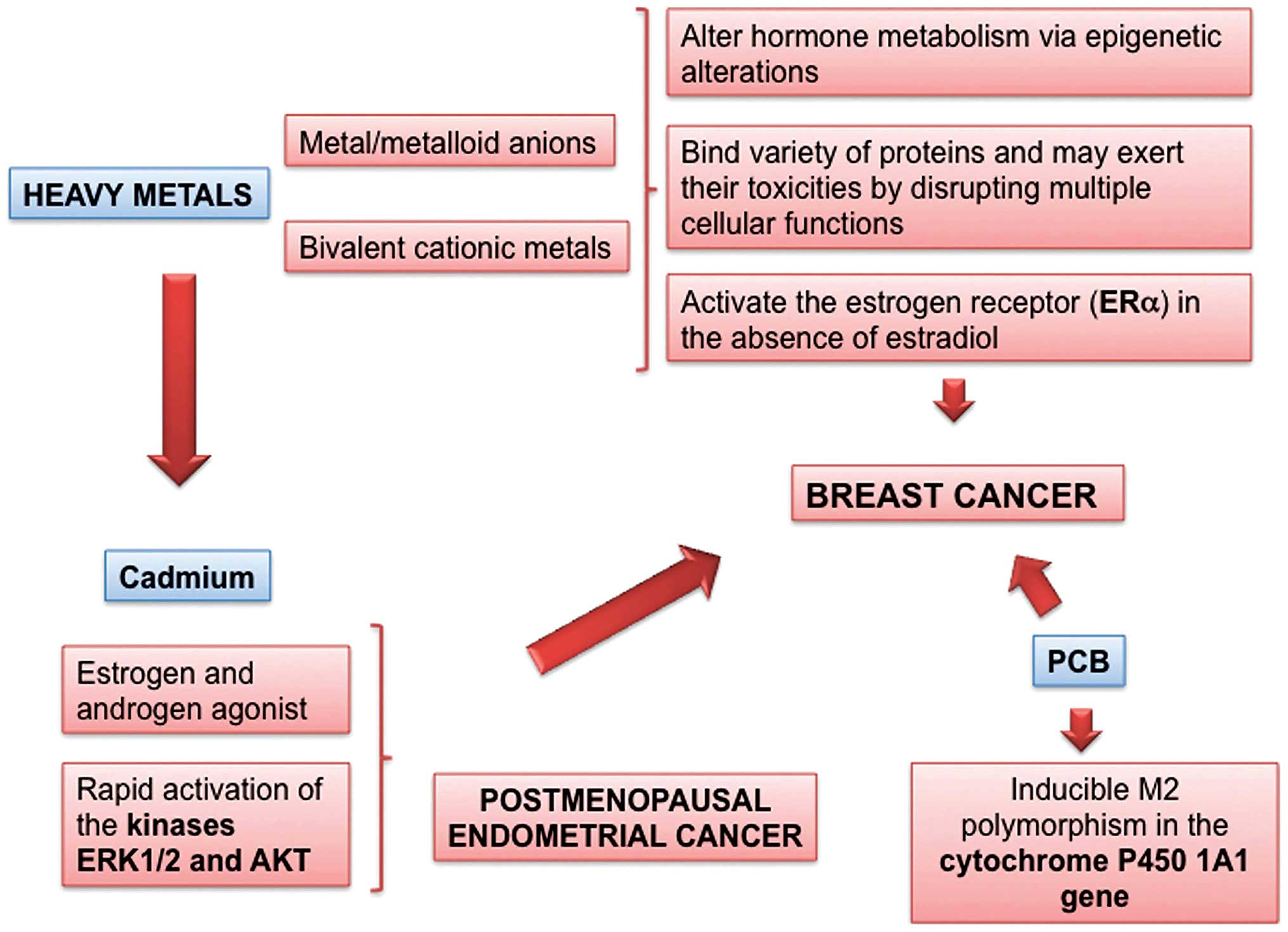
Cadmium and heavy metals
Cadmium has been classified as a human carcinogen by
the International Agency for Research on Cancer (IARC, Lyon,
France) (53). Cadmium is widely
dispersed into the environment through industrial emission, waste
incineration, and combustion of fossil fuels. Even in industrially
non-polluted areas, farmland may become contaminated by atmospheric
deposition and by the use of cadmium-containing fertilizers and
sewage sludge. The highest concentration of cadmium in food can be
found in shellfish, offal products, and certain seeds; however,
because of a comparatively high accumulation of cadmium in
agricultural crops and a high consumption of these products, the
main sources of dietary cadmium exposure (80%) are bread and other
cereals, potatoes, root crops, and vegetables. Cadmium may induce
cancer by several mechanisms, such as aberrant gene expression,
inhibition of DNA damage repair, induction of oxidative stress and
inhibition of apoptosis. Cadmium has features of an ED, as it is an
estrogen, and even an androgen mimetic that may promote the
development of estrogen-dependent malignancies, such as breast and
endometrial cancer (Fig. 5). Both
in vitro and in vivo studies have provided evidence
that cadmium may act as a metalloestrogen. The estrogen-mimicking
effects of cadmium on mammary gland are associated with the
interaction to nuclear ERα and its hormone-binding domain, as well
as with the activation of membrane-bound estrogen receptors.
Cadmium exposure is positively associated with the
risk of breast cancer and increased mammographic density. Cadmium
is a slow-acting, pro-oxidant toxic metal which is also an ED; it
acts as an estrogen and androgen agonist in vivo and in
vitro and can cause rapid activation of the kinases ERK1/2 and
AKT in human breast cancer-derived cells, like estradiol (54). There are epidemiological indications
linking the exposure to cadmium with breast cancer (55) and postmenopausal endometrial cancer
(56). Oily seeds like flaxseed are
a dietary source of cadmium: interestingly, rat offspring exposed
in utero and during lactation to a diet high (up to 10%) in
flaxseed were more susceptible (shorter latency, more tumors per
rat) to the rat mammary carcinogen 7,12-dimethylbenz(a)anthracene
(DMBA). Flaxseed exposure did not alter the mammary gland
differentiation, but altered the ER expression in gland structures,
namely, it increased ERα and especially reduced ERβ. Although the
link between the effects and cadmium exposure was inferential, the
observed changes were strongly suggestive of an effect by an
estrogen-active ED; since flaxseed is naturally rich in lignans,
which are considered as phytoestrogens, a contribution by the high
intake of these compounds may not be ruled out.
The role for dietary cadmium in postmenopausal
breast cancer development was confirmed by a prospective study
cohort of 55,987 postmenopausal women (57). During an average of 12.2 years of
follow-up, 2,112 incident cases of invasive breast cancer were
ascertained (1,626 ER+ and 290 ER-). After
adjusting for confounders, including consumption of whole grains
and vegetables (which accounted for 40% of the dietary exposure,
but also contained putative anticarcinogenic phytochemicals),
dietary cadmium intake was positively associated with overall
breast cancer tumors [rate ratio (RR), 1.21; 95% confidence
interval (CI), 1.07–1.36; Ptrend =0.02]. Among lean and normal
weight women, statistically significant associations were observed
for all tumors (RR, 1.27; 95% CI, 1.07–1.50) and for ER+
tumors (RR, 1.25; 95% CI, 1.03–1.52) and similar, but no
statistically significant associations were found for
ER− tumors (RR, 1.22; 95% CI, 0.76–1.93).
In a large population-based prospective cohort of
women, the Swedish Mammography Cohort (56) found a statistically significant
positive association between dietary cadmium exposure and risk of
endometrial cancer. During 16.0 years (484,274 person-years) of
follow-up there were 378 incident cases of endometrioid
adenocarcinoma. The average estimated dietary cadmium intake was 15
µg/day (80% from cereals and vegetables). Cadmium intake was
statistically significantly associated with increased risk of
endometrial cancer in all women; the multivariate relative risk was
1.39 [95% CI, 1.04–1.86; Ptrend =0.019], comparing highest fertile
versus lowest. Among never-smoking women with body mass index (BMI)
of <27 kg/m2, the relative risk was 1.86 (95% CI,
1.13–3.08; Ptrend =0.009). We observed a 2.9-fold increased risk
(95% CI, 1.05–7.79) associated with long-term cadmium intake
consistently above the median at both baseline and in never-smoking
women with low bio-available estrogen (BMI of <27
kg/m2 and nonusers of postmenopausal hormones).
Heavy metals present in cigarettes may substantially
contribute to tumorigenesis by inducing intercellular ROS
accumulation, increased expression of oncogenic and anti-apoptotic
markers (58).
A recent meta-analysis and systematic review
(59) based on population-based
studies, concluded that the frequency of breast cancer may be an
indicator of the early genetic effects for cadmium-exposed
populations. A meta-analysis based on individual data might provide
more precise and reliable results. Therefore, it is necessary to
construct an international database on genetic damage among
populations exposed to cadmium that may contain all raw data of
studies examining genetic toxicity
Polychlorinated biphenyls
Polychlorinated biphenyls (PCBs) are a large group
of different persistent organic pollutants (POPs) that could induce
cytochrome P4501A1 (CYP1A1), which is involved in the metabolism of
steroid hormones and polycyclic aromatic hydrocarbons in humans
(Fig. 5) (60).
The cytochrome P450 1A1 (CYP1A1) is a member of the
CYP1 family. It participates in the metabolism of a vast number of
xenobiotics, as well as endogenous substrates, and the A2455G G
allele is a risk factor for breast cancer among Caucasian subjects
(61).
Discrete windows of susceptibility to toxicants have
been identified for the breast, including in utero, puberty,
pregnancy, and postpartum. PCB measured during the early postpartum
predicts an increased risk of maternal breast cancer diagnosed
before age 50. In the Child Health and Development Studies cohort
(62), PCB 167 was associated with
a lower risk [odds ratio (OR), 75th vs. 25th percentile =0.2, 95%
CI, 0.1–0.8] as was PCB 187 (OR, 75th vs. 25th percentile =0.4, 95%
CI, 0.1–1.1). In contrast, PCB 203 was associated with a 6-fold
increased risk (OR, 75th vs. 25th percentile =6.3, 95% CI,
1.9–21.7). The net association of PCB exposure, estimated by a
post-hoc score, was nearly a 3-fold increase in risk for breast
cancer (OR, 75th vs. 25th percentile =2.8, 95% CI, 1.1–7.1) among
women with a higher proportion of PCB 203 in relation to the sum of
PCBs 167 and 187. Postpartum PCB exposure as well as pregnancy
exposure, may predict increased risk for early breast cancer,
depending on the mixture that represents the internal dose. It
remains unclear whether individual differences in exposure,
response to exposure, or both, explain the risk patterns
observed.
Phytoestrogens
Studies on the relationship between soy consumption
and risk of breast cancer are discordant: foods rich in
phytoestrogens (PEs) may have complex actions exerting both
preventive and promoting effects (63). Ingestion before puberty, when the
mammary gland is relatively immature seems protective (64). A meta-analysis indicates that high
soy intake might reduce the risk of developing premenopausal breast
cancer but has no effect on post-menopausal breast cancer risk
(65). The effects of ingestion of
dietary PEs by breast cancer patients and survivors is also
controversial (66,67). High consumption of soy products and
other legumes is associated with a decreased risk of endometrial
cancer for the highest compared with the lowest quartile of soy
intake (68). However, a randomized
doubled-bind, placebo-controlled study on 298 post-menopausal women
showed an increased incidence of endometrial hyperplasia following
5 years of treatment with 50 mg of soy isoflavones (69). Thus, PE supplements should be
reconsidered, particularly in women at high risk for endometrial
cancer. Indeed, PE can cause both proliferative and
anti-proliferative effects, depending on tumor cell type,
concentrations, timing of phytoestrogen exposure and type of PE
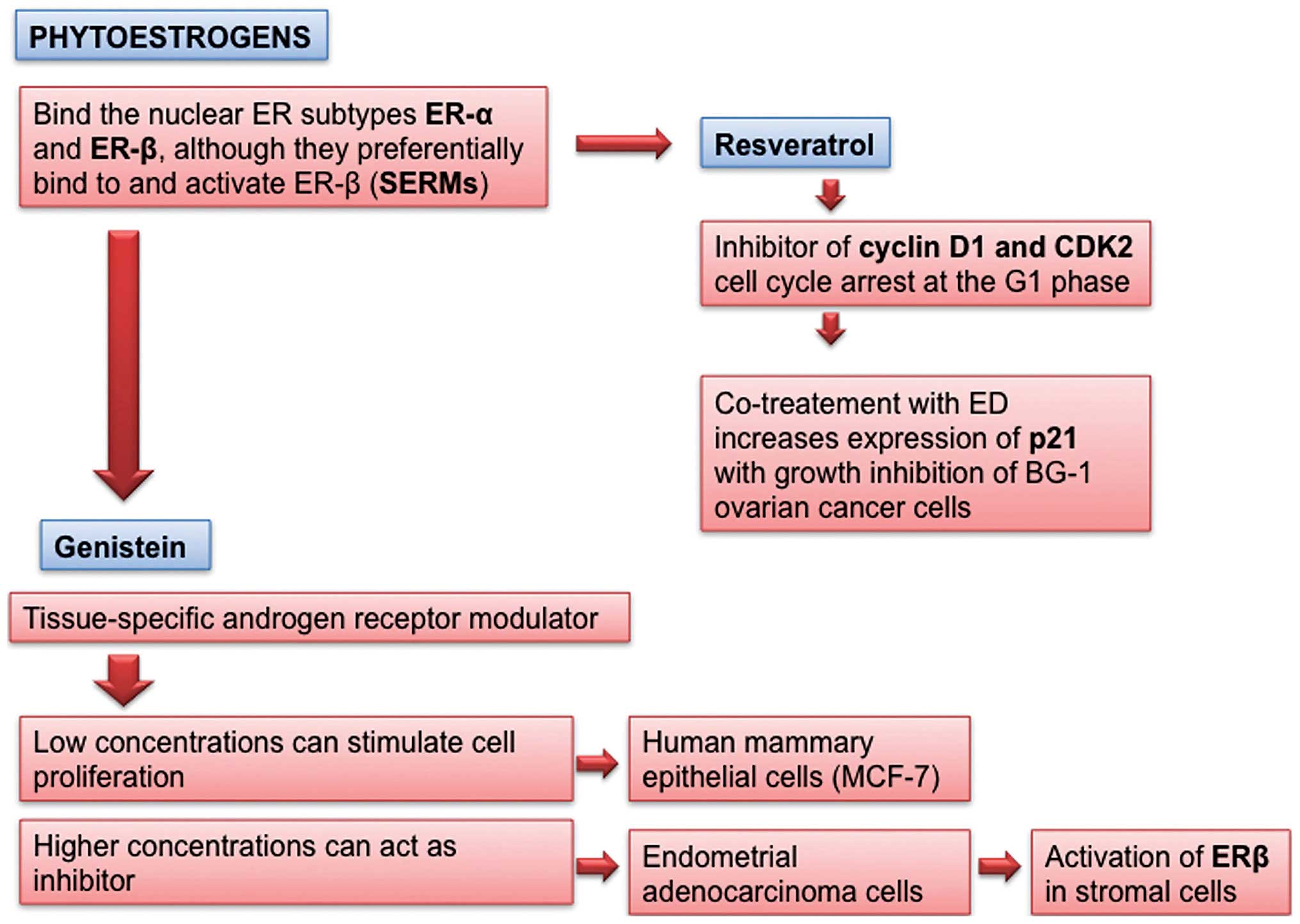
given. They bind to nuclear ER subtypes ERα, but they
preferentially bind to and activate ERβ (Fig. 6).
Genistein, the main soy isoflavone, stimulates the
growth of estrogen-sensitive mammary cancer cells in vitro
at low concentrations (0.1–10 mM), whereas at higher concentrations
(≥10 mM) it is an inhibitor. It is not only a selective estrogen
receptor modulator, but also a tissue-specific androgen receptor
modulator as it affects androgen receptor (AR)-mediated gene
expression.
The interactions with genistein and ED xenobiotics
may be quite complex, ranging from antagonism to additivity
depending on thr substances and experimental systems.
Several experimental studies have explored the
complex links between PE action and endometrial proliferation.
While daidzein potentiates estrogen-induced endometrial cell
proliferation (70), genistein
inhibits it, possibly through activation of ERβ in stromal cells at
low concentrations (nM), while much higher (µM)
concentrations increase endometrial proliferation or uterine
leiomyomas, through non-genomic ER signaling, which determines
epigenetic changes (71). Overall,
the findings suggest some caution towards high intakes of
isoflavones through, e.g., supplements or 'healthy' products.
Phytoestrogens could also have other complex,
indirect, interfering, or protective mechanisms, in particular,
suppression of the activity of the aromatase enzymes, which are
responsible for conversion of androgens to estrogens; induction of
apoptosis in human breast cancer cells, also in ER-negative cell
lines, which supports the occurrence also of hormone-independent
mechanisms of action; inhibition of tyrosine kinase activity,
involved in a number of growth factor signaling pathways,
implicated in the control of cell growth and differentiation;
antioxidant activity; stimulation of the immune system and
inhibition of angiogenesis.
Resveratrol (trans-3,4,5-trihydroxystilbene;
RES), a naturally occurring PE, has a growth inhibitory effect on
the cell viability effect of BPA on BG-1 human ovarian cancer
cells. It could be a candidate for prevention of tumor progression
caused by EDs, including BPA via effective inhibition of the
crosstalk of ERα and IGF-1R signaling pathways (Fig. 6) (72).
4. Conclusion
Various carcinogenetic mechanisms of main endocrine
disruptors have been documented, yet we are still far from the full
knowledge of the female cancer effects of the multitude of complex
interacting pollutants. EDs can dysregulate hormone signaling and
cell function through multifaceted molecular, cellular and
biochemical mechanisms (73).
Chronic exposure to EDs can permanently alter physiological hormone
signaling (74) and it may provoke
epigenetic and genetic modifications in tissue stem cells that can
lead to cancer (75).
The research findings can help us to better
understand ED-related cancer mechanisms (76) and to identify possible ways to
prevent cancer through fundamental changes in lifestyle that can
effectively counter bio-accumulation of certain EDs (77).
Although we do not know the clinical impact of the
potential carcinogenetic effects of each ED (76), also invoking the precautionary
principle, these findings support the urgent need for health and
environmental policies aimed at protecting the public in general,
and, in particular, the developing fetus and women of reproductive
age.
References
|
1
|
Macon MB and Fenton SE: Endocrine
disruptors and the breast: Early life effects and later life
disease. J Mammary Gland Biol Neoplasia. 18:43–61. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
US Environmental Protection Agency:
Endocrine Disruptors Research.
|
|
3
|
Maqbool F, Mostafalou S, Bahadar H and
Abdollahi M: Review of endocrine disorders associated with
environmental toxicants and possible involved mechanisms. Life Sci.
145:265–273. 2016. View Article : Google Scholar
|
|
4
|
Soto AM and Sonnenschein C: Environmental
causes of cancer: Endocrine disruptors as carcinogens. Nat Rev
Endocrinol. 6:363–370. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hilakivi-Clarke L, de Assis S and Warri A:
Exposures to synthetic estrogens at different times during the
life, and their effect on breast cancer risk. J Mammary Gland Biol
Neoplasia. 18:25–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Caserta D, Maranghi L, Mantovani A, Marci
R, Maranghi F and Moscarini M: Impact of endocrine disruptor
chemicals in gynaecology. Hum Reprod Update. 14:59–72. 2008.
View Article : Google Scholar
|
|
7
|
Brody JG, Moysich KB, Humblet O, Attfield
KR, Beehler GP and Rudel RA: Environmental pollutants and breast
cancer: Epidemiologic studies. Cancer. 109(Suppl 12): 2667–2711.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Collins LL, Lew BJ and Lawrence BP: TCDD
exposure disrupts mammary epithelial cell differentiation and
function. Reprod Toxicol. 28:11–17. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warner M, Eskenazi B, Mocarelli P,
Gerthoux PM, Samuels S, Needham L, Patterson D and Brambilla P:
Serum dioxin concentrations and breast cancer risk in the Seveso
Women's Health Study. Environ Health Perspect. 110:625–628. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bertazzi A, Pesatori AC, Consonni D,
Tironi A, Landi MT and Zocchetti C: Cancer incidence in a
population accidentally exposed to
2,3,7,8-tetrachlorodibenzo-para-dioxin. Epidemiology. 4:398–406.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pesatori AC, Consonni D, Bachetti S,
Zocchetti C, Bonzini M, Baccarelli A and Bertazzi PA: Short- and
long-term morbidity and mortality in the population exposed to
dioxin after the 'Seveso accident'. Ind Health. 41:127–138. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Laden F, Collman G, Iwamoto K, Alberg AJ,
Berkowitz GS, Freudenheim JL, Hankinson SE, Helzlsouer KJ, Holford
TR, Huang HY, et al: 1,1-Dichloro-2,2-bis(p-chlorophenyl)ethylene
and polychlorinated biphenyls and breast cancer: Combined analysis
of five U.S. studies. J Natl Cancer Inst. 93:768–776. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wolff MS, Toniolo PG, Lee EW, Rivera M and
Dubin N: Blood levels of organochlorine residues and risk of breast
cancer. J Natl Cancer Inst. 85:648–652. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hardell L, van Bavel B, Lindström G,
Björnfoth H, Orgum P, Carlberg M, Sörensen CS and Graflund M:
Adipose tissue concentrations of p,p′-DDE and the risk for
endometrial cancer. Gynecol Oncol. 95:706–711. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pavuk M, Cerhan JR, Lynch CF, Kocan A,
Petrik J and Chovancova J: Case-control study of PCBs, other
organo-chlorines and breast cancer in Eastern Slovakia. J Expo Anal
Environ Epidemiol. 13:267–275. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Calle EE, Frumkin H, Henley SJ, Savitz DA
and Thun MJ: Organochlorines and breast cancer risk. CA Cancer J
Clin. 52:301–309. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
López-Cervantes M, Torres-Sánchez L,
Tobías A and López-Carrillo L: Dichlorodiphenyldichloroethane
burden and breast cancer risk: A meta-analysis of the epidemiologic
evidence. Environ Health Perspect. 112:207–214. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ibarluzea JmJ, Fernández MF, Santa-Marina
L, Olea-Serrano MF, Rivas AM, Aurrekoetxea JJ, Expósito J, Lorenzo
M, Torné P, Villalobos M, et al: Breast cancer risk and the
combined effect of environmental estrogens. Cancer Causes Control.
15:591–600. 2004. View Article : Google Scholar
|
|
19
|
Cassidy RA, Natarajan S and Vaughan GM:
The link between the insecticide heptachlor epoxide, estradiol, and
breast cancer. Breast Cancer Res Treat. 90:55–64. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Charlier C, Albert A, Herman P, Hamoir E,
Gaspard U, Meurisse M and Plomteux G: Breast cancer and serum
organochlorine residues. Occup Environ Med. 60:348–351. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Engel LS, Hill DA, Hoppin JA, Lubin JH,
Lynch CF, Pierce J, Samanic C, Sandler DP, Blair A and Alavanja MC:
Pesticide use and breast cancer risk among farmers' wives in the
agricultural health study. Am J Epidemiol. 161:121–135. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cohn BA, Wolff MS, Cirillo PM and Sholtz
RI: DDT and breast cancer in young women: New data on the
significance of age at exposure. Environ Health Perspect.
115:1406–1414. 2007.PubMed/NCBI
|
|
23
|
Ingber SZ, Buser MC, Pohl HR, Abadin HG,
Murray HE and Scinicariello F: DDT/DDE and breast cancer: A
meta-analysis. Regul Toxicol Pharmacol. 67:421–433. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tiemann U: In vivo and in vitro effects of
the organochlorine pesticides DDT, TCPM, methoxychlor, and lindane
on the female reproductive tract of mammals: A review. Reprod
Toxicol. 25:316–326. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gore AC, Walker DM, Zama AM, Armenti AE
and Uzumcu M: Early life exposure to endocrine-disrupting chemicals
causes lifelong molecular reprogramming of the hypothalamus and
premature reproductive aging. Mol Endocrinol. 25:2157–2168. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wetzel LT, Luempert LG III, Breckenridge
CB, Tisdel MO, Stevens JT, Thakur AK, Extrom PJ and Eldridge JC:
Chronic effects of atrazine on estrus and mammary tumor formation
in female Sprague-Dawley and Fischer 344 rats. J Toxicol Environ
Health. 43:169–182. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Young HA, Mills PK, Riordan DG and Cress
RD: Triazine herbicides and epithelial ovarian cancer risk in
central California. J Occup Environ Med. 47:1148–1156. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ventura C, Nieto MR, Bourguignon N,
Lux-Lantos V, Rodriguez H, Cao G, Randi A, Cocca C and Núñez M:
Pesticide chlorpyrifos acts as an endocrine disruptor in adult rats
causing changes in mammary gland and hormonal balance. J Steroid
Biochem Mol Biol. 156:1–9. 2016. View Article : Google Scholar
|
|
29
|
Teitelbaum SL, Gammon MD, Britton JA,
Neugut AI, Levin B and Stellman SD: Reported residential pesticide
use and breast cancer risk on Long Island, New York. Am J
Epidemiol. 165:643–651. 2007. View Article : Google Scholar
|
|
30
|
Alavanja MC, Sandler DP, Lynch CF, Knott
C, Lubin JH, Tarone R, Thomas K, Dosemeci M, Barker J, Hoppin JA,
et al: Cancer incidence in the agricultural health study. Scand J
Work Environ Health. 31(Suppl 1): 39–45; discussion 5–7.
2005.PubMed/NCBI
|
|
31
|
Thongprakaisang S, Thiantanawat A,
Rangkadilok N, Suriyo T and Satayavivad J: Glyphosate induces human
breast cancer cells growth via estrogen receptors. Food Chem
Toxicol. 59:129–136. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pestana D, Teixeira D, Faria A, Domingues
V, Monteiro R and Calhau C: Effects of environmental organochlorine
pesticides on human breast cancer: Putative involvement on invasive
cell ability. Environ Toxicol. 30:168–176. 2015. View Article : Google Scholar
|
|
33
|
Lovekamp-Swan T and Davis BJ: Mechanisms
of phthalate ester toxicity in the female reproductive system.
Environ Health Perspect. 111:139–145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen FP and Chien MH: Lower concentrations
of phthalates induce proliferation in human breast cancer cells.
Climacteric. 17:377–384. 2014. View Article : Google Scholar
|
|
35
|
Hunt PA, Sathyanarayana S, Fowler PA and
Trasande L: Female reproductive disorders, diseases, and costs of
exposure to endocrine disrupting chemicals in the European Union. J
Clin Endocrinol Metab. 101:1562–1570. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rossing MA, Cushing-Haugen KL, Wicklund
KG, Doherty JA and Weiss NS: Risk of epithelial ovarian cancer in
relation to benign ovarian conditions and ovarian surgery. Cancer
Causes Control. 19:1357–1364. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Johnson NP, Hummelshoj L, Abrao MS,
Adamson GD, Allaire C, Amelung V, Andersson E, Becker C, Birna
Ardal KB, Bush D, et al: World Endometriosis Society Montpellier
Consortium: Consensus on current management of endometriosis. Hum
Reprod. 28:1552–1568. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Roy D, Morgan M, Yoo C, Deoraj A, Roy S,
Yadav VK, Garoub M, Assaggaf H and Doke M: Integrated
bioinformatics, environmental epidemiologic and genomic approaches
to identify environmental and molecular links between endometriosis
and breast cancer. Int J Mol Sci. 16:25285–25322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
European Food Safety Authority: Scientific
opinion on the risks to public health related to the presence of
bisphenol A (BPA) in foodstuffs. EFSA J. 13:39782015. View Article : Google Scholar
|
|
40
|
Caserta D, Ciardo F, Bordi G, Guerranti C,
Fanello E, Perra G, Borghini F, La Rocca C, Tait S, Bergamasco B,
et al: Correlation of endocrine disrupting chemicals serum levels
and white blood cells gene expression of nuclear receptors in a
population of infertile women. Int J Endocrinol. 2013:5107032013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hengstler JG, Foth H, Gebel T, Kramer P-J,
Lilienblum W, Schweinfurth H, Völkel W, Wollin KM and Gundert-Remy
U: Critical evaluation of key evidence on the human health hazards
of exposure to bisphenol A. Crit Rev Toxicol. 41:263–291. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Soto AM, Brisken C, Schaeberle C and
Sonnenschein C: Does cancer start in the womb? altered mammary
gland development and predisposition to breast cancer due to in
utero exposure to endocrine disruptors. J Mammary Gland Biol
Neoplasia. 18:199–208. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Durando M, Kass L, Piva J, Sonnenschein C,
Soto AM, Luque EH and Muñoz-de-Toro M: Prenatal bisphenol A
exposure induces preneoplastic lesions in the mammary gland in
Wistar rats. Environ Health Perspect. 115:80–86. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Murray TJ, Maffini MV, Ucci AA,
Sonnenschein C and Soto AM: Induction of mammary gland ductal
hyperplasias and carcinoma in situ following fetal bisphenol A
exposure. Reprod Toxicol. 23:383–390. 2007. View Article : Google Scholar :
|
|
45
|
van der Ven LT, van de Kuil T, Leonards
PE, Slob W, Lilienthal H, Litens S, Herlin M, Håkansson H, Cantón
RF, van den Berg M, et al: Endocrine effects of
hexabromocyclododecane (HBCD) in a one-generation reproduction
study in Wistar rats. Toxicol Lett. 185:51–62. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jenkins S, Raghuraman N, Eltoum I,
Carpenter M, Russo J and Lamartiniere CA: Oral exposure to
bisphenol a increases dimethylbenzanthracene-induced mammary cancer
in rats. Environ Health Perspect. 117:910–915. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Acevedo N, Davis B, Schaeberle CM,
Sonnenschein C and Soto AM: Perinatally administered bisphenol a as
a potential mammary gland carcinogen in rats. Environ Health
Perspect. 121:1040–1046. 2013.PubMed/NCBI
|
|
48
|
Dairkee SH, Seok J, Champion S, Sayeed A,
Mindrinos M, Xiao W, Davis RW and Goodson WH: Bisphenol A induces a
profile of tumor aggressiveness in high-risk cells from breast
cancer patients. Cancer Res. 68:2076–2080. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Montes-Grajales D, Bernardes GJ and
Olivero-Verbel J: Urban endocrine disruptors targeting breast
cancer proteins. Chem Res Toxicol. 29:150–161. 2016. View Article : Google Scholar
|
|
50
|
Swan SH: Intrauterine exposure to
diethylstilbestrol: Long-term effects in humans. APMIS.
108:793–804. 2000. View Article : Google Scholar
|
|
51
|
International Agency for Research on
Cancer (IARC): Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Humans. 72. Hormonal Contraception and
Post-Menopausal Hormonal Therapy; Lyon: pp. 291–294. 1999
|
|
52
|
Cook JD, Davis BJ, Goewey JA, Berry TD and
Walker CL: Identification of a sensitive period for developmental
programming that increases risk for uterine leiomyoma in Eker rats.
Reprod Sci. 14:121–136. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
World Health Organization (WHO);
International Agency for Research on Cancer (IARC): Monographs on
the Evaluation of the Carcinogenic Risk of Chemicals to Humans.
Beryllium, cadmium, mercury, and exposures in the glass
manufacturing industry. 58. IARC; Lyon: 1993
|
|
54
|
Liu Z, Yu X and Shaikh ZA: Rapid
activation of ERK1/2 and AKT in human breast cancer cells by
cadmium. Toxicol Appl Pharmacol. 228:286–294. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
McElroy JA, Shafer MM, Trentham-Dietz A,
Hampton JM and Newcomb PA: Cadmium exposure and breast cancer risk.
J Natl Cancer Inst. 98:869–873. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Akesson A, Julin B and Wolk A: Long-term
dietary cadmium intake and postmenopausal endometrial cancer
incidence: A population-based prospective cohort study. Cancer Res.
68:6435–6441. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Julin B, Wolk A, Bergkvist L, Bottai M and
Åkesson A: Dietary cadmium exposure and risk of postmenopausal
breast cancer: A population-based prospective cohort study. Cancer
Res. 72:1459–1466. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mohapatra P, Preet R, Das D, Satapathy SR,
Siddharth S, Choudhuri T, Wyatt MD and Kundu CN: The contribution
of heavy metals in cigarette smoke condensate to malignant
transformation of breast epithelial cells and in vivo initiation of
neoplasia through induction of a PI3K-AKT-NFκB cascade. Toxicol
Appl Pharmacol. 274:168–179. 2014. View Article : Google Scholar
|
|
59
|
Rahim F, Jalali A and Tangestani R: Breast
cancer frequency and exposure to cadmium: A meta-analysis and
systematic review. Asian Pac J Cancer Prev. 14:4283–4287. 2013.
View Article : Google Scholar
|
|
60
|
Moysich KB, Shields PG, Freudenheim JL,
Schisterman EF, Vena JE, Kostyniak P, Greizerstein H, Marshall JR,
Graham S and Ambrosone CB: Polychlorinated biphenyls, cytochrome
P4501A1 polymorphism, and postmenopausal breast cancer risk. Cancer
Epidemiol Biomarkers Prev. 8:41–44. 1999.PubMed/NCBI
|
|
61
|
Sergentanis TN and Economopoulos KP: Four
polymorphisms in cytochrome P450 1A1 (CYP1A1) gene and breast
cancer risk: A meta-analysis. Breast Cancer Res Treat. 122:459–469.
2010. View Article : Google Scholar
|
|
62
|
Cohn BA, Terry MB, Plumb M and Cirillo PM:
Exposure to polychlorinated biphenyl (PCB) congeners measured
shortly after giving birth and subsequent risk of maternal breast
cancer before age 50. Breast Cancer Res Treat. 136:267–275. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bouker KB and Hilakivi-Clarke L:
Genistein: Does it prevent or promote breast cancer? Environ Health
Perspect. 108:701–708. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Shu XO, Jin F, Dai Q, Wen W, Potter JD,
Kushi LH, Ruan Z, Gao YT and Zheng W: Soyfood intake during
adolescence and subsequent risk of breast cancer among Chinese
women. Cancer Epidemiol Biomarkers Prev. 10:483–488.
2001.PubMed/NCBI
|
|
65
|
Trock B, White BL, Clarke R and
Hilakivi-Clarke L: Meta-analysis of soy intake and breast cancer
risk. Third International Symposium on the Role of Soy in
Preventing and Treating Chronic Disease. J Nutr. 130:653S–680S.
2000.
|
|
66
|
Messina MJ and Loprinzi CL: Soy for breast
cancer survivors: A critical review of the literature. J Nutr.
131(Suppl 11): 3095S–3108S. 2001.PubMed/NCBI
|
|
67
|
This P, De La Rochefordière A, Clough K,
Fourquet A and Magdelenat H; Breast Cancer Group of the Institut
Curie: Phytoestrogens after breast cancer. Endocr Relat Cancer.
8:129–134. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Horn-Ross PL, John EM, Canchola AJ,
Stewart SL and Lee MM: Phytoestrogen intake and endometrial cancer
risk. J Natl Cancer Inst. 95:1158–1164. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Unfer V, Casini ML, Costabile L, Mignosa
M, Gerli S and Di Renzo GC: Endometrial effects of long-term
treatment with phytoestrogens: A randomized, double-blind,
placebo-controlled study. Fertil Steril. 82:145–148; quiz 265.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gaete L, Tchernitchin AN, Bustamante R,
Villena J, Lemus I, Gidekel M, Cabrera G and Astorga P:
Daidzein-estrogen interaction in the rat uterus and its effect on
human breast cancer cell growth. J Med Food. 15:1081–1090. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Greathouse KL, Bredfeldt T, Everitt JI,
Lin K, Berry T, Kannan K, Mittelstadt ML, Ho SM and Walker CL:
Environmental estrogens differentially engage the histone
methyltransferase EZH2 to increase risk of uterine tumorigenesis.
Mol Cancer Res. 10:546–557. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kang NH, Hwang KA, Lee HR, Choi DW and
Choi KC: Resveratrol regulates the cell viability promoted by
17β-estradiol or bisphenol A via down-regulation of the cross-talk
between estrogen receptor α and insulin growth factor-1 receptor in
BG-1 ovarian cancer cells. Food Chem Toxicol. 59:373–379. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Reaves DK, Ginsburg E, Bang JJ and Fleming
JM: Persistent organic pollutants and obesity: Are they potential
mechanisms for breast cancer promotion? Endocr Relat Cancer.
22:R69–R86. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Patterson AR, Mo X, Shapiro A, Wernke KE,
Archer TK and Burd CJ: Sustained reprogramming of the estrogen
response after chronic exposure to endocrine disruptors. Mol
Endocrinol. 29:384–395. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Burgio E and Migliore L: Towards a
systemic paradigm in carcinogenesis: Linking epigenetics and
genetics. Mol Biol Rep. 42:777–790. 2015. View Article : Google Scholar
|
|
76
|
Berretta M, Di Francia R and Tirelli U:
The new oncologic challenges in the 3RD millennium. WCRJ.
1:e1332014.
|
|
77
|
Del Buono A, D'Orta A, Del Buono R, Del
Buono MG, De Monaco A and Marullo MN: Relationship between diet and
heavy metals in high risk of the environmental toxicity areas.
Implication for cancer prevention. WCRJ. 1:e4112014.
|