Introduction
Casticin prepared from the fruit of Vitex
trifolia L. has a wide range of activities, including antitumor
effects in a variety of cancers such as leukemia and uterine
cervix, lung, and colon cancers (1–3).
Casticin induces G2/M growth arrest and apoptosis in breast cancer
cells regardless of p53 expression, and Bcl-2 depletion is a key
event in casticin-induced apoptosis (4). Casticin also triggers tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis
in human colon cancer cells through downregulation of Bcl-2 and
X-linked inhibitor of apoptosis protein (XIAP) (3). Although wild-type p53 mediates
increased sensitivity to radiation or chemotherapeutic agents
(5), casticin induces apoptosis in
human cervical cancer HeLa cells lacking wild-type p53 expression
(6). Therefore, understanding the
role of p53 or related proteins in casticin-treated human cancer
cells is important for elucidation of the signaling pathway leading
to apoptosis.
The p53 family proteins, p53, p63, and p73, play
important roles in development and tumorigenesis (7). The transcriptionally active (TA)
isoforms of p63 and p73 can substitute for p53 in the context of
inducing apoptosis or cell cycle arrest (8,9). p73
is expressed as two major isoform classes, TAp73 and ΔNp73
(N-terminal deleted form), which have distinct functions (10). Following induction of DNA damage by
genotoxic drugs, TAp73 can bind to the same set of p53-responsive
elements and activate downstream target genes of p53 to provoke the
cell cycle arrest or induce apoptosis (11). The expression of XIAP-associated
factor 1 (XAF1), as a novel target of p53, increases the activity
of p53-dependent apoptosis (12).
XAF1 as a tumor suppressor is also markedly reduced in
muscle-invasive bladder tumors (13,14).
However, the relationship between XAF1 and TAp73 and the detailed
mechanisms of XAF1 induction in bladder cancer are still not fully
elucidated.
Intracellular production of reactive oxygen species
(ROS) triggers apoptosis of EBV-infected human B cells through
disruption of the mitochondria membrane potential resulting from
upregulation of TAp73 and XAF1 (15). Casticin also induces apoptosis of
cancer cells through the generation of ROS and activation of
caspase-9 (6). Receptor-interacting
protein (RIP) kinase family members have emerged as essential
sensors of intracellular and extracellular stresses (16), and the interaction of phosphorylated
RIP1 and RIP3 generates downstream ROS production to induce cell
death (17). In this study, we
examined the role and mechanism of ROS production in
casticin-treated bladder cancer cells and investigated whether
ROS-mediated TAp73 expression regulates the expression of XAF1 in
bladder cancer after treatment with casticin.
Materials and methods
Cell culture
The human bladder cell line T24 was purchased from
the American Type Culture Collection (Manassas, VA, USA). Cells
were maintained in RPMI-1640 medium (Hyclone, Logan, UT, USA)
supplemented with 10% FBS (Hyclone) and antibiotics under a
humidified atmosphere with 5% CO2.
Drug and chemicals
Casticin was purchased from Santa Cruz Biotechnology
(Santa Cruz, CA, USA), dissolved in sterile dimethyl sulfoxide
(DMSO) as a 200-mM stock solution, and diluted in medium to the
indicated concentration before use. z-LEHD-fmk
(z-Leu-Glu(OMe)-His-Asp-(OMe)-fluoremethylketone), z-DEVD-fmk
(N-benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone), z-VAD-fmk
(N-benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethyl ketone),
SB203580, and SP600125 were purchased from Calbiochem (San Diego,
CA, USA). N-acetylcysteine (NAC) was obtained from Sigma-Aldrich
(St. Louis, MO, USA). Necrostatin-1 was purchased from Selleckchem
(Houston, TX, USA).
Analysis of apoptotic cells by flow
cytometry
The percentage of T24 cells undergoing apoptosis was
determined by flow cytometry using FITC-labeled Annexin-V (BD
Biosciences, San Diego, CA, USA) and 7-aminoactinomycin D (7-AAD)
(BD Biosciences). To determine optimal conditions, experiments were
performed using different concentrations of casticin (0, 10, 20,
50, 100 and 200 µM) and different periods of incubation (2,
4, 8, 24 and 48 h). DMSO (0.05%) was used as a vehicle control. To
examine the role of caspases, cells were pretreated with z-LEHD-fmk
(20 µM, caspase-9 inhibitor), z-DEVD-fmk (20 µM,
caspase-3 inhibitor) or z-VAD-fmk (20 µM, pan-caspase
inhibitor) for 2 h before casticin treatment. Cells were harvested,
rinsed with PBS, and resuspended in 100 µl of 1X Annexin-V
binding buffer [10 mM HEPES/NaOH (pH 7.4), 140 mM NaCl, 2.5 mM
CaCl2]. After addition of 3 µl of Annexin-V-FITC
and 3 µl of 7-AAD the cells were incubated at room
temperature for 15 min in the dark with gentle vortexing. Stained
cells were analyzed using a FACSCalibur flow cytometer (BD
Biosciences) equipped with CellQuestpro software (BD
Biosciences).
Measurement of mitochondria membrane
potential (Δψm) and intracellular reactive oxygen
species (ROS) production
Changes in mitochondrial membrane potential were
measured using DiOC6 (3,3′-dihexyloxacarboxyanine
iodide; Molecular Probes, Eugene, OR, USA). Cells were treated with
casticin or DMSO for 24 h, washed twice in PBS, resuspended in PBS
supplemented with DiOC6 (20 nM), incubated at 37°C for
15 min in the dark, and immediately analyzed with a flow cytometer
using the FL-1 filter.
Intracellular accumulation of ROS was monitored by
flow cytometry after staining with the fluorescent probe DCFH-DA
(10 µM, 2′,7′-dichlorodihydro-fluorescein diacetate;
Molecular Probes) as previously described (15) with slight modification. DCFH-DA is
deacetylated in cells by esterases to yield a non-fluorescent
compound, DCFH, which remains trapped within the cell and is
cleaved and oxidized by ROS in the presence of endogenous
peroxidase to yield a highly fluorescent compound, DCF
(2′,7′-dichlorofluorescein). Cells were preincubated with 10
µM DCFH-DA for 30 min at 37°C and then seeded in 6-well
plates (5×105 cells/ml) and treated with or without
casticin for 24 h. Cells were washed, resuspended in PBS, and ROS
levels were determined using a FACSCalibur flow cytometer.
Western blot analysis
Cells were washed in PBS and lysed in NP-40 buffer
(Elpis Biotech, Daejeon, Korea) supplemented with a protease
inhibitor cocktail (Sigma-Aldrich). To address phosphorylation
events, an additional set of phosphatase inhibitors (Cocktail II,
Sigma-Aldrich) was added to the NP-40 buffer. Protein concentration
was determined using a BCA assay kit (Pierce, Rockford, IL, USA).
Proteins (10 µg/sample) were resolved by SDS-PAGE and
transferred to nitrocellulose (Millipore Corp., Billerica, MA,
USA). The membranes were blocked with 5% skim milk and western blot
analysis was performed using a standard commercial method.
Immunoreactivity was detected using an enhanced chemiluminescence
(ECL) kit (Advansta Corp., Menlo Park, CA, USA) and Multiple Gel
DOC system (Fujifilm). Primary antibodies (Abs) against the
following proteins were used: caspase-8, caspase-3, caspase-9,
PARP, β-actin, Bcl-2, Bax, Mcl-1, Bak, p53, XIAP, RIP, phospho-JNK
(Thr183/Tyr185), JNK, phospho-p38-MAPK
(Thr180/Tyr182), p38-MAPK, phospho-ERK1/2
(Thr202/Tyr204), and ERK1/2 (Cell Signaling
Technology, Beverly, MA, USA); XAF1 and TAp73 (Abcam, Cambridge,
UK); TAp63 (BioLegend, San Diego, CA, USA).
Small interfering RNA (siRNA)
transfection
Experimentally verified human TAp73-siRNA duplex and
negative control-siRNA were obtained from Bioneer (Daejeon, Korea).
Cells were transiently transfected under optimized conditions.
Briefly, cells were seeded at a concentration of 1×105
cells per well in a 6-well plate and grown overnight. Cells in each
well were transfected with 200 nM siRNA using Lipofectamine RNAiMAX
Reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's instructions. Cells were used for further
experiments 48 h after transfection.
Statistical analysis
Data were expressed as mean ± standard deviation
(SD). Statistical analysis was conducted using one-way analysis of
the variance (ANOVA). P-values <0.05 were considered
statistically significant.
Results
Casticin induces apoptosis of bladder
cancer cells in a time-and dose-dependent manner
To evaluate whether casticin affects cell survival
through regulation of the mitochondria membrane potential, the
percentage of apoptotic cells in casticin-treated T24 cells was
determined by flow cytometry using FITC-labeled Annexin-V and 7-AAD
and the mitochondria membrane potential was measured by staining
with DiOC6. Casticin treatment of T24 cells resulted in
a dose- and time-dependent increase in the
Annexin-V+/7-AAD+ population (Fig. 1A and B). Casticin also significantly
disrupted the integrity of the mitochondria membrane after 8 h of
treatment (Fig. 1A and B). We
further analyzed the activity of caspases by immunoblotting. Levels
of cleaved forms of caspase-8, caspase-9 and caspase-3 were
increased in T24 cells after treatment with casticin. Cleavage of
PARP was also detected in the presence of casticin (Fig. 1C). Treatment with specific caspase
inhibitors z-LEHD-fmk (caspase-9 inhibitor) or z-DEVD-fmk
(caspase-3 inhibitor) or the pan-caspases inhibitor z-VAD,
effectively blocked casticin-mediated apoptosis (Fig. 1D). These results suggest that
casticin-induced apoptosis of bladder cancer cells involves
interruption of the mitochondria membrane potential and activation
of caspases.
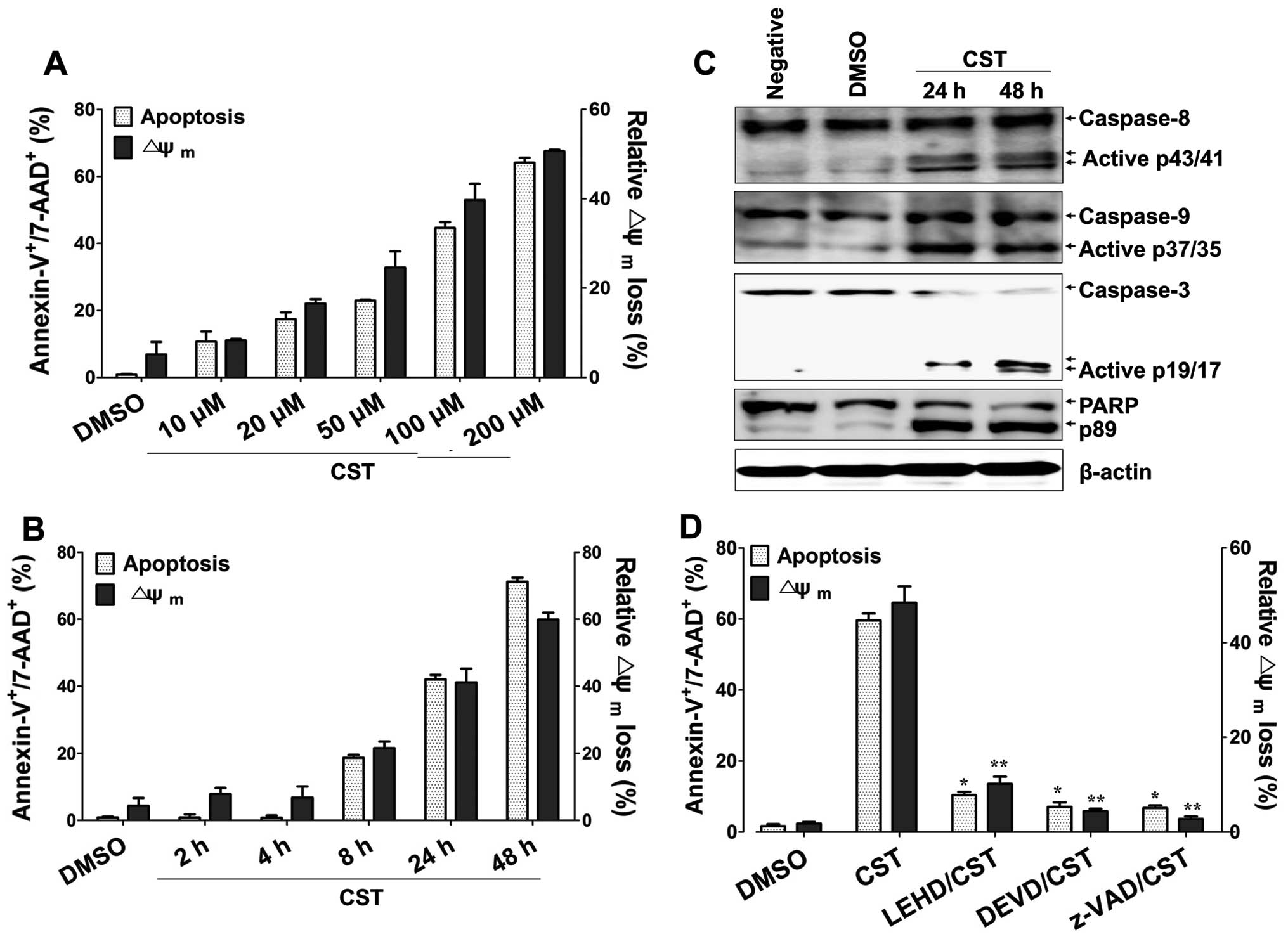 | Figure 1Casticin (CST) induces
caspase-mediated apoptosis in human bladder cancer cells. (A) Cells
were treated with 10, 20, 50, 100 or 200 µM casticin for 24
h. (B) Cells were treated with 100 µM casticin for 2, 4, 8,
24 or 48 h. (C) Cells were treated with 100 µM casticin for
the indicated times. (D) Cells were preincubated with z-LEHD-fmk
(20 µM), z-DEVD-fmk (20 µM) or z-VAD-fmk (20
µM) for 2 h and then treated with 100 µM casticin for
24 h. Western blot analysis of active casapse-8, -9 and -3 or PARP
cleavage was performed to characterize the apoptotic response.
β-actin was used to normalize protein content. To detect the degree
of apoptosis, cells were stained with Annexin-V-FITC and 7-AAD and
analyzed by flow cytometry. The percentage of apoptotic cells was
determined by the proportion of
Annexin-V+/7-AAD+ cells. To measure
disruption of Δψm, cells were stained with
DiOC6. Diminished DiOC6 fluorescence (%)
indicates Δψm disruption. Each value represents the mean
± SD of three determinations. *p<0.01 (apoptosis of
T24 treated with casticin versus T24 treated with inhibitor and
casticin); **p<0.01 (Δψm of T24 treated
with casticin versus T24 treated with inhibitor and casticin). |
MAPK-mediated expression of TAp73 and
XAF1 regulates casticin-induced apoptosis
On the basis of previous results we investigated the
expression of XAF1, apoptosis-related proteins, and p53 family
proteins to elucidate the underlying mechanism of casticin-induced
T24 cell apoptosis. Although casticin treatment had no influence on
the expression level of p53 and induction of TAp63, the level of
TAp73 was markedly elevated in casticin-exposed T24 cells. XAF1
expression was also upregulated, whereas XIAP was downregulated
(Fig. 2A). The levels of
antiapoptotic proteins, including Mcl-2 and Bcl-2, were decreased,
whereas expression of proapoptotic proteins (Bak and Bax) was
increased (Fig. 2B). Because JNK
and p38 MAPK regulate phosphorylation and activation of TAp73
(18,19), we next examined whether activation
of mitogen-activated protein kinases (MAPKs) is correlated with the
induction of TAp73 in casticin-treated T24 cells. Casticin
significantly induced the phosphorylation of JNK and p38 MAPK,
whereas activation of ERK was blocked (Fig. 2C). Pharmacological inhibition of JNK
and p38 with SP600125 and SB203580, respectively, not only
suppressed the activation of caspase-9 and caspase-3 (Fig. 2D), but also attenuated the
casticin-induced expression of XAF1, TAp73, Bak and Bax (Fig. 2E and F). These results suggest that
MAPKs are involved in the upregulation of XAF1 and TAp73 in T24
cells after treatment with casticin.
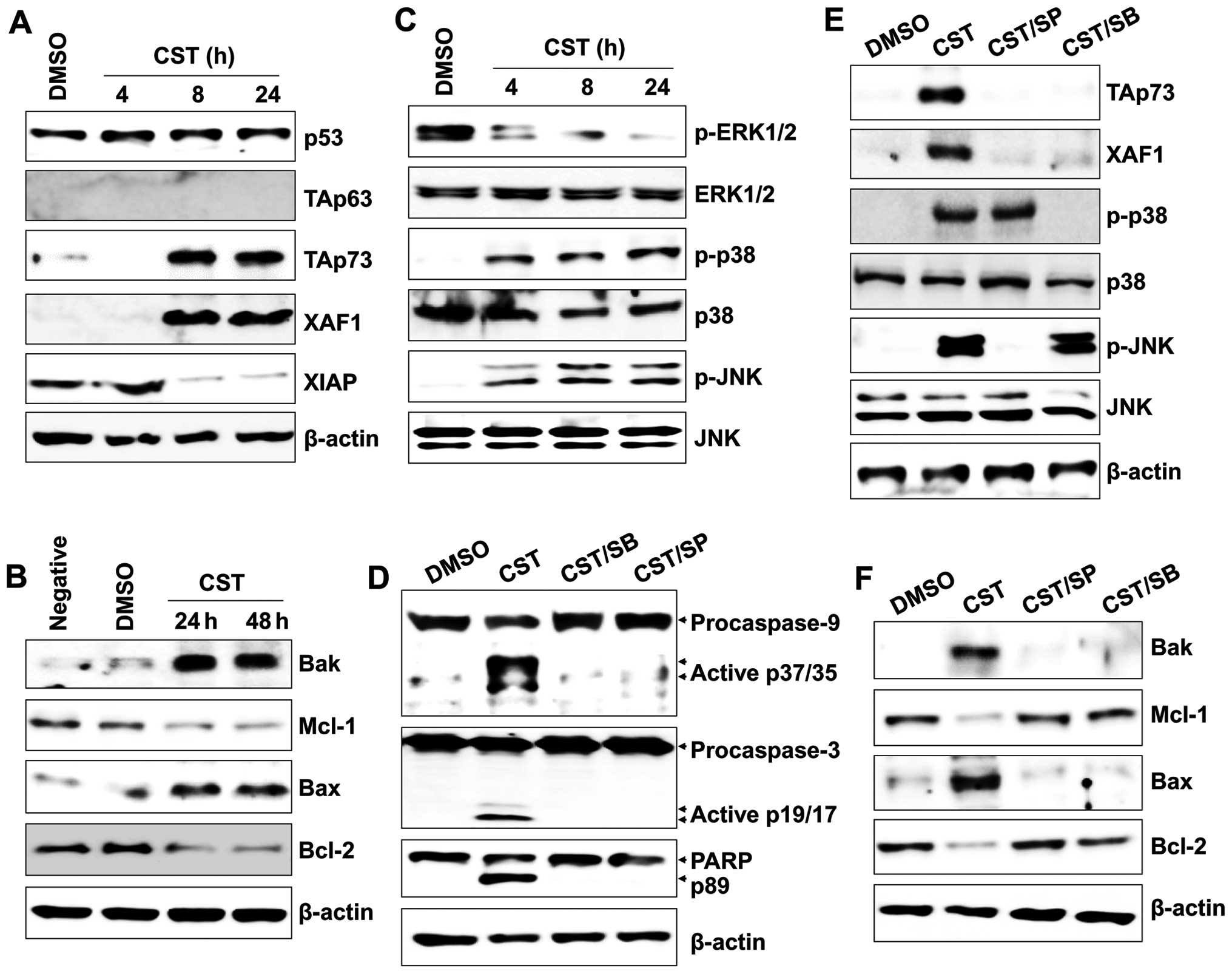 | Figure 2The role of JNK and p38-MAPK in TAp73
expression in human bladder cancer cells after casticin (CST)
treatment. Cells were treated with 100 µM casticin for the
indicated times. Total proteins were extracted from cell lysates
and western blot analysis was performed for (A) p53, TAp63, TAp73,
XAF1 or XIAP protein (B) and Bax, Bcl-2, Mcl-1 or Bak protein. (C)
Total cell lysates were immunoblotted with antibody against p-ERK,
ERK, p-JNK, JNK, p-p38-MAPK, and p38-MAPK. (D and E) Cells were
pretreated with 25 µM SP600125 (SP) or 10 µM SB203580
(SB) for 1 h. Cells were then washed with PBS and treated with 100
µM casticin for 24 h. Total cell lysates were immunoblotted
with antibody against (D) caspase-9, caspase-3 or PARP, (E) XAF1,
TAp73, p-p38-MAPK, MAPK, p-JNK or JNK, and (F) Bak, Bax, Bcl-2 or
Mcl-1. β-actin was used to normalize protein content. Results are
representative of three independent experiments. |
ROS-modulated TAp73 controls XAF1
expression and activation of caspases after treatment with
casticin
We next investigated whether the apoptotic effect of
XAF1 is regulated by the expression and activation of TAp73 in
casticin-exposed T24 cells. To define the relationship between
TAp73 activation and induction of XAF1, we analyzed the expression
of XAF1 and activation of MAPKs in casticin-treated T24 cells after
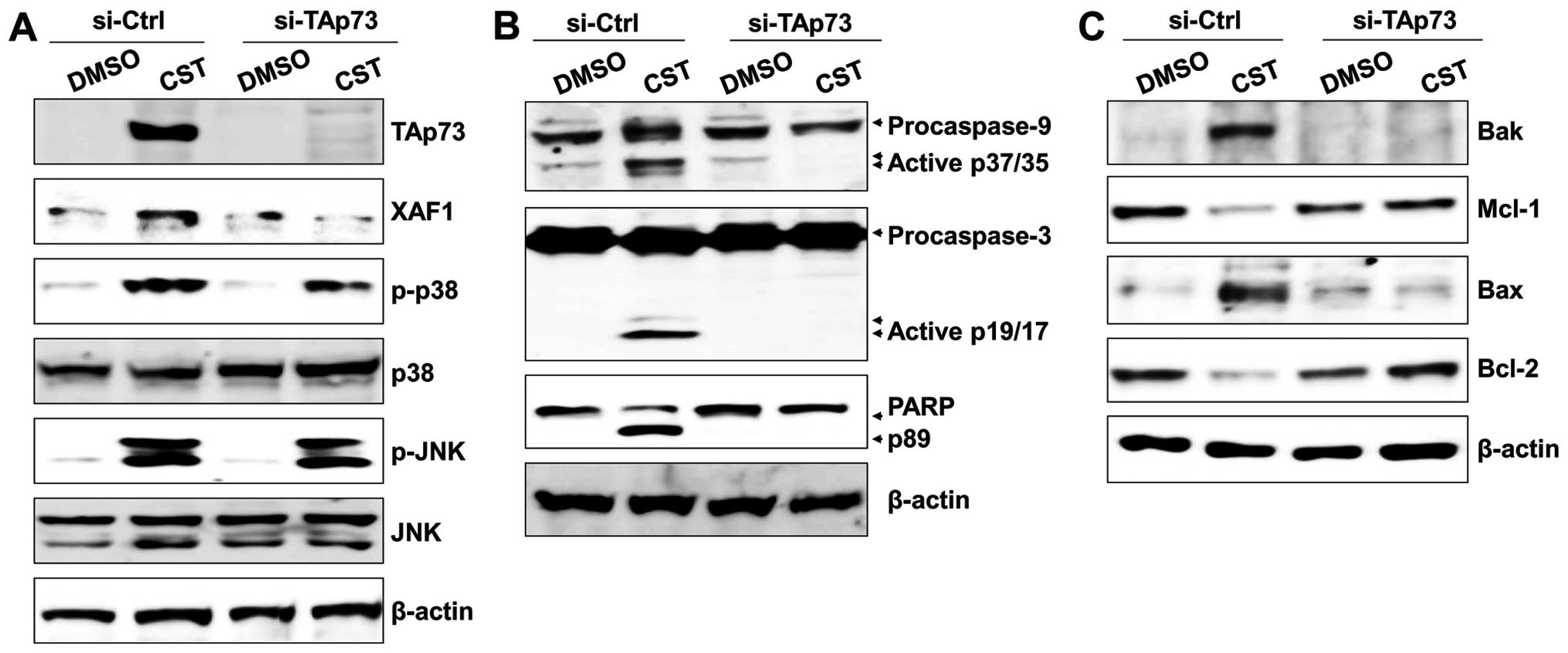
silencing TAp73 with siRNA. Knockdown of TAp73 with siRNA resulted
in downregulation of XAF1 but had no effect on the activation of
JNK and p38 MAPK (Fig. 3A). Gene
silencing of TAp73 effectively blocked the cleavage of caspase-9,
caspase-3, and PARP in casticin-treated T24 cells (Fig. 3B). In addition, the level of
proapoptotic proteins (Bak and Bax) was suppressed in
TAp73-knockdown T24 cells after treatment with casticin (Fig. 3C). As ROS production is an early
signal mediator for apoptosis induction after treatment with
casticin (3,6,20) we
examined the effect of ROS on XAF1 and TAp73 activation in
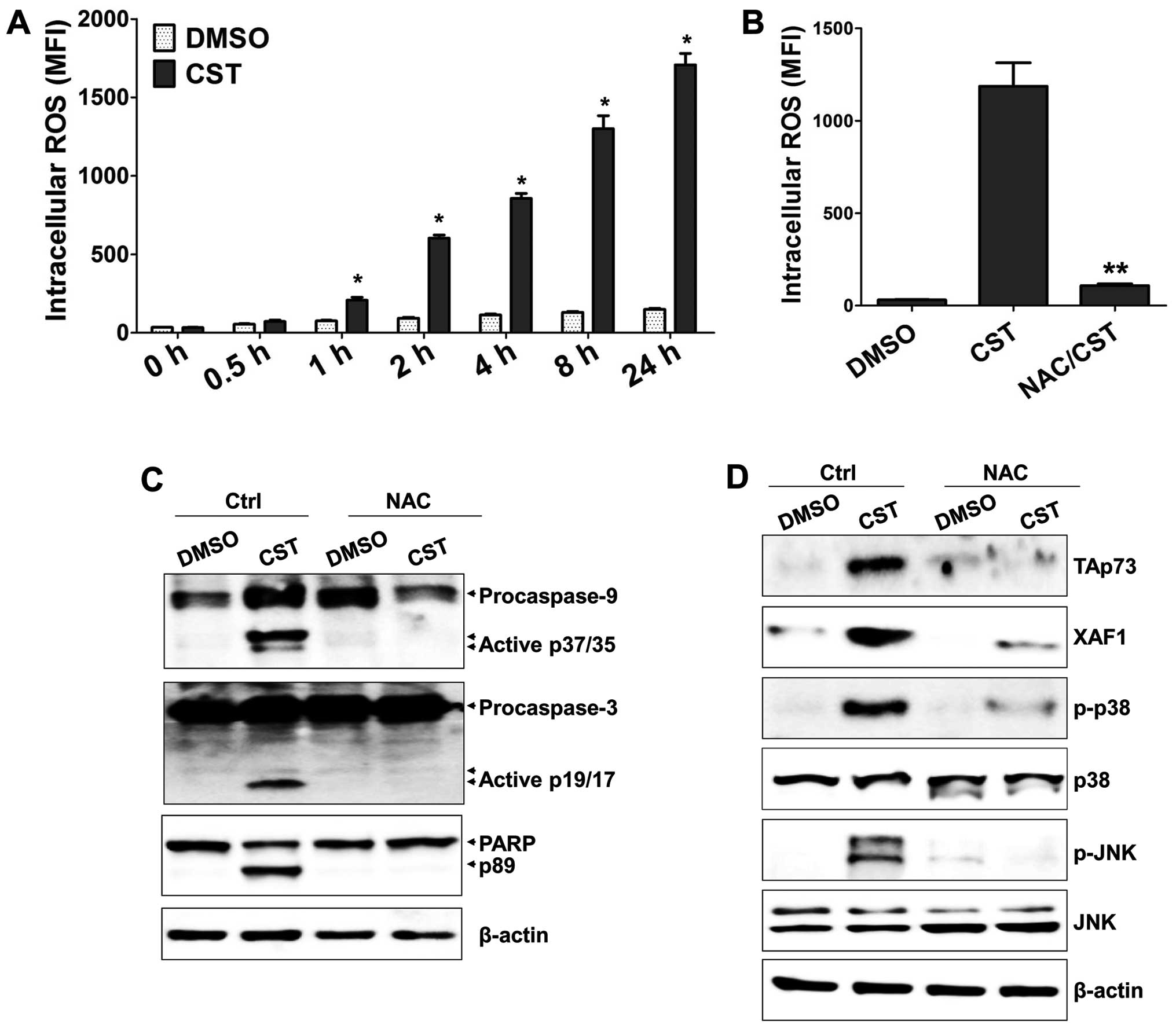
casticin-treated T24 cells. Casticin upregulated intracellular ROS
levels within 1 h (Fig. 4A).
Moreover, NAC (a ROS scavenger) not only efficiently inhibited ROS
production (Fig. 4B), but also
blocked generation of the cleaved forms of caspase-9, caspase-3,
and PARP (Fig. 4C). Furthermore,
expression of XAF1, TAp73, phosphorylated p38, and phosphorylated
JNK was suppressed by NAC treatment (Fig. 4D). These results suggest that
ROS-mediated TAp73 expression is one of the critical factors for
activation of XAF1 and induction of apoptosis in casticin-treated
T24 cells.
RIP-mediated ROS generation initiates the
TAp73/XAF1 apoptosis pathway in casticin-treated T24 cells
RIP kinases mediate apoptosis or necrosis induced by
tumor necrosis factor (TNF)-α or genotoxic stresses through the
accumulation of ROS (21,22). ROS are also closely associated with
the caspase cascade and activation of MAPKs (23). Our previous results showed that
casticin promoted ROS-induced MAPK phosphorylation to activate
caspases and induce apoptosis of T24 cells. Therefore, we examined
whether RIP kinases are involved in ROS production for activation
of the TAp73/XAF1 signaling pathway in casticin-treated T24 cells.
Casticin elicited RIP kinase expression (Fig. 5A). Treatment with necrostatin-1 (a
RIP kinase inhibitor) blocked the cleavage of caspases (Fig. 5B), attenuated ROS generation
(Fig. 5C), and suppressed
MAPK-mediated TAp73/XAF1 activation (Fig. 5D) in T24 cells after treatment with
casticin. However, gene silencing of TAp73 with siRNA and treatment
with MAPK inhibitors (SP600125 and SB203580) failed to block the
expression of RIP kinase in casticin-treated T24 cells (Fig. 5E and F). In addition, NAC treatment
of casticin-exposed T24 cells had no influence on the activation of
RIP kinase (Fig. 5G). These results
suggest that the activation of RIP kinase plays a critical role in
casticin-induced apoptosis through ROS production.
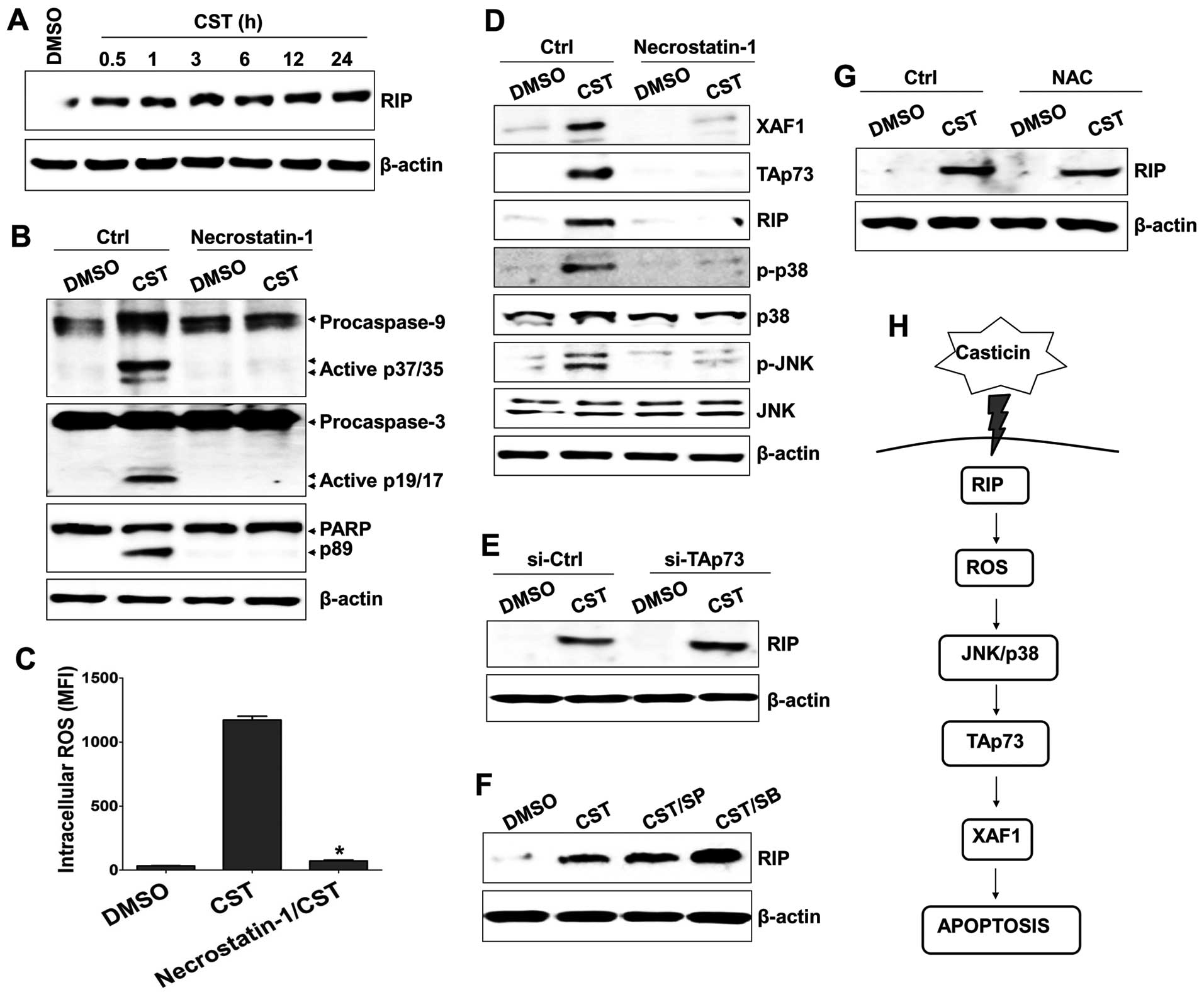 | Figure 5Receptor-interacting protein (RIP) is
an upstream signaling molecule in casticin (CST)-induced ROS
generation and TAp73 expression. (A) Cells were treated with 100
µM casticin for the indicated times. Total proteins were
extracted from cell lysates and subjected to western blotting for
RIP. (B–D) Cells were pretreated with 30 µM necrostatin-1
for 1 h to block RIP expression and then treated with 100 µM
casticin or DMSO at 37°C for 24 h. (B) Whole cell lysates were
subjected to western blot analysis using antibodies to caspase-9,
-3 or PARP. (C) Effects of necrostatin-1 on casticin-induced ROS
production was measured using flow cytometry. The numbers in
histograms indicate the mean fluorescence intensity (MFI) for
DCFH-DA. *p<0.01 (ROS production of T24 treated with
casticin versus T24 treated with necrostatin-1 and casticin). (D)
Whole cell lysates were subjected to western blot analysis using
antibodies against RIP, XAF1, TAp73, p-p38-MAPK, MAPK, p-JNK or
JNK. (E) Cells were treated with 100 µM casticin for 6 h and
then transfected with 200 nM si-TAp73. Cells were used for further
experiments 48 h after transfection. Whole cell lysates were
subjected to western blot analysis of RIP. (F) Cells were
pretreated with 25 µM SP600125 (SP) or 10 µM SB203580
(SB) for 1 h, washed with PBS, and treated with 100 µM
casticin for 24 h. Total cell lysates were immunoblotted with
antibody against RIP. (G) Cells were pretreated with 10 mM
N-acetylcysteine for 1 h, washed with PBS, and treated with 100
µM casticin for 24 h. Total cell lysates were immunoblotted
with antibody against RIP. β-actin was used to normalize protein
content. (H) Schematic diagram of the intracellular signaling
mechanism during casticin-induced apoptosis in human bladder cancer
cells. |
Discussion
Wild-type p53 functions as a cancer suppressor by
increasing the sensitivity of cells to radiation or
chemotherapeutic agents (5);
however, casticin induces apoptotic cell death in p53 mutant or
null breast and cervical cancer cells (1,4).
Previous studies have shown that casticin enhances TRAIL-induced
apoptosis and suppresses cell survival proteins, including Bcl-2
and XIAP (3). Melphalan, a
therapeutic agent for multiple myeloma, promotes TAp73-mediated
XAF1 expression via ROS generation and activates the p38 MAPK
pathway to induce apoptosis (15).
Based on these results, we investigated whether casticin
potentiates the ROS-mediated apoptosis pathway in a p53-independent
manner. We also examined the underlying mechanism of XAF1
activation and the relationship between TAp73 and XAF in
casticin-treated T24 cells.
The XAF1 gene, which is located at 17p13.2,
was first identified as a novel negative regulator of XIAP
(24). XAF1 binds to XIAP and
antagonizes its ability to mediate apoptosis through activation of
the caspase cascade in cancer cells (25). XAF1 levels are significantly lower
in cancer tissues than in normal tissues (26); however, XAF1 expression is
significantly increased in colorectal cancer cells after treatment
with cisplatin (27). A reduction
in XAF1 expression was reported in 50% (10 of 20) of transitional
cell carcinomas and 60% (12 of 20) of renal cell carcinomas
(14). In addition, this reduction
of XAF1 was significantly greater in invasive and high-grade tumors
(14). Because XAF1 expression is
involved in activation of p53 and expression of its downstream
target genes (14) it is essential
to understand the changes in p53 family proteins, including p63 and
p73. As a proapoptotic protein, XAF1 promotes the translocation of
Bax to the mitochondria and the release of cytochrome c from
mitochondria to trigger apoptosis (28). In this study, we showed that
casticin induced apoptosis of T24 bladder cancer cells through
activation of caspases and disruption of the mitochondria membrane
potential. ROS-mediated MAPK activation triggered expression of
TAp73 and activation of XAF1 in casticin-treated T24 cells
(Fig. 5H). Loss of TAp73 expression
led to downregulation of XAF1 and suppression of casticin-induced
apoptosis. These results suggest that the TAp73/XAF1 signaling
pathway is a new therapeutic target for modulating carcinogenesis
in the bladder.
The TAp73 protein, which is activated by Ras,
controls the expression of glucose-6-phosphate dehydrogenase for
glucose metabolism in bladder cancer cells (29). In addition to its role in tumor
suppression and promotion, p73 is required for neuronal
differentiation and development of the central nervous system
(8). TAp73-knockdown mammary
epithelial cells lose their epithelial characteristics and express
mesenchymal markers, including Snail-1, Slug, and Twist (30). The expression of fibroblast growth
factor receptor 3 (FGFR3), a transcriptional target of TAp73, is
closely related to the transition of superficial bladder cancers to
an invasive phenotype (31). These
results suggest that TAp73 is not only a key regulatory molecule in
the control of cancer cell survival and carcinogenesis but also an
alternative target for blocking metastasis in bladder cancer.
In addition to their role in inflammation and other
immune responses, RIP kinases also have an important function in
death-inducing processes (32).
RIP1 binds to several death receptors, such as the death domain of
receptor Fas (CD95), TNF receptor 1 (TNF-R1), TRAIL receptor 1
(TRAIL-R1), and TRAIL-R2 (33,34).
RIP3 can interact with phosphorylated RIP1 upon TNF stimulation,
leading to the generation of necrosomes and cell death (17). RIP1 plays an important role in
TNF-R1-mediated NF-κB activation for cell survival (35,36).
In contrast, casticin enhances the TRAIL-induced death of colon
cancer cells (3). In this study we
showed that casticin-treated bladder cancer cells produce ROS
through the activation of RIP kinase and thereby initiate the
apoptosis signaling pathway. Although necrostatin-1 (an inhibitor
of RIP kinase) blocked the entire apoptotic process of
casticin-treated T24 cells, treatment with MAPK inhibitors or gene
silencing of TAp73 with siRNA failed to suppress the activation of
RIP kinase after treatment with casticin. These results suggest
that ROS generation induced by RIP kinase activation is a critical
upstream event in casticin-induced apoptosis in T24 bladder cancer
cells.
Urothelial carcinoma is the fifth most common cancer
in developed countries (37). XAF1
levels in bladder cancer cells are significantly decreased compared
with those in normal cells (14).
Casticin has previously been shown to exert anti-tumor activity by
activating proapoptotic pathways in various cancers (22). The results of this study implicate
casticin as a new candidate therapeutic drug for the control of
bladder cancer through enhancement of the TAp73-induced XAF1
apoptosis signaling pathway.
Acknowledgments
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(NRF-2015R1D1A1A01056672).
References
|
1
|
Csupor-Löffler B, Hajdú Z, Zupkó I, Réthy
B, Falkay G, Forgo P and Hohmann J: Antiproliferative effect of
flavonoids and sesquiterpenoids from Achillea millefolium s.1. on
cultured human tumour cell lines. Phytother Res. 23:672–676. 2009.
View Article : Google Scholar
|
|
2
|
Shen JK, Du HP, Yang M, Wang YG and Jin J:
Casticin induces leukemic cell death through apoptosis and mitotic
catastrophe. Ann Hematol. 88:743–752. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang SY, Zhong MZ, Yuan GJ, Hou SP, Yin
LL, Jiang H and Yu ZY: Casticin, a flavonoid, potentiates
TRAIL-induced apoptosis through modulation of anti-apoptotic
proteins and death receptor 5 in colon cancer cells. Oncol Rep.
29:474–480. 2013.
|
|
4
|
Haïdara K, Zamir L, Shi QW and Batist G:
The flavonoid Casticin has multiple mechanisms of tumor
cytotoxicity action. Cancer Lett. 242:180–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pirollo KF, Bouker KB and Chang EH: Does
p53 status influence tumor response to anticancer therapies?
Anticancer Drugs. 11:419–432. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen D, Cao J, Tian L, Liu F and Sheng X:
Induction of apoptosis by casticin in cervical cancer cells through
reactive oxygen species-mediated mitochondrial signaling pathways.
Oncol Rep. 26:1287–1294. 2011.PubMed/NCBI
|
|
7
|
Deyoung MP and Ellisen LW: p63 and p73 in
human cancer: Defining the network. Oncogene. 26:5169–5183. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang A, Walker N, Bronson R, Kaghad M,
Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, et
al: p73-deficient mice have neurological, pheromonal and
inflammatory defects but lack spontaneous tumours. Nature.
404:99–103. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Melino G, De Laurenzi V and Vousden KH:
p73: Friend or foe in tumorigenesis. Nat Rev Cancer. 2:605–615.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dötsch V, Bernassola F, Coutandin D, Candi
E and Melino G: p63 and p73, the ancestors of p53. Cold Spring Harb
Perspect Biol. 2:a0048872010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jost CA, Marin MC and Kaelin WG Jr: p73 is
a simian [correction of human] p53-related protein that can induce
apoptosis. Nature. 389:191–194. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zou B, Chim CS, Pang R, Zeng H, Dai Y,
Zhang R, Lam CS, Tan VP, Hung IF, Lan HY, et al: XIAP-associated
factor 1 (XAF1), a novel target of p53, enhances p53-mediated
apoptosis via post-translational modification. Mol Carcinog.
51:422–432. 2012. View
Article : Google Scholar
|
|
13
|
Liston P, Fong WG, Kelly NL, Toji S,
Miyazaki T, Conte D, Tamai K, Craig CG, McBurney MW and Korneluk
RG: Identification of XAF1 as an antagonist of XIAP anti-Caspase
activity. Nat Cell Biol. 3:128–133. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee MG, Huh JS, Chung SK, Lee JH, Byun DS,
Ryu BK, Kang MJ, Chae KS, Lee SJ, Lee CH, et al: Promoter CpG
hyper-methylation and downregulation of XAF1 expression in human
urogenital malignancies: Implication for attenuated p53 response to
apoptotic stresses. Oncogene. 25:5807–5822. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park GB, Kim YS, Kim D, Kim S, Lee HK, Cho
DH, Lee WJ and Hur DY: Melphalan-induced apoptosis of
EBV-transformed B cells through upregulation of TAp73 and XAF1 and
nuclear import of XPA. J Immunol. 191:6281–6291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Declercq W, Vanden Berghe T and
Vandenabeele P: RIP kinases at the crossroads of cell death and
survival. Cell. 138:229–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FK: Phosphorylation-driven assembly of the
RIP1-RIP3 complex regulates programmed necrosis and virus-induced
inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang P, Yu W, Hu Z, Jia L, Iyer VR,
Sanders BG and Kline K: Involvement of JNK/p73/NOXA in vitamin E
analog-induced apoptosis of human breast cancer cells. Mol
Carcinog. 47:436–445. 2008. View
Article : Google Scholar
|
|
19
|
Sanchez-Prieto R, Sanchez-Arevalo VJ,
Servitja JM and Gutkind JS: Regulation of p73 by c-Abl through the
p38 MAP kinase pathway. Oncogene. 21:974–979. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang J, Yang Y, Tian L, Sheng XF, Liu F
and Cao JG: Casticin-induced apoptosis involves death receptor 5
upregulation in hepatocellular carcinoma cells. World J
Gastroenterol. 17:4298–4307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin Y, Choksi S, Shen HM, Yang QF, Hur GM,
Kim YS, Tran JH, Nedospasov SA and Liu ZG: Tumor necrosis
factor-induced nonapoptotic cell death requires
receptor-interacting protein-mediated cellular reactive oxygen
species accumulation. J Biol Chem. 279:10822–10828. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kaiser WJ and Offermann MK: Apoptosis
induced by the toll-like receptor adaptor TRIF is dependent on its
receptor interacting protein homotypic interaction motif. J
Immunol. 174:4942–4952. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Müller M, Schilling T, Sayan AE, Kairat A,
Lorenz K, Schulze-Bergkamen H, Oren M, Koch A, Tannapfel A,
Stremmel W, et al: TAp73/Delta Np73 influences apoptotic response,
chemosensitivity and prognosis in hepatocellular carcinoma. Cell
Death Differ. 12:1564–1577. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liston P, Roy N, Tamai K, Lefebvre C,
Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda JE,
MacKenzie A, et al: Suppression of apoptosis in mammalian cells by
NAIP and a related family of IAP genes. Nature. 379:349–353. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Arora V, Cheung HH, Plenchette S, Micali
OC, Liston P and Korneluk RG: Degradation of survivin by the
X-linked inhibitor of apoptosis (XIAP)-XAF1 complex. J Biol Chem.
282:26202–26209. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fong WG, Liston P, Rajcan-Separovic E, St
Jean M, Craig C and Korneluk RG: Expression and genetic analysis of
XIAP-associated factor 1 (XAF1) in cancer cell lines. Genomics.
70:113–122. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ju WC, Huang GB, Luo XY, Ren WH, Zheng DQ,
Chen PJ, Lou YF and Li B: X-linked inhibitor of
apoptosis-associated factor l (XAFl) enhances the sensitivity of
colorectal cancer cells to cisplatin. Med Oncol. 31:2732014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Straszewski-Chavez SL, Visintin IP,
Karassina N, Los G, Liston P, Halaban R, Fadiel A and Mor G: XAF1
mediates tumor necrosis factor-alpha-induced apoptosis and X-linked
inhibitor of apoptosis cleavage by acting through the mitochondrial
pathway. J Biol Chem. 282:13059–13072. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X, Wu G, Cao G, Yang L, Xu H, Huang J
and Hou J: Zoledronic acid inhibits the pentose phosphate pathway
through attenuating the Ras-TAp73-G6PD axis in bladder cancer
cells. Mol Med Rep. 12:4620–4625. 2015.PubMed/NCBI
|
|
30
|
Zhang Y, Yan W, Jung YS and Chen X:
Mammary epithelial cell polarity is regulated differentially by p73
isoforms via epithelial-to-mesenchymal transition. J Biol Chem.
287:17746–17753. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sayan AE, D'Angelo B, Sayan BS, Tucci P,
Cimini A, Cerù MP, Knight RA and Melino G: p73 and p63 regulate the
expression of fibroblast growth factor receptor 3. Biochem Biophys
Res Commun. 394:824–828. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang D, Lin J and Han J:
Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol.
7:243–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stanger BZ, Leder P, Lee TH, Kim E and
Seed B: RIP: A novel protein containing a death domain that
interacts with Fas/APO-1 (CD95) in yeast and causes cell death.
Cell. 81:513–523. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chaudhary PM, Eby M, Jasmin A, Bookwalter
A, Murray J and Hood L: Death receptor 5, a new member of the TNFR
family, and DR4 induce FADD-dependent apoptosis and activate the
NF-kappaB pathway. Immunity. 7:821–830. 1997. View Article : Google Scholar
|
|
35
|
Festjens N, Vanden Berghe T, Cornelis S
and Vandenabeele P: RIP1, a kinase on the crossroads of a cell's
decision to live or die. Cell Death Differ. 14:400–410. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin Y, Devin A, Cook A, Keane MM, Kelliher
M, Lipkowitz S and Liu ZG: The death domain kinase RIP is essential
for TRAIL (Apo2L)-induced activation of IkappaB kinase and c-Jun
N-terminal kinase. Mol Cell Biol. 20:6638–6645. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|