Introduction
Characterized by high mortality and poor prognosis,
esophageal cancer is one of the most common malignant tumors
worldwide, and the 5-year survival rate of advanced stage
esophageal squamous cell carcinoma (ESCC) patients is only 10%. In
China, ESCC is the main histologic subtype, and the Henan Province
of Northern China, Linzhou and its adjacent counties have the
highest incidence of ESCC worldwide (1,2).
Furthermore, previous studies have revealed that ESCC development
is closely associated with various risk factors, such as heredity,
environment and diet (3). Such as
other solid tumors, the occurrence of ESCC is a long process with
numerous genetic alterations, including oncogene activation and
tumor-suppressor gene inactivation. As detected by comparative
genome hybridization and loss of heterozygosity, 3p deletion is one
of the most common genetic changes in ESCC, therefore there may be
one or more tumor-suppressor genes in 3p (4–7). A
number of genes located in 3p have already been considered as
candidate tumor-suppressor genes in ESCC, such as FHIT, RASSF1A,
CACNA2D2, DLEC1 and PLCδ1 (8–11).
As a member of the semaphorin family, semaphorin 3B
(SEMA3B) is located in the 3p21.3 region. SEMA3B encodes a secreted
protein consisting of 749 amino acids, including a signal peptide,
a highly conservative Sema structure which has an immune globulin
structure domain. Identified as a candidate tumor-suppressor gene,
SEMA3B exhibits frequent allelic loss and downregulation in lung
and breast cancer, ovarian and hepatocellular carcinoma and
cholangiocarcinomas (12–15). However, the relationship between
SEMA3B and ESCC occurrence and development has not been explored.
In our previous study, SEMA3B was found to be frequently
downregulated in ESCC specimens, which was detected by cDNA
microarray analysis (16). In the
present study, reverse transcription-polymerase chain reaction
(RT-PCR) and immunohistochemical (IHC) staining were employed to
determine SEMA3B expression at the mRNA and protein levels in
clinical ESCC samples. Moreover, in vitro assays were
adopted to study the tumor-suppressive function of SEMA3B. In
addition, the tumor-suppressive mechanism of SEMA3B and its
clinical significance in ESCC were investigated as well.
Materials and methods
Cell lines and primary tumor
specimens
Three Chinese ESCC cell lines (HKESC1, EC18 and
EC109), six Japanese ESCC cell lines (KYSE30, KYSE140, KYSE180,
KYSE410, KYSE510 and KYSE520; reviewed in ref. 17) and two immortalized esophageal
epithelial cell lines (NE1 and NE3) were kindly provided by
Professor Srivastava (Department of Pathology, University of Hong
Kong). After surgical resection at Linzhou Cancer Hospital, a total
of 300 primary ESCC tumor tissue samples and their paired
non-tumorous tissue samples were immediately collected from the
proximal resection margins. Some were quickly put into vials and
stored in liquid nitrogen and other tumor tissues were routinely
formalin-fixed and paraffin-embedded. Before operative treatment,
no patients recruited in the study received chemotherapy or
radiotherapy and the follow-up period was 60 months. Samples used
in the present study were approved by the Individual Institutional
Committees for Ethical Review of Research Involving Human
Subjects.
Semi-quantitative reverse
transcription-PCR
Through the application of TRIzol (Invitrogen,
Carlsbad, CA, USA), total RNA was extracted from the cell lines and
frozen ESCC tissues. Advantage RT for PCR kit (Clontech, Mountain
View, CA, USA) was employed to conduct reverse transcription of
total RNA (2 μg) and cDNA was subjected to PCR for 30 cycles
of amplification with the following pairs of primers: SEMA3B Fw,
5′-CCAGTGCCAAGAGGCGGTTC and SEMA3B Rv, 5′-AGCACCTGGGTGTGGGCTGT. The
GAPDH gene served as a control with the pair of primers: GAPDH Fw,
5′-CGGGAAGCTTGTCATCAATGG and GAPDH Rv, 5′-GGCAGTGATGGCATGGACTG. In
order to analyze the relative grey value, the mRNA expression was
quantified by Quantity One software, during which GAPDH was
regarded as an internal control.
Tissue microarrays and
immunohistochemistry
As previously described, tissue microarrays (TMA)
containing 300 pairs of primary ESCC tissue samples and their
corresponding non-tumorous tissues were constructed (19). In short, tissue sections with a
thickness of 5 μm were cut from tissue microarray blocks and
mounted on microscope slides. By adopting the standard
streptavidin-biotin-peroxidase complex method, immunohistochemistry
(IHC) was performed (19). TMA
slides were deparaffinized in xylene, rehydrated through a graded
alcohol series and incubated with 3% hydrogen peroxide. For antigen
retrieval, TMA slides were boiled in 10 mM sodium citrate buffer
(pH 6.0) for 15 min with the help of a pressure cooker. Blocked by
10% normal rabbit serum at room temperature for 30 min, the slides
were then incubated with primary anti-SEMA3B monoclonal antibody
(1:50 dilution; Lifespan) at 4°C overnight. At a concentration of
1:75, the TMA sections were incubated with biotinylated goat
anti-rabbit immunoglobulin for 30 min at 37°C. Through the
application of ImmPRESS™ Universal Antibody kit, primary antibody
staining was visualized with NovaRed (both from Vector
Laboratories, Burlingame, CA, USA) as a substrate. In the next
step, the sections were counterstained with hematoxylin, dehydrated
and mounted. As previously described, an immunoreactivity scoring
(IRS) system was applied (19). The
percentage of SEMA3B-positive cells was scored as 0, <5%,
negative; 1, 5–25%, sporadic; 2, >25–50%, focal; 3, >50%,
diffuse; and the intensity of SEMA3B-positive staining was scored
as 0, negative; 1, weak; 2, moderate; and 3, strong. Both the
percentage of positive cells and cell staining intensity were
determined in a double-blinded manner. According to the following
formula, the total score was determined: Staining index = intensity
× positive rate. In the present study, a staining index of ≤4 was
considered to be downregulated expression and a staining index of
>4 was considered as normal expression.
SEMA3B-30-KYSE30 (SEMA3B-30) esophageal
cancer cell line construction with SEMA3B overexpression
In order to test the function and mechanism
underlying growth inhibition by SEMA3B, SEMA3B was PCR amplified,
sequence-verified, cloned into pcDNA3.1(+) vector (Invitrogen) and
transfected into ESCC, KYSE30 cells which lacked SEMA3B expression.
A stable SEMA3B-expressing clone was selected and SEMA3B cDNA was
re-sequenced. Blank vector-transfected KYSE30 cells (Vec-30) were
used as control. RT-PCR and western blot analysis were employed to
detect SEMA3B expression. Taking GAPDH as an internal control, mRNA
and protein expression was quantified by Quantity One software, to
analyze the relative grey value.
Tumor-suppressive function of SEMA3B
In order to test the tumor-suppressive function of
the SEMA3B gene, foci formation assay was performed. Into a 6-well
plate, 1×103 SEMA3B-30 and Vec-30 cells were planted.
After being cultured for 10 days, surviving colonies (>50
cells/colony) were counted with Giemsa staining and triplicate
independent experiments were carried out. Moreover, cell growth
rates of the SEMA3B-30 and Vec-30 cells were detected by MTT assay.
At a density of 1×103/well, the cells were seeded in a
96-well plate. According to the manufacturer's instructions, the
cell growth rate was aassessed using cell proliferation MTT kit
(Sigma). The experiment was conducted in triplicate.
Migration and invasion assays
For the cell migration assays, SEMA3B-30 or Vec-30
cells were cultured in a 6-well plate until confluent and the cell
layer was wound using a sterile tip. After being incubated for 36
h, the cells were photographed under a phase-contrast microscope.
The experiment was conducted in triplicate. For the invasion assay,
SEMA3B-30 or Vec-30 cells were starved of serum-free medium for 24
h before the assay. After suspension in 500-μl serum-free
medium, the cells (5×104) were loaded onto the upper
compartment of an invasion chamber coated with Matrigel (BD
Biosciences). However, the lower compartment was filled with
500-μl complete medium as chemoattractant. After 48 h, the
invasive cells were fixed, stained and counted, and independent
experiments were carried out in triplicate.
Cell cycle analysis
SEMA3B-30 or Vec-30 cells (1×106) were
cultured in RPMI-1640 medium containing 10% fetal bovine serum
(FBS). When the cells reached 70% confluency, the serum was
withdrawn from the culture medium. After 72 h, 10% FBS was added to
the medium for an additional 12 h. The cells were fixed in 70%
ethanol and stained with propidium iodide. In addition, DNA content
was analyzed by applying Cytomics FC (Beckman Coulter).
Western blot analysis
SEMA3B-30 and Vec-30 cell lysates were obtained.
Followed by protein transfer to polyvinylidene fluoride membranes
(PVDF), equal amounts of protein from each sample were diluted with
loading buffer, denatured and separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After being
incubated in a blocking solution containing 5% non-fat milk powder
in Tris-buffered saline and Tween-20 (TBST) buffer (10 mM Tris-HCl,
pH 8.0, 150 mM NaCl and 0.1% Tween-20) for 1 h at room temperature,
the membranes were immunoblotted overnight with primary monoclonal
antibodies for GAPDH, p53, p21, cyclin D1, total Akt and Akt
(Ser473; Cell Signaling Technology) at a dilution of 1:1,000 at 4°C
and then incubated with the secondary antibody (1:1,000 dilution)
for 2 h at room temperature. Enhanced chemiluminescence detection
system was adopted to detect the protein antibody complex. The
protein expression was quantified by Quantity One software, so as
to analyze relative grey value, during which GAPDH served as an
internal control.
Statistical analysis
SPSS standard version 16.0 was used to carry out the
statistical analysis. Correlations between SEMA3B expressions and
clinicopathological characteristics were assessed by χ2
or Fisher's exact tests. In addition, disease-specific survival
(DSS) was calculated from the date of diagnosis to the date of
cancer-related death or last follow-up. According to the
Kaplan-Meier method, survival curves were generated and statistical
analysis was performed by making use of the log-rank test. However,
the Cox proportional hazards regression model was employed to
identify independent prognostic factors. A P-value of <0.05 was
considered to indicate a statistically significant result.
Results
Frequent downregulation of SEMA3B in
ESCCs
Our previous study showed that SEMA3B was
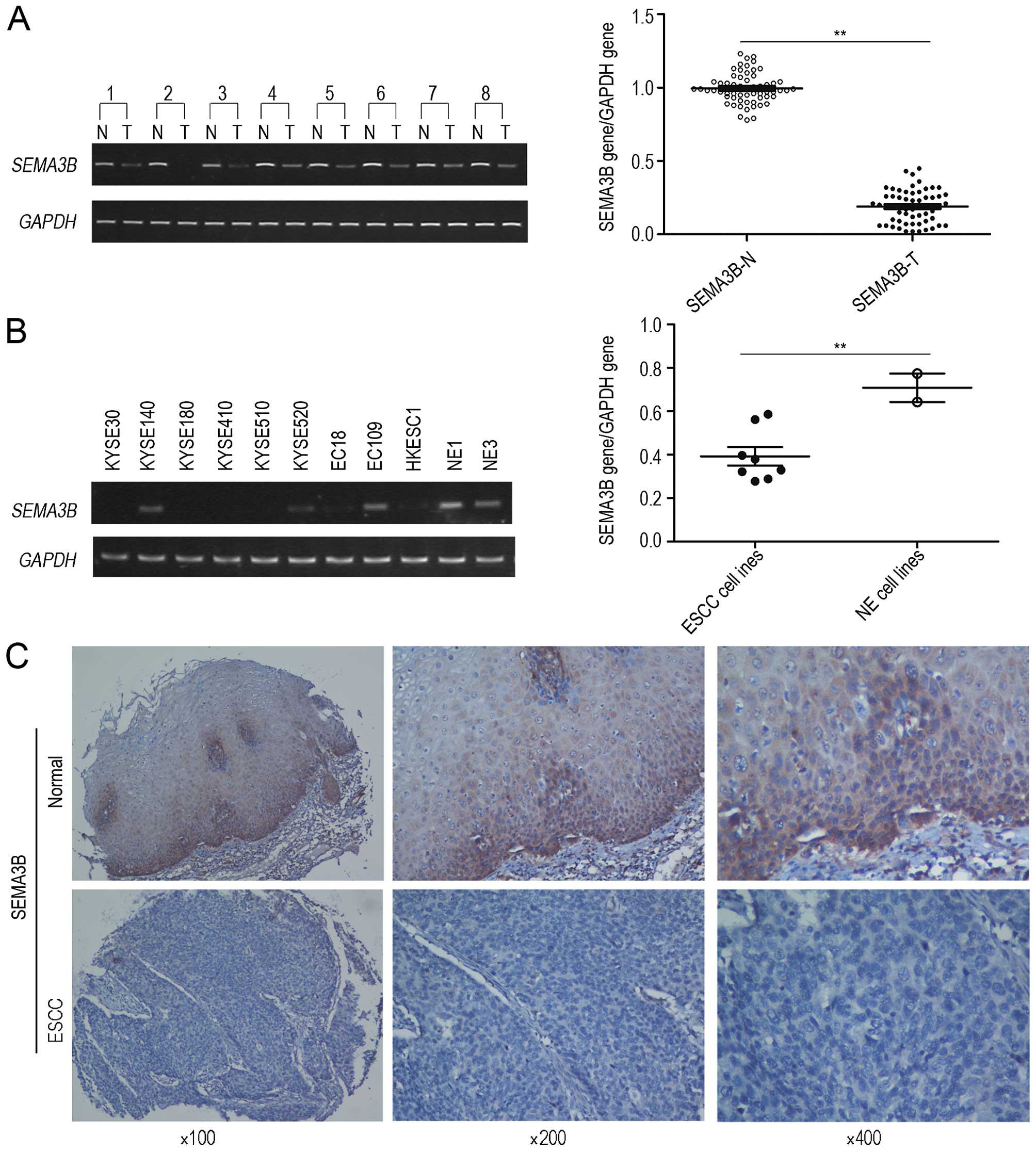
significantly downregulated in ESCC by cDNA microarray (16), thus the expression of SEMA3B at the
mRNA and protein levels was explored in the present study. The
expression of SEMA3B at the mRNA level was assessed by
semi-quantitative RT-PCR in 60 primary ESCC tumors as well as their
paired non-tumorous tissues and 9 ESCC cell lines. The results
showed that the downregulation of SEMA3B was detected in 34/60
(56.7%) ESCC tumor tissues in comparison with their paired
non-tumorous tissues. Ratios of SEMA3B gene/GAPDH expression for
ESCC tissues and their paired non-tumor tissues were 0.19±0.11 and
0.99±0.10, respectively (Fig. 1A).
In comparison with the non-tumor tissues, lower expression of
SEMA3B (P<0.05) was found in the ESCC tissues. Meanwhile, the
downregulation of the expression of SEMA3B was detected in 6/9
(66.7%) (EC18, HYESC1, KYSE30, KYS180, KYS410 and KYS510) ESCC cell
lines. Ratios of SEMA3B gene/GAPDH expression for ESCC cell lines
and immortalized esophageal epithelial cell lines were 0.39±0.12
and 0.71±0.09, respectively (P<0.05; Fig. 1B). Furthermore, the quantity of
samples was expanded to verify its expression at the protein level.
A consistent result was detected in the expression of SEMA3B at the
protein level by IHC through application of TMA. Informative
results were obtained from 222 pairs of ESCCs. Non-informative
samples including lost samples, unrepresentative samples, samples
with too few tumor cells, and samples with inappropriate staining
were not involved in data analysis. Downregulation of SEMA3B at the
protein level was found in 125/222 (56.3%) informative ESCC cases
(Fig. 1C).
Clinical significance of SEMA3B
downregulation in ESCC
Afterwards, we examined the clinical association
between the expression of SEMA3B protein and the
clinicopathological characteristics of the ESCC cases. The analysis
demonstrated that downregulation of SEMA3B protein was
significantly correlated with lymph node metastasis (P=0.000) and
advanced clinical stage (P=0.001; Table
I), but was not correlated with gender, age, cell
differentiation and general classification. The univariate survival
analysis revealed that downregulation of SEMA3B, poor
differentiation, status quo of lymph node metastasis and advanced
tumor-node-metastasis (TNM) stage were significantly correlated
with poor overall survival (P<0.05; Table II). All these variables which
showed statistical significance in the univariate analysis were
further examined by multivariate analysis (Table III). The results revealed that
SEMA3B downregulation was an independent risk factor for overall
patient survival (P=0.017), tumor cell differentiation (P=0.005)
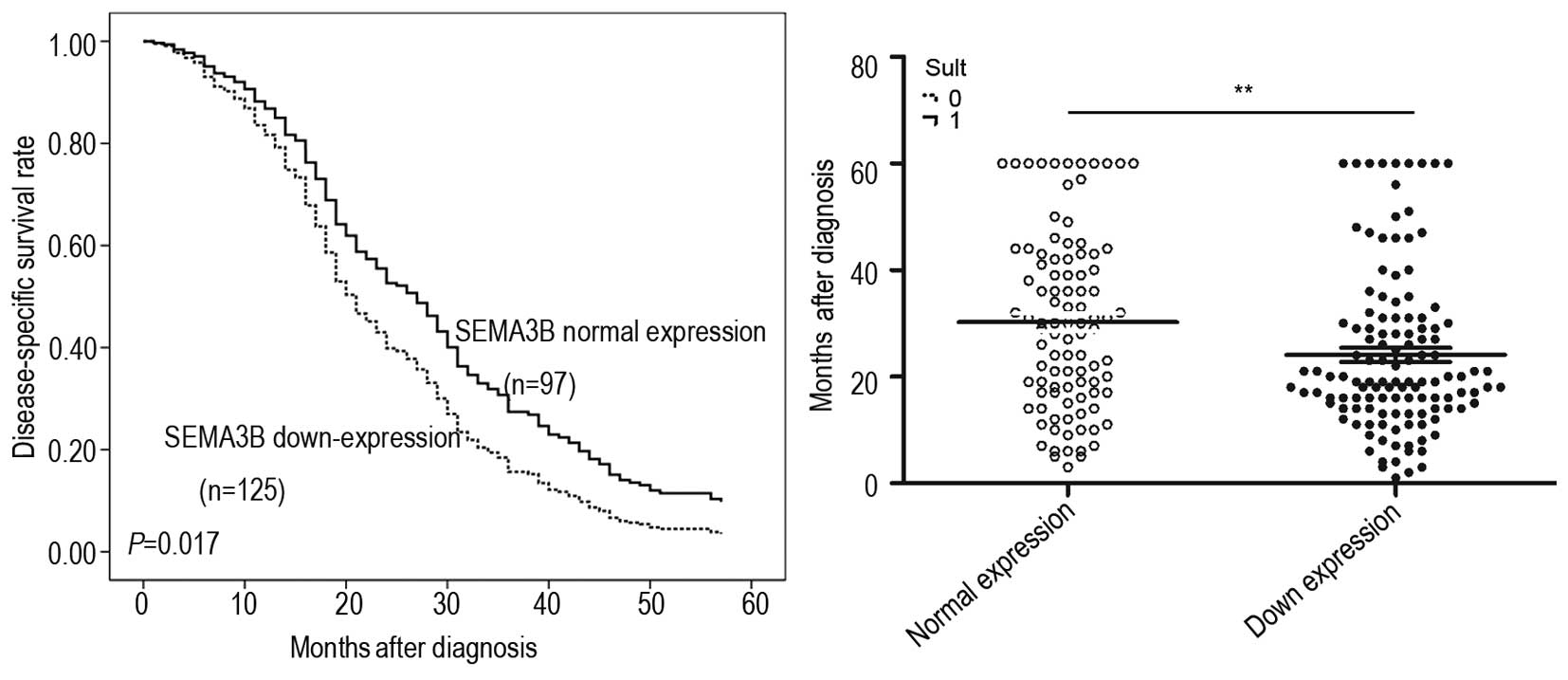
and TNM stage (P=0.005). The results of Kaplan-Meier analysis
demonstrated that ESCC patients with SEMA3B downregulation (median
survival time, 19 months) had a shorter disease-specific survival
(DSS) than patients with normal SEMA3B expression (median survival
time, 30 months; P=0.017; Fig.
2).
 | Table IAssociation of the downregulation of
SEMA3B expression with clinicopathological characteristics of the
patients with ESCC (n=222). |
Table I
Association of the downregulation of
SEMA3B expression with clinicopathological characteristics of the
patients with ESCC (n=222).
| Clinicopathological
characteristics | n | SEMA3B expression,
n (%)
| χ2 | P-value |
|---|
| Downregulated
expression | Normal
expression |
|---|
| Age (years) |
| ≤60 | 102 | 58 (56.9) | 44 (4.1) | 0.024 | 0.893 |
| >60 | 120 | 67 (55.8) | 53 (44.2) | | |
| Gender |
| Male | 126 | 70 (55.6) | 56 (44.4) | 0.067 | 0.891 |
| Female | 96 | 55 (57.3) | 41 (42.7) | | |
| Tumor cell
differentiation |
| Well | 29 | 15 (51.7) | 14 (48.3) | 0.290 | 0.865 |
| Moderate | 146 | 83 (56.8) | 63 (43.2) | | |
| Poor | 47 | 27 (57.4) | 20 (42.6) | | |
| Lymph node
metastasis (N) |
| N0 | 125 | 55 (44.0) | 70 (56.0) | 1.76E1 | 0.000a |
| N1 | 97 | 70 (72.2) | 27 (27.8) | | |
| TNM stage |
| I | 12 | 3 (25.0) | 9 (75.0) | 1.49E1 | 0.001a |
| II | 134 | 67 (50.0) | 67 (50.0) | | |
| III | 76 | 55 (72.4) | 21 (27.6) | | |
| General
classification |
| Medullar type | 120 | 64 (53.3) | 56 (46.7) | 1.316 | 0.725 |
| Ulcerative
type | 65 | 39 (60.0) | 26 (40.0) | | |
| Sclerotic
type | 12 | 8 (66.7) | 4 (33.3) | | |
| Mushroom type | 25 | 14 (56.0) | 11 (44.0) | | |
| Table IIUnivariate Cox regression analysis of
factors possibly influencing disease-specific survival in patients
with ESCC. |
Table II
Univariate Cox regression analysis of
factors possibly influencing disease-specific survival in patients
with ESCC.
| Variables | Hazard ratio | 95% CI | P-value |
|---|
| SEMA3B
expression | 0.697 | 0.527–0.924 | 0.012a |
| Age (years) | 1.263 | 0.958–1.667 | 0.098 |
| Gender | 1.052 | 0.794–1.392 | 0.725 |
| Tumor cell
differentiation | 1.826 | 1.159–2.877 | 0.009a |
| pN factor | 1.697 | 1.284–2.243 | 0.000a |
| TNM stage | 2.093 | 1.563–2.802 | 0.000a |
| Table IIIMultivariate Cox regression analysis
of factors possibly influencing disease-specific survival in
patients with ESCC. |
Table III
Multivariate Cox regression analysis
of factors possibly influencing disease-specific survival in
patients with ESCC.
| Variables | Hazard ratio | 95% CI | P-value |
|---|
| SEMA3B
expression | 0.698 | 0.520–0.938 | 0.017a |
| Tumor cell
differentiation | 1.944 | 1.227–3.082 | 0.005a |
| pN factor | 0.897 | 0.536–1.500 | 0.679 |
| TNM stage | 2.143 | 1.257–3.653 | 0.005a |
Establishment of a stable
SEMA3B-expressing cell line
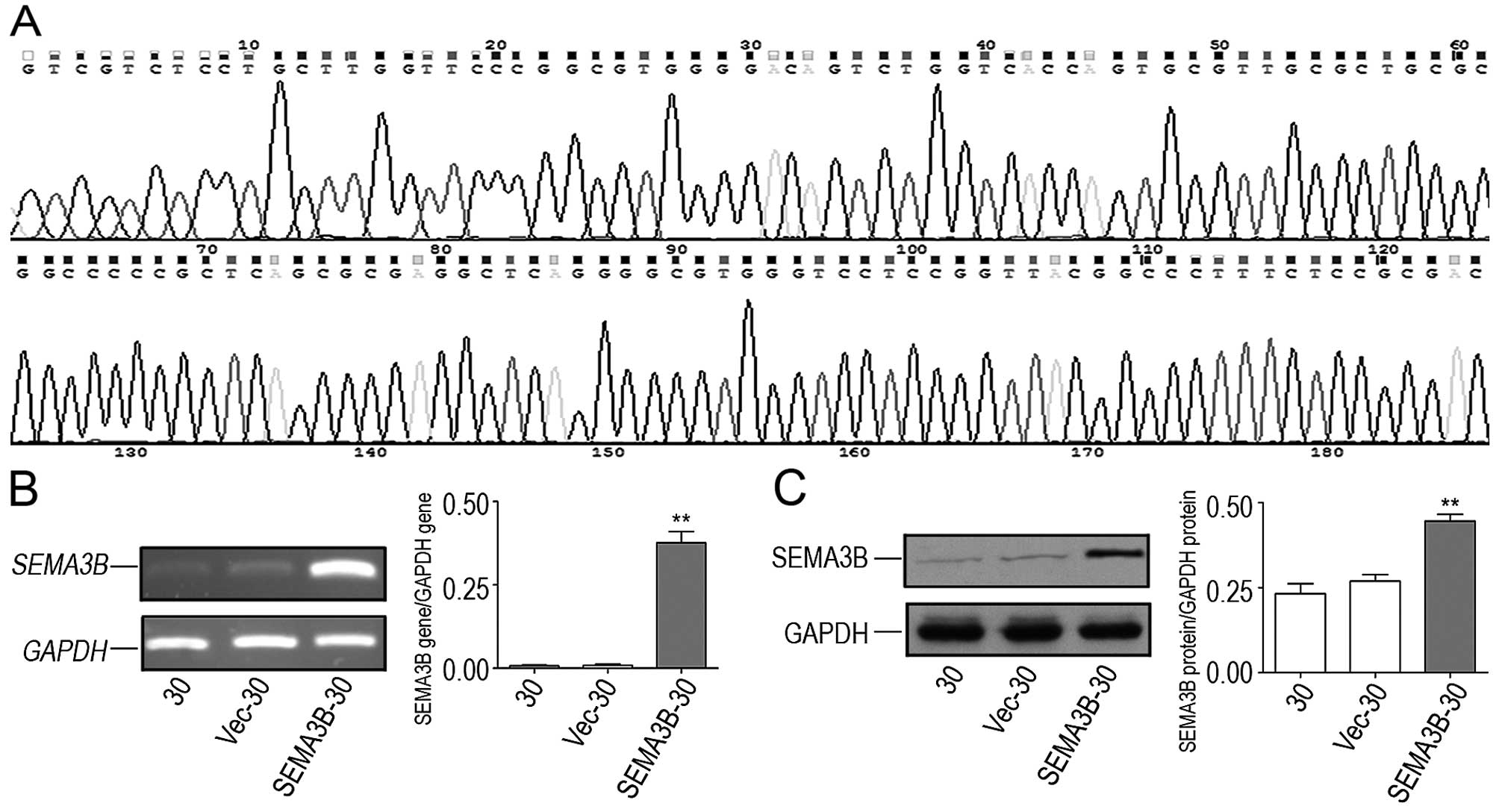
The pcDNA3.1(+)-SEMA3B plasmid was constructed and
its sequence was subsequently confirmed (Fig. 3A). Both the two plasmids,
pcDNA3.1(+)-SEMA3B and blank vector were transfected into KYSE30
cells separately. In order to detect the transfection efficiency of
the plasmids in the SEMA3B-30 cells, the SEMA3B gene and protein
expression in the SEMA3B-30 cells were confirmed by RT-PCR and
western blot analysis. Ratios for SEMA3B gene/GAPDH gene expression
for the KYSE30, Vec-30 and SEMA3B-30 cells were 0.0068±0.0047,
0.0084±0.0069 and 0.3766±0.0574, respectively (Fig. 3B; P<0.05). Rates for SEMA3B
protein/GAPDH protein expression were 0.2321±0.0510, 0.2691±0.0333
and 0.4460±0.0343, respectively (Fig.
3C; P<0.05). In comparison with the KYSE30 and blank
vector-transfected KYSE30 (Vec-30) cells, the expression of SEMA3B
was higher in the SEMA3B-30 cells.
SEMA3B blocks proliferation and migration
of ESCC cells
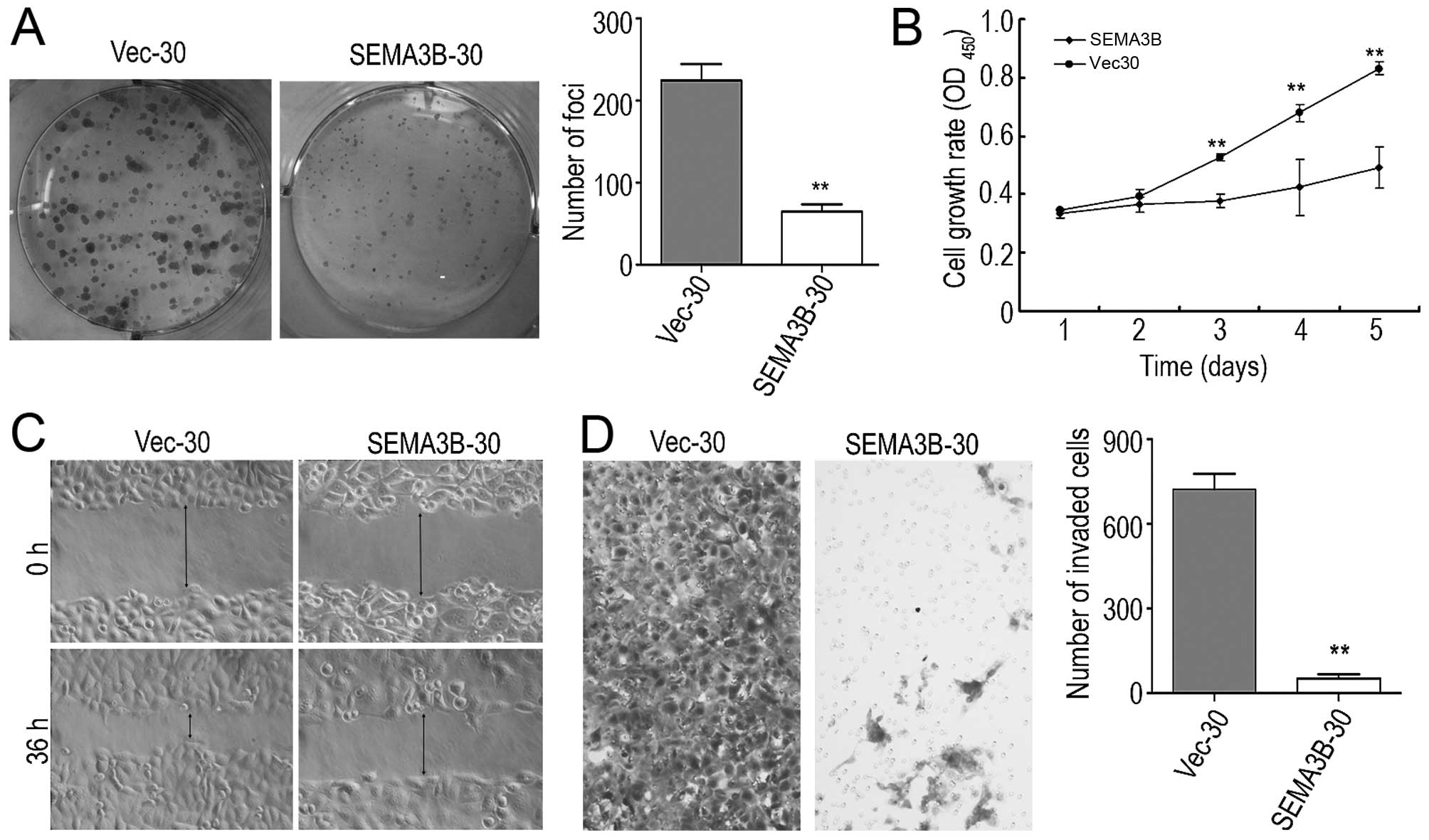
The tumor-suppressive function of SEMA3B was
assessed by foci formation and cell growth assays. The mean value
of the colony formation quantity of the SEMA3B-30 cells was
55.5±10.60, which was lower than that of the Vec-30 cells
(220±46.32). The foci formation assay showed that the efficiency of
foci formation was highly inhibited (P<0.05) in the SEMA3B-30
cells in comparison with the Vec-30 cells (Fig. 4A). As confirmed in the cell growth
assay, the cell proliferation rate in the SEMA3B-30 cells was
significantly inhibited by SEMA3B (P<0.05) in comparison with
the Vec-30 cells (Fig. 4B) after
five successive days of the measurement of OD values.
The TMA results indicated that the downregulation of
the protein expression of SEMA3B was significantly correlated with
lymph node metastasis, thus the effects by SEMA3B on cell migration
and invasion were studied by wound-healing and cell invasion
assays. The wound-healing assay indicated that SEMA3B inhibited
cell mobility (Fig. 4C). The
Matrigel invasion assay also showed that the number of migrating
SEMA3B-30 cells (75±30.61) was lower than that of the Vec-30 cells
(768.33±94.73; Fig. 4D). SEMA3B
inhibited ESCC cell invasiveness, as the number of invasive
SEMA3B-30 cells was significantly decreased in comparison with the
Vec-30 cells (P<0.05).
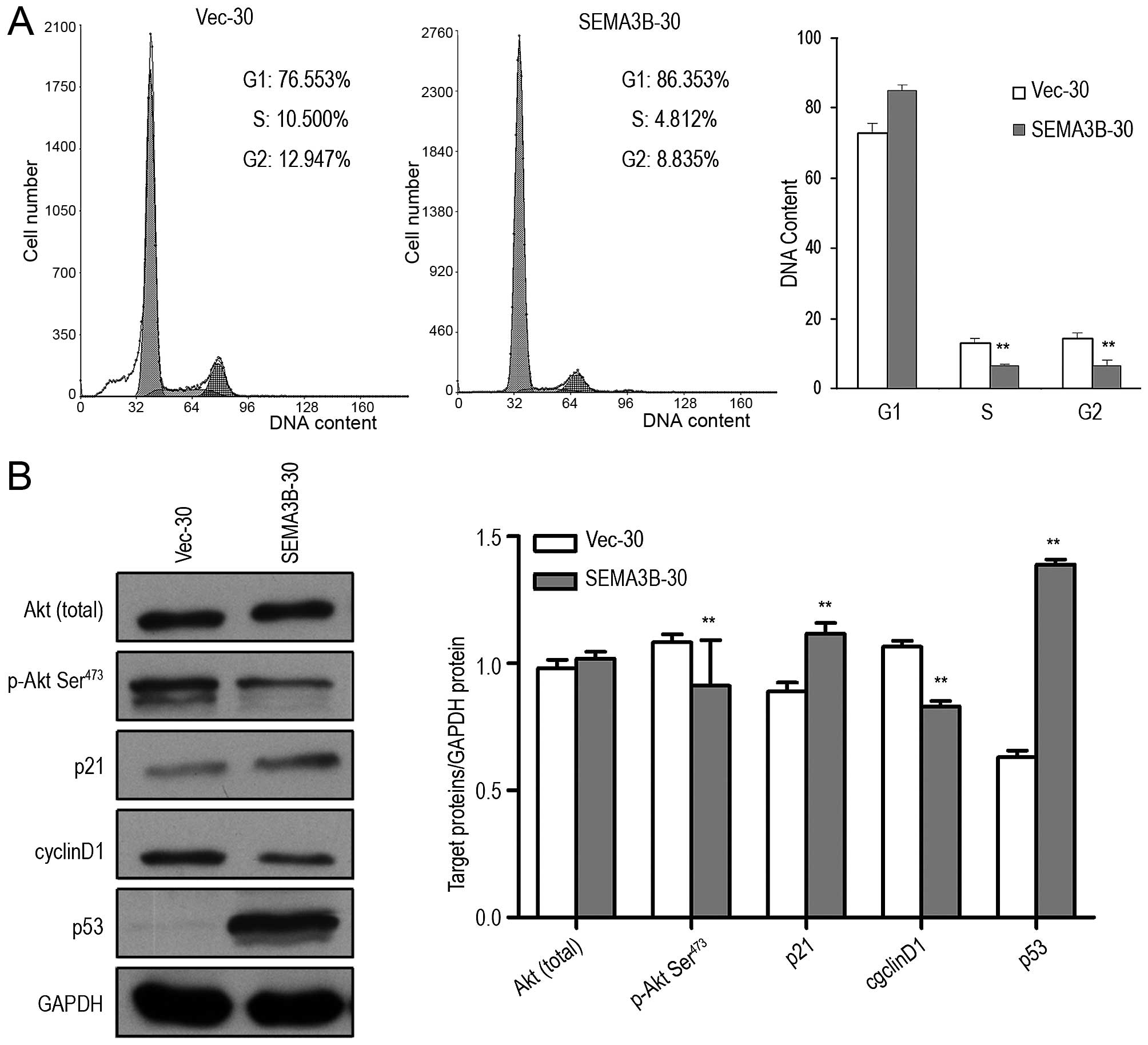
SEMA3B arrests the cell cycle at the G1/S
checkpoint by upregulating p21 and p53 and inhibits the
phosphorylation of Akt
To explore the mechanism underlying the growth
inhibition of SEMA3B, flow cytometry was used to compare the cell
cycle distribution in the SEMA3B-30 cells and the control Vec-30
cells. After 3 days of serum starvation followed by addition of 10%
serum for 12 h, significant G1/S phase arrest was found in the
SEMA3B-30 cells in comparison with the control Vec-30 cells. The
percentage of SEMA3B-30 cells in the G1 phase was 86.35%, while
this percentage in the Vec-30 cells was 76.553%. The percentage of
SEMA3B-30 cells in the S phase was significantly decreased in
comparison with that in the control Vec-30 cells (4.812 and
10.500%; P<0.05; Fig. 5A).
To elucidate the potential molecular mechanism of
SEMA3B in cell cycle arrest, the effects of SEMA3B were examined on
several key cell cycle regulators including p21, p53, cyclin D1,
and Akt from the phosphatidylinositol 3-kinase/Akt pathway. Total
Akt and phosphorylated Akt (Ser473) proteins were determined by
western blotting with isotype-specific antibodies. According to
Fig. 5B, downregulation of
phosphorylated Akt at Ser473 was significantly downregulated by
SEMA3B in the SEMA3B-30 cells in comparison with that in the Vec-30
cells (P<0.05), while the total Akt level was not changed
(P>0.05). Furthermore, the expression of p21, p53 and cyclin D1
in the SEMA3B-30 cells was found to be increased in comparison with
the Vec-30 cells (P<0.05; Fig.
5B).
Discussion
Located at the chromosomal region 3p21.3, SEMA3B is
considered as a member of the class 3 Semaphorin family, a group of
secreted proteins that repulse axonal extension (20). In the present study, downregulation
of SEAM3B was detected in 56.7% of 60 pairs of primary ESCC cases
at the mRNA level, and 66.7% of 9 ESCC cell lines, respectively.
After that, the quantity of samples was expanded, so as to verify
its expression at the protein level. The tissue microarray study
indicated that the absence of expression of SEMA3B at the protein
level was detected in 56.3% of primary ESCCs. These consistent
observations indicated the downregulation of SEMA3B in ESCC.
Furthermore, the relationship between gene and clinical
pathological characteristics indicated that the downregulation of
SEMA3B was more frequently observed in patients with lymph node
metastasis (P=0.000), advanced clinical stage (P=0.001) and poor
survival (P=0.017). The Kaplan-Meier analysis showed that the
overall survival rate of ESCC patients decreased as SEMA3B was
downregulated in the tumor tissues. The multivariate analysis
indicated that downregulation of SEMA3B could be used as an
independent prognostic predictor for ESCC patients. Chromosome 3
allelic loss and promoter hypermethylation may be the main cause of
SEMA3B downregulation (21–26).
It is well known that SEMA3B has marked ability to
induce tumor cell apoptosis and regulate cell growth in lung and
breast cancer, and other solid tumors (13,14,21,27,28).
In order to explore the function and mechanism of antitumorigenic
properties of SEMA3B in ESCC cells, a stable SEMA3B-expressing ESCC
cell line, SEMA3B-30, was established. The tumor-suppressive
function of SEMA3B was investigated in our in vitro assay.
The result indicated that SEMA3B could significantly suppress cell
growth, decrease foci formation and inhibit ESCC cell migration and
invasion. Further study revealed that SEMA3B could inhibit the
phosphorylation of Akt expression, indicating that the
tumor-suppressive function of SEMA3B in ESCC may be correlated with
the PI3K/AKT signaling transduction pathway. Known as protein
kinase B, Akt is activated downstream of PI3K in response to
receptor stimulation and modulates the function of numerous
substrates involved in the regulation of cell survival, cell cycle
progression and cell growth (29–34).
Phospho-Akt leads to activation of MDM-2, which targets p53 for
degradation (35–38), whereas inactivation of apoptotic
regulators and apoptotic factors may conversely take place. Akt
activation promotes cell cycle progression and cell growth by
impeding nuclear localization of p21 and p27 which are
cyclin-dependent kinase inhibitors; inhibiting glycogen synthase
kinase-3 (GSK-3) so as to lead to the stabilization of expression
of cyclin D1; maintaining levels of the anti-apoptotic protein
survivin (39–42). Our results revealed that SEMA3B was
able to arrest ESCC cells at the G1/S checkpoint through inhibition
of the phosphatidylinositol 3-kinase/AKT signaling transduction
pathway, upregulation of p53 and p21 expression and downregulation
of cyclin D1 expression.
Moreover, it has been confirmed that SEMA3B induced
IL-8 secretion from tumor cell by initiating the
p38-mitogen-activated protein kinase pathway. The release of IL-8
induced the recruitment of tumor-associated macrophages which may
promote cancer progression and metastasis (43,44).
Recent studies indicate that SEMA3B reduces invasive properties by
inhibiting MMP-2, MMP-9, αvβ3 and pro-angiogenesis genes and by
upregulating anti-angiogenesis genes in endometrial cancer cells
(45). Previous studies indicated
that TIMP3, which is an inhibitor of MMP and also a key player in
tumor cell invasion, angiogenesis and cell growth processes, was
found to be upregulated under SEMA restoration (46). In contrast, SEMA3B shares similar
binding sites on NP-1 and NP-2 proteins with VEFG165, acting as an
autocrine survival factor (28,47).
SEMA3B competes with VEFG165 for binding on the tumor cell surface.
Therefore, SEMA3B mediates its tumor-suppressing effects, at least
in part, by blocking VEGF autocrine activity.
In summary, for the first time, the results of the
present study demonstrated that downregulation of SEMA3B was
frequently found in ESCC tumor specimens and predicted a worse
prognosis of ESCC patients. Our results indicate that SEMA3B may be
an important tumor-suppressor gene in the malignant progression of
ESCC and a valuable prognostic marker for ESCC patients. Further
research needs to be conducted in in vivo studies and the
reasons for SEMA3B downregulation require further
investigation.
References
|
1
|
Ke L: Mortality and incidence trends from
esophagus cancer in selected geographic areas of China circa
1970–90. Int J Cancer. 102:271–274. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parkin DM, Bray FI and Devesa SS: Cancer
burden in the year 2000. The global picture. Eur J Cancer. 37(Suppl
8): S4–66. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A1, Bray F, Center MM, Ferlay J,
Ward E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yen CC, Chen YJ, Chen JT, Hsia JY, Chen
PM, Liu JH, Fan FS, Chiou TJ, Wang WS and Lin CH: Comparative
genomic hybridization of esophageal squamous cell carcinoma:
Correlations between chromosomal aberrations and disease
progression/prognosis. Cancer. 92:2769–2777. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kwong D, Lam A, Guan X, Law S, Tai A, Wong
J and Sham J: Chromosomal aberrations in esophageal squamous cell
carcinoma among Chinese: Gain of 12p predicts poor prognosis after
surgery. Hum Pathol. 35:309–316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Qin YR, Wang LD, Kwong D, Gao SS, Guan XY,
Zhuang ZH, Fan ZM, Deng W and Hu L: Comparative genomic
hybridization: The profile of chromosomal imbalances in esophageal
squamous cell carcinoma. Zhonghua Bing Li Xue Za Zhi. 34:80–83.
2005.In Chinese. PubMed/NCBI
|
|
7
|
Ogasawara S, Maesawa C, Tamura G and
Satodate R: Frequent microsatellite alterations on chromosome 3p in
esophageal squamous cell carcinoma. Cancer Res. 55:891–894.
1995.PubMed/NCBI
|
|
8
|
Dammann R, Li C, Yoon JH, Chin PL, Bates S
and Pfeifer GP: Epigenetic inactivation of a RAS association domain
family protein from the lung tumour suppressor locus 3p21.3. Nat
Genet. 25:315–319. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ji L, Nishizaki M, Gao B, Burbee D, Kondo
M, Kamibayashi C, Xu K, Yen N, Atkinson EN, Fang B, et al:
Expression of several genes in the human chromosome 3p21.3
homozygous deletion region by an adenovirus vector results in tumor
suppressor activities in vitro and in vivo. Cancer Res.
62:2715–2720. 2002.PubMed/NCBI
|
|
10
|
Daigo Y, Nishiwaki T, Kawasoe T, Tamari M,
Tsuchiya E and Nakamura Y: Molecular cloning of a candidate tumor
suppressor gene, DLC1, from chromosome 3p21.3. Cancer Res.
59:1966–1972. 1999.PubMed/NCBI
|
|
11
|
Fu L, Qin YR, Xie D, Hu L, Kwong DL,
Srivastava G, Tsao SW and Guan XY: Characterization of a novel
tumor-suppressor gene PLC delta 1 at 3p22 in esophageal squamous
cell carcinoma. Cancer Res. 67:10720–10726. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Castro-Rivera E, Ran S, Thorpe P and Minna
JD: Semaphorin 3B (SEMA3B) induces apoptosis in lung and breast
cancer, whereas VEGF165 antagonizes this effect. Proc
Natl Acad Sci USA. 101:11432–11437. 2004. View Article : Google Scholar
|
|
13
|
Tomizawa Y, Sekido Y, Kondo M, Gao B,
Yokota J, Roche J, Drabkin H, Lerman MI, Gazdar AF and Minna JD:
Inhibition of lung cancer cell growth and induction of apoptosis
after reexpression of 3p21.3 candidate tumor suppressor gene
SEMA3B. Proc Natl Acad Sci USA. 98:13954–13959. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tse C, Xiang RH, Bracht T and Naylor SL:
Human Semaphorin 3B (SEMA3B) located at chromosome 3p21.3
suppresses tumor formation in an adenocarcinoma cell line. Cancer
Res. 62:542–546. 2002.PubMed/NCBI
|
|
15
|
Tischoff I, Markwarth A, Witzigmann H,
Uhlmann D, Hauss J, Mirmohammadsadegh A, Wittekind C, Hengge UR and
Tannapfel A: Allele loss and epigenetic inactivation of 3p21.3 in
malignant liver tumors. Int J Cancer. 115:684–689. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qin YR1, Tang H, Xie F, Liu H, Zhu Y, Ai
J, Chen L, Li Y, Kwong DL, Fu L and Guan XY: Characterization of
tumor-suppressive function of SOX6 in human esophageal squamous
cell carcinoma. Clin Cancer Res. 17:46–55. 2011. View Article : Google Scholar
|
|
17
|
Shimada Y, Imamura M, Wagata T, Yamaguchi
N and Tobe T: Characterization of 21 newly established esophageal
cancer cell lines. Cancer. 69:277–284. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie D, Sham JS, Zeng WF, Lin HL, Che LH,
Wu HX, Wen JM, Fang Y, Hu L and Guan XY: Heterogeneous expression
and association of beta-catenin, p16 and c-myc in multistage
colorectal tumorigenesis and progression detected by tissue
microarray. Int J Cancer. 107:896–902. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brown RS and Wahl RL: Overexpression of
Glut-1 glucose transporter in human breast cancer. An
immunohistochemical study. Cancer. 72:2979–2985. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zou Y, Stoeckli E, Chen H and
Tessier-Lavigne M: Squeezing axons out of the gray matter: A role
for slit and semaphorin proteins from midline and ventral spinal
cord. Cell. 102:363–375. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Loginov VI, Dmitriev AA, Senchenko VN,
Pronina IV, Khodyrev DS, Kudryavtseva AV, Krasnov GS, Gerashchenko
GV, Chashchina LI, Kazubskaya TP, et al: Tumor suppressor function
of the SEMA3B gene in human lung and renal cancers. PLoS One.
10:e01233692015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Senchenko VN, Liu J, Loginov W, Bazov I,
Angeloni D, Seryogin Y, Ermilova V, Kazubskaya T, Garkavtseva R,
Zabarovska VI, et al: Discovery of frequent homozygous deletions in
chromosome 3p21.3 LUCA and AP20 regions in renal, lung and breast
carcinomas. Oncogene. 23:5719–5728. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ito M, Ito G, Kondo M, Uchiyama M, Fukui
T, Mori S, Yoshioka H, Ueda Y, Shimokata K and Sekido Y: Frequent
inactivation of RASSF1A, BLU, and SEMA3B on 3p21.3 by promoter
hypermethylation and allele loss in non-small cell lung cancer.
Cancer Lett. 225:131–139. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Loginov VI, Khodyrev DS, Pronina IV,
Maliukova AV, Kazubskaia TP, Ermilova VD, Gar'kavtseva RF,
Zabarovskiĭ ER and Braga EA: Two CpG-islands of SEMA3B gene:
Methylation in clear cell renal cell carcinoma. Mol Biol.
43:1088–1092. 2009.In Russian. View Article : Google Scholar
|
|
25
|
Chen R, Zhuge X, Huang Z, Lu D, Ye X, Chen
C, Yu J and Lu G: Analysis of SEMA3B methylation and expression
patterns in gastric cancer tissue and cell lines. Oncol Rep.
31:1211–1218. 2014.PubMed/NCBI
|
|
26
|
Wang K, Ling T, Wu H and Zhang J:
Screening of candidate tumor-suppressor genes in 3p21.3 and
investigation of the methylation of gene promoters in oral squamous
cell carcinoma. Oncol Rep. 29:1175–1182. 2013.PubMed/NCBI
|
|
27
|
Kuroki T, Trapasso F, Yendamuri S,
Matsuyama A, Alder H, Williams NN, Kaiser LR and Croce CM: Allelic
loss on chromosome 3p21.3 and promoter hypermethylation of
semaphorin 3B in non-small cell lung cancer. Cancer Res.
63:3352–3355. 2003.PubMed/NCBI
|
|
28
|
Castro-Rivera E, Ran S, Brekken RA and
Minna JD: Semaphorin 3B inhibits the phosphatidylinositol
3-kinase/Akt pathway through neuropilin-1 in lung and breast cancer
cells. Cancer Res. 68:8295–8303. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dudek H, Datta SR, Franke TF, Birnbaum MJ,
Yao R, Cooper GM, Segal RA, Kaplan DR and Greenberg ME: Regulation
of neuronal survival by the serine-threonine protein kinase Akt.
Science. 275:661–665. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kennedy SG, Wagner AJ, Conzen SD, Jordán
J, Bellacosa A, Tsichlis PN and Hay N: The PI 3-kinase/Akt
signaling pathway delivers an anti-apoptotic signal. Genes Dev.
11:701–713. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Carnero A, Blanco-Aparicio C, Renner O,
Link W and Leal JF: The PTEN/PI3K/AKT signalling pathway in cancer,
therapeutic implications. Curr Cancer Drug Targets. 8:187–198.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Janku F, Stewart DJ and Kurzrock R:
Targeted therapy in non-small-cell lung cancer - is it becoming a
reality? Nat Rev Clin Oncol. 7:401–414. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Reungwetwattana T, Weroha SJ and Molina
JR: Oncogenic pathways, molecularly targeted therapies, and
highlighted clinical trials in non-small-cell lung cancer (NSCLC).
Clin Lung Cancer. 13:252–266. 2012. View Article : Google Scholar
|
|
35
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell.
91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo
P, Hu LS, Anderson MJ, Arden KC, Blenis J and Greenberg ME: Akt
promotes cell survival by phosphorylating and inhibiting a Forkhead
transcription factor. Cell. 96:857–868. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cardone MH, Roy N, Stennicke HR, Salvesen
GS, Franke TF, Stanbridge E, Frisch S and Reed JC: Regulation of
cell death protease caspase-9 by phosphorylation. Science.
282:1318–1321. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sherr CJ and Weber JD: The ARF/p53
pathway. Curr Opin Genet Dev. 10:94–99. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shin I, Yakes FM, Rojo F, Shin NY, Bakin
AV, Baselga J and Arteaga CL: PKB/Akt mediates cell-cycle
progression by phosphorylation of p27Kip1 at threonine
157 and modulation of its cellular localization. Nat Med.
8:1145–1152. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Diehl JA, Cheng M, Roussel MF and Sherr
CJ: Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis
and subcellular localization. Genes Dev. 12:3499–3511. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou BP, Liao Y, Xia W, Spohn B, Lee MH
and Hung MC: Cytoplasmic localization of p21Cip1/WAF1 by
Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat
Cell Biol. 3:245–252. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Oliveira CS, de Bock CE, Molloy TJ,
Sadeqzadeh E, Geng XY, Hersey P, Zhang XD and Thorne RF: Macrophage
migration inhibitory factor engages PI3K/Akt signalling and is a
prognostic factor in metastatic melanoma. BMC Cancer. 14:6302014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rolny C, Capparuccia L, Casazza A, Mazzone
M, Vallario A, Cignetti A, Medico E, Carmeliet P, Comoglio PM and
Tamagnone L: The tumor suppressor semaphorin 3B triggers a
prometastatic program mediated by interleukin 8 and the tumor
microenvironment. J Exp Med. 205:1155–1171. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami
S, Stanley ER, Segall JE, Pollard JW and Condeelis J: Direct
visualization of macrophage-assisted tumor cell intravasation in
mammary tumors. Cancer Res. 67:2649–2656. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nguyen H, Ivanova VS, Kavandi L, Rodriguez
GC, Maxwell GL and Syed V: Progesterone and 1,25-dihydroxyvitamin
D3 inhibit endometrial cancer cell growth by
upregulating semaphorin 3B and semaphorin 3F. Molecular cancer
research. Mol Cancer Res. 9:1479–1492. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tunuguntla R, Ripley D, Sang QX and
Chegini N: Expression of matrix metalloproteinase-26 and tissue
inhibitors of metalloproteinases TIMP-3 and -4 in benign
endometrium and endometrial cancer. Gynecol Oncol. 89:453–459.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Staton CA, Shaw LA, Valluru M, Hoh L, Koay
I, Cross SS, Reed MW and Brown NJ: Expression of class 3
semaphorins and their receptors in human breast neoplasia.
Histopathology. 59:274–282. 2011. View Article : Google Scholar : PubMed/NCBI
|