Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common form of cancer worldwide (1). It is estimated that more than 35,600
new cases of HCC were reported and 24,550 people died due to HCC in
2015 in the USA (2). Since most HCC
patients are diagnosed at a late stage, surgical resection and
liver transplantation fail to significantly increase the 5-year
survival rate (3). Chemotherapeutic
drugs are useful for advanced stage HCC patients, and clinical
trials have revealed that advanced HCC patients treated with
sorafenib have extended median survival by 3 months (4,5). One
recent study demonstrated that fluorouracil, leucovorin, and
oxaliplatin (FOLFOX4) treatment conferred some benefit to patients
with advanced HCC (6). Recently,
one study validated gemcitabine and oxaliplatin (GEMOX) as
effective with manageable toxicity in advanced HCC patients
(7). However, due to the fact that
many patients develop drug resistance to standard chemotherapy,
these treatments often do not improve patient outcomes (8,9).
Therefore, it is important to develop new therapeutic agents to
increase drug sensitivity to chemotherapy and achieve better
treatment outcomes for HCC patients.
Emerging evidence has demonstrated that
epithelial-mesenchymal transition (EMT) is critically involved in
chemoresistance in human cancers including HCC (10). It has been well documented that
during the EMT process, epithelial cells convert into mesenchymal
cells due to loss of epithelial cell-cell junction (11). Mechanistically, expression of
epithelial markers including E-cadherin and γ-catenin is
downregulated, whereas the mesenchymal markers such as vimentin,
Twist, Slug, and Snail are upregulated. EMT-type cells acquire
enhanced migration and invasion leading to more enhanced metastasis
(12). In addition, there appears
to be a direct connection between EMT and the development of drug
resistance (13). For example,
multiple studies have revealed that gemcitabine-resistant (GR)
cells acquired EMT characteristics in pancreatic cancer and HCC
cells (14–17). Specifically, several signaling
pathways have been identified to be involved in GR-mediated EMT
including Notch-1, platelet-derived growth factor-D (PDGF-D), and
hypoxia inducible factor-1α (HIF-1α) pathways (14–17).
Thus, targeting these pathways could reverse EMT and increase
gemcitabine sensitivity in human cancers. Therefore, in-depth
investigation of the molecular mechanisms governing GR-induced EMT
would aid the discovery of novel strategies for the treatment of
HCC patients.
Previous studies have implicated the p53 pathway as
an essential regulator of EMT and drug resistance (18–20).
For instance, p53 inactivation in breast cancer cells promoted EMT
and increased the susceptibility to 3-bromopyruvate (18). Lin et al also identified that
p53 modulated NF-κB-mediated EMT in head and neck squamous cell
carcinoma (19). Another study
identified that targeting ephrin-B2 might enhance the therapeutic
potential of DNA-damaging chemotherapeutic agents in human tumors
harboring mutant p53 (20). It is
well established that p53 is negatively regulated by the murine
double minute 2 (MDM2) oncoprotein (21). Thus, it was logical to develop small
molecules that selectively disrupt the interaction between MDM2 and
p53, leading to a restoration of p53 function in tumor cells with
wild-type p53 (22). To this end,
Nutlin-3 was identified as a selective inhibitor of the p53-MDM2
interaction and exhibited antitumor activities in a variety of
human malignances (23). By
activating the p53 pathway, Nutlin-3 enhances cellular apoptosis
and inhibits cell growth (24).
Recent studies indicate that Nutlin-3 also exerts its anticancer
functions partly through negative regulation of migration, invasion
and drug resistance (25–27), however further studies are required
to fully understand the molecular mechanisms driving
Nutlin-3-induced inhibition of tumorigenesis.
Here we report that Nutlin-3 inhibited cell
migration and invasion in GR HCC cells, which acquired EMT
features. Moreover, to further define the mechanisms behind the
tumor-suppressive functions of Nutlin-3, we identified that
treatment with Nutlin-3 led to upregulation of E-cadherin and
downregulation of mesenchymal markers including vimentin, Snail and
Slug in GR HCC cells. Mechanistically, our results revealed that
Smad2 was upregulated in GR HCC cells compared with their parental
cells, and that Nutlin-3 inhibited the expression of Smad2 in GR
HCC cells. Consistent with this notion, depletion of Smad2
suppressed cell migration and regulated the expression of EMT
markers in GR HCC cells. These results indicate that Nutlin-3 could
reverse EMT in GR HCC cells, providing the rational for the use of
Nutlin-3 as a potential treatment for HCC patients.
Materials and methods
Cell culture, reagents and
antibodies
Human HepG2 and SMMC-7721 cell lines were cultured
at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium
(DMEM; Gibco, Gaithersburg, MD, USA) supplemented with 10% fetal
bovine serum. HepG2 GR and SMMC-7721 GR cell lines were established
in our laboratory as described previously (14). Gemcitabine,
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]
(MTT), and Nutlin-3 were obtained from Sigma (St. Louis, MO, USA).
Antibodies against vimentin and E-cadherin were obtained from
Abcam. Antibodies against Snail, Slug, Smad2, and GAPDH were
obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
MTT assay
Cells were seeded at equal densities into 96-well
culture plates and incubated overnight. Cells were then treated
with the indicated concentrations of Nutlin-3 for 48 h. MTT assay
was conducted as previously described (28). Cell growth inhibition curves were
generated using the cell survival rates. The concentrations of
Nutlin-3 including IC30, IC50, and IC70 were calculated and used in
the subsequent experiments.
Wound healing assay
HepG2 GR and SMMC-7721 GR cells were cultured in
6-well plates and grown to confluency. After cells converged almost
100%, monolayers of cells were scratched with yellow pipette tips.
Then the cells were washed with PBS and medium was added with
different concentrations of Nutlin-3. The scratched area was
photographed with a microscope at 0 and 20 h, respectively, to
observe the cell motility ability.
Transwell migration and invasion
assays
The migration assay was carried out in chambers
without matrix gel and the invasion assay was performed in a
chamber with 50 µl matrix gel. Briefly, the HepG2 GR and
SMMC-7721 GR cells were seeded in the upper chamber with 200
µl serum-free medium and the indicated concentrations of
Nutlin-3 and 500 µl complete medium in the lower chamber
with the same concentration of Nutlin-3. After incubation for 24 h,
the membrane of the chamber was immobilized with methanol and
strained with 1% crystal violet. Cells were counted and
photographed with a microscope.
Cell attachment and detachment
assays
For the attachment assay, HepG2 GR and SMMC-7721 GR
cells, treated with the indicated concentrations of Nutlin-3, were
seeded in 24-well plates at 5×104 cells per well.
Unattached cells were removed after a 1-h incubation, and the
attached cells were counted after 0.25% trypsinization. The data
are presented as a percentage of the attached cells compared to
total cells. For the cell detachment assay, after a 24-h
incubation, the cells were incubated with 0.05% trypsin for 3 min
to detach the cells. Culture medium was then added to inactivate
the trypsin and the detached cells were collected. The remaining
cells were incubated with 0.25% trypsin to detach and counted. The
data are presented as a percentage of the detached cells to total
cells.
Real-time RT-PCR
Total RNA from the HepG2 GR and SMMC-7721 GR cells
treated with Nutlin-3 was isolated with TRIzol or with the RNeasy
Mini kit and RNase-free DNase set according to the manufacturer's
protocols. RNA was reversed-transcribed into cDNA by RevertAid
First Strand cDNA Synthesis kit (Thermofisher Scientific, Inc.)
according to the manufacturer's protocol. The primers used in the
PCR reactions are listed in Table
I. The expression of GAPDH was used as internal control. RT-PCR
amplifications were performed as previously described (28).
 | Table IPrimer sequences used for RT-PCR. |
Table I
Primer sequences used for RT-PCR.
| Name | Sequence |
|---|
| E-cadherin | Sense:
5′-GAAGTGTCCGAGGACTTTGG-3′ |
| Antisense:
5′-CAGTGTCTCTCCAAATCCGATA-3′ |
| Vimentin | Sense:
5′-TGTCCAAATCGATGTGGATGTTTC-3′ |
| Antisense:
5′-TTGTACCATTCTTCTGCCTCCTG-3′ |
| Slug | Sense:
5′-CATGCCTGTCATACCACAAC-3′ |
| Antisense:
5′-GGTGTCAGATGGAGGAGGG-3′ |
| Snail | Sense:
5′-CGGAAGCCTAACTACAGCGA-3′ |
| Antisense:
5′-GGACAGAGTCCCAGATGAGC-3′ |
| GAPDH | Sense:
5′-CAGCCTCAAGATCATCAGCA-3′ |
| Antisense:
5′-TGTGGTCATGAGTCCTTCCA-3′ |
Western blot analysis
Harvested cells were washed by PBS and lysed with
protein lysis buffer, and protein concentrations were measured by
Bradford assay reagent. Equal amounts of protein were separated by
sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis
(SDS-PAGE) and then transferred onto polyvinylidene fluoride (PVDF)
membranes. The membranes where incubated with the primary antibody
at 4°C overnight. After washing with TBS-Tween-20, the membranes
were incubated with the secondary antibody conjugated to HRP at
room temperature for 1 h. The membranes were washed again in
TBS-Tween-20, and protein was detected by electrochemiluminescence
(ECL) assay.
Transfection
Cells were seeded in 6-well plates and transfected
with control siRNA or Smad2 siRNAs (GenePharma, Shanghai, China)
using Lipofectamine 2000 as previously described (28).
Statistical analysis
All statistical analyses were conducted using
SPSS17.0. Student's t-test was performed to evaluate statistical
significance. Results are presented as mean ± SD. P<0.05 was
considered as statistically significant.
Results
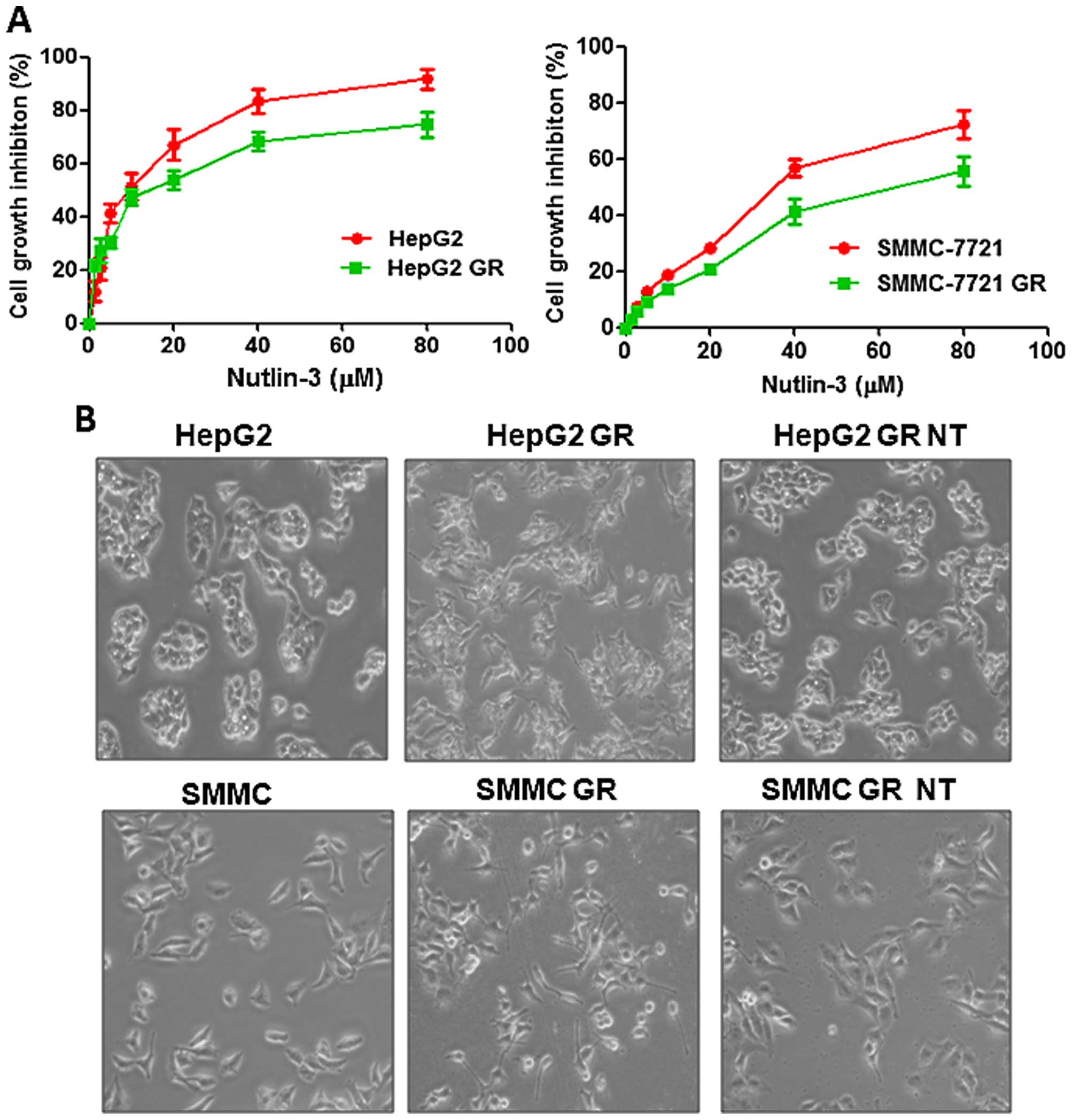
Nutlin-3 inhibits cell proliferation and
alters the morphology of GR HCC cells
To determine whether Nutlin-3 inhibits cell
proliferation in HCC cells, we performed MTT assays following
Nutlin-3 treatment. We found that Nutlin-3 significantly inhibited
cell proliferation in both the HepG2 and SMMC-7721 cells (Fig. 1A). Moreover, we observed that
Nutlin-3 also suppressed cell proliferation in both the GR HepG2
and GR SMMC-7721 cells (Fig. 1A).
Our results indicate that HCC cells are more sensitive to Nutlin-3
treatment compared with GR HCC cells (Fig. 1A). Specifically, IC30, IC50 and IC70
of Nutlin-3 in the GR HepG2 cells were 5, 18 and 70 µM,
respectively. Whereas the IC70 of Nutlin-3 was 25 µM in the
HepG2 cells, suggesting that HepG2 cells were more sensitive to
Nutlin-3 treatment (Fig. 1A, left
panel). Similar results were observed in the SMMC-7721 cells
(Fig. 1A, right panel). IC30, IC50
and IC70 of Nutlin-3 in the GR SMMC-7721 cells were 40, 59 and 80
µM, respectively. In subsequent experiments, we used IC30
and IC70 to explore the function of Nutlin-3 in the GR HCC cells.
Our previous study demonstrated that GR HCC cells acquired an EMT
phenotype (14). In line with this
report, we observed the morphological change of GR HCC cells, which
is indicative of EMT, such as increased pseudopodia, and longer,
narrower and a more disperse morphology (Fig. 1B). Moreover, we found that Nutlin-3
treatment reversed this EMT phenotype (Fig. 1B). Specifically, in GR HCC cells,
pseudopodia were reduced and cells became ovoid and clustered
during growth following Nutlin-3 treatment (Fig. 1B). These results indicate that
Nutlin-3 may regulate GR-induced EMT in HCC cells.
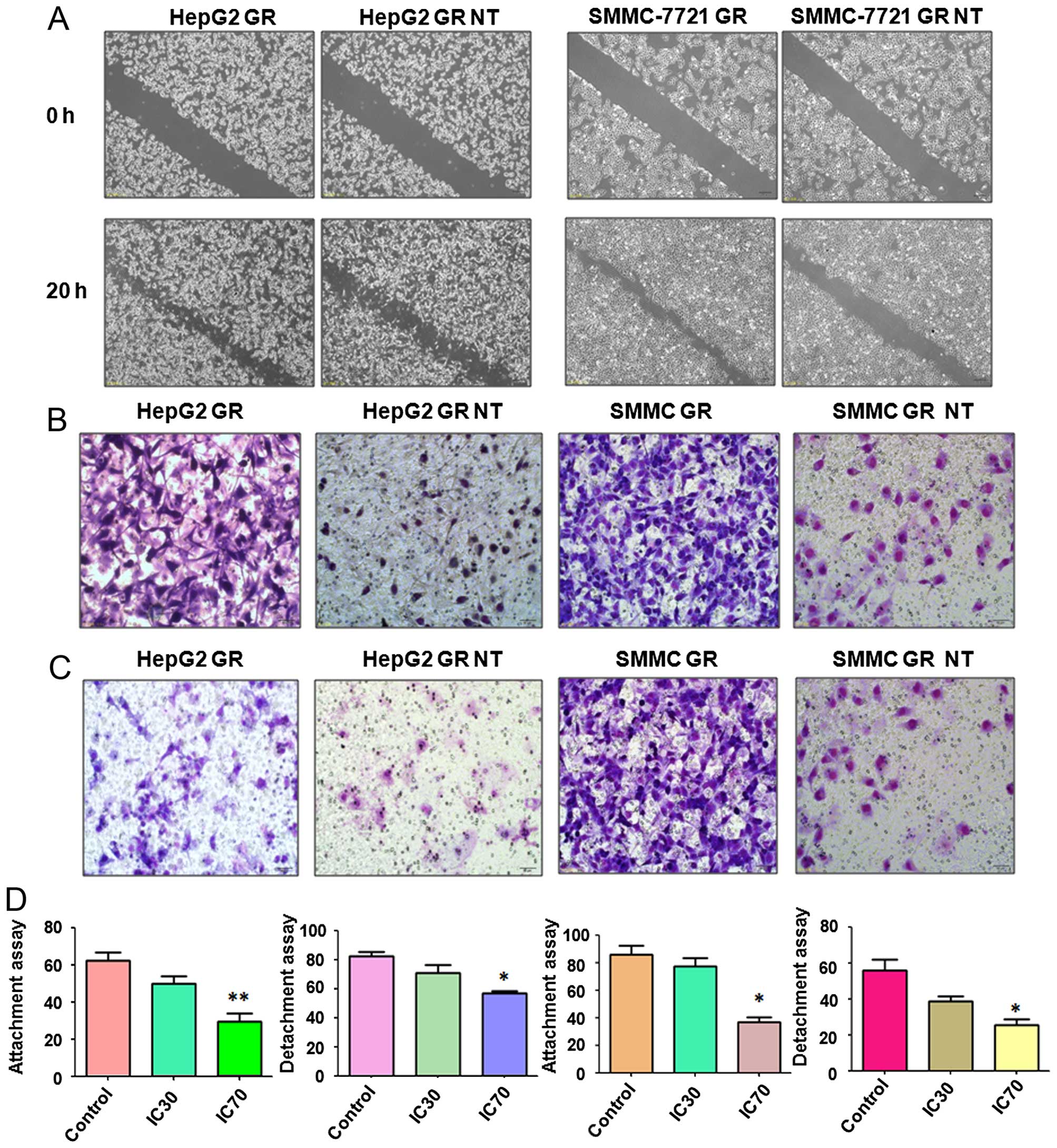
Nutlin-3 suppresses motility in GR HCC
cells
To explore whether Nutlin-3 suppresses the motility
of GR HCC cells, we performed wound healing assays to investigate
the migration of GR HepG2 and GR SMMC-7721 cells following Nutlin-3
treatment. Our results from the wound healing assays demonstrated
that Nutlin-3 markedly decreased cell migration in both the GR
HepG2 and GR SMMC-7721 cells (Fig.
2A). To further validate this finding, migration and invasion
assays were conducted to measure the cell migratory and invasive
activity of the GR HCC cells treated with Nultin-3. We observed
that Nutlin-3 significantly inhibited the migration (Fig. 2B) and invasion (Fig. 2C) in the GR HCC cells. Cell
detachment and attachment are associated with cell motility,
therefore we measured the cell attachment and detachment in the GR
HCC cells following Nutlin-3 treatment. As demonstrated in Fig. 2D, Nutlin-3 treatment suppressed both
cell attachment and detachment in the GR HepG2 and GR SMMC-7721
cells. Altogether, Nutlin-3 treatment may inhibit motility in GR
HCC cells.
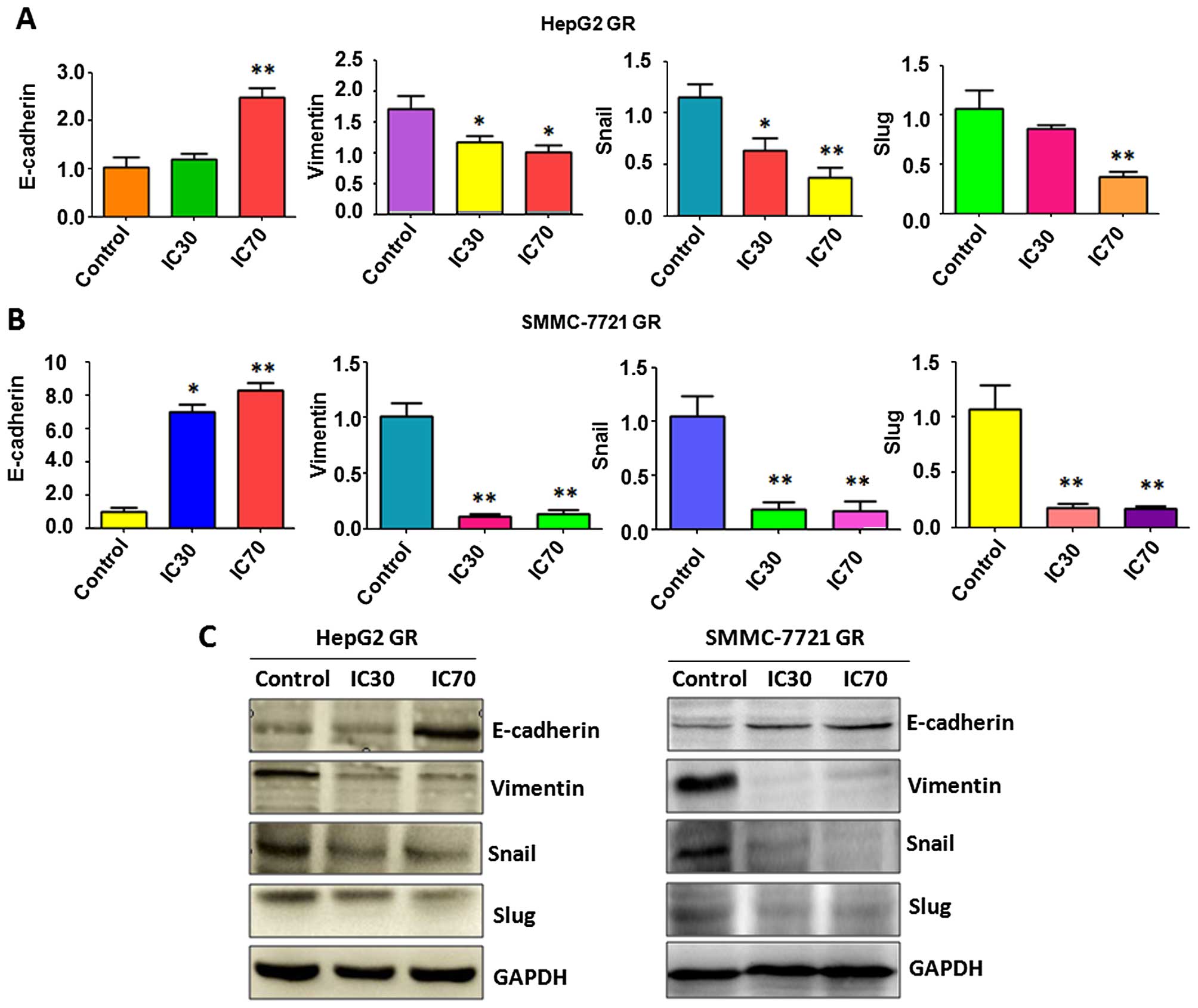
Nutlin-3 reverses EMT to MET in GR HCC
cells
To further identify whether Nutlin-3 regulates
specific EMT molecules in GR HCC cells, we measured the expression
of markers of epithelial and mesenchymal phenotypes using RT-PCR
and western blot analysis, respectively. Our RT-PCR results
indicated that Nutlin-3 treatment increased E-cadherin mRNA levels,
but decreased several mesenchymal markers at the mRNA level
including vimentin, Snail and Slug in both GR HepG2 (Fig. 3A) and GR SMMC-7721 cells (Fig. 3B). Importantly, our western blot
analysis confirmed that the protein level of E-cadherin was
increased, whereas the expressions of vimentin, Snail and Slug were
downregulated in the GR HCC cells following Nutlin-3 treatment
(Fig. 3C). These observations
indicated that Nutlin-3 reversed EMT to MET phenotype, suggesting
that Nutlin-3 could be a potential treatment agent to target
GR-triggered EMT in HCC cells.
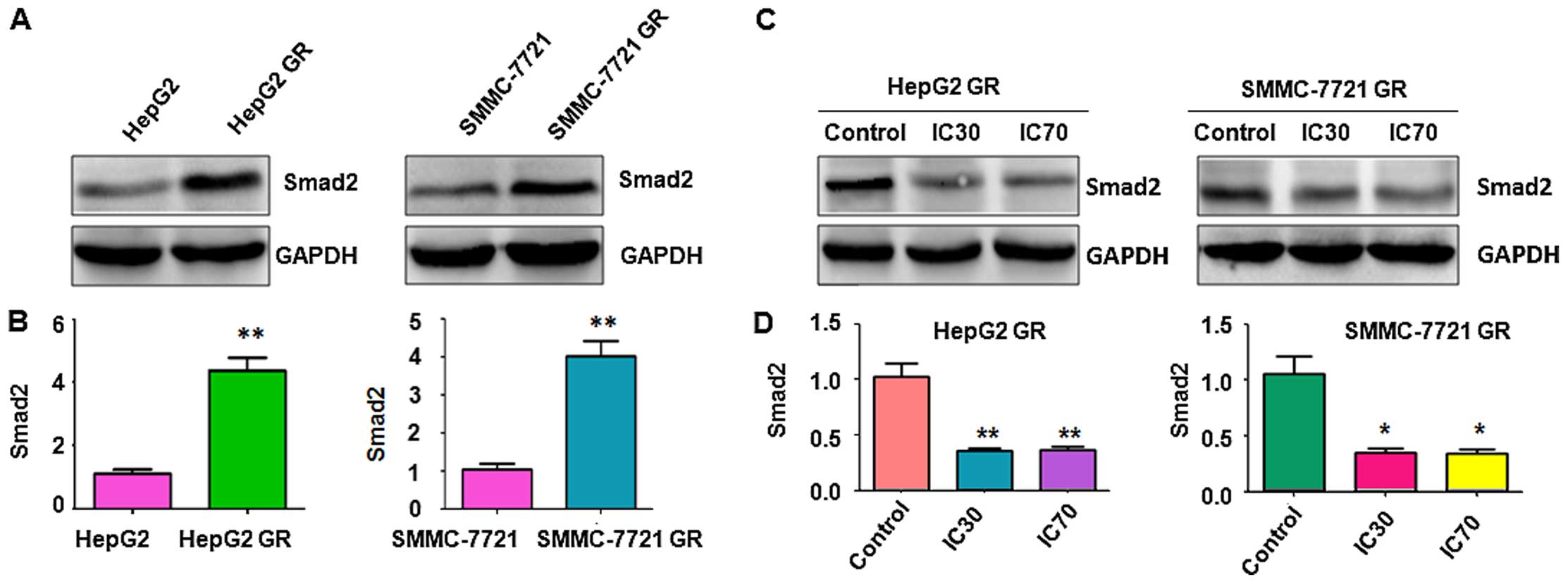
Activation of Smad2 is observed in GR HCC
cells
Since Smad2 was previously reported to be involved
in the EMT process (29), we
measured the expression of Smad2 by western blot analysis. We found
higher expression of Smad2 in GR HepG2 and GR SMMC-7721 cells
compared with their parental control cells (Fig. 4A and B), suggesting that Smad2 might
be involved in GR-induced EMT in HCC cells. It has been reported
that Nutlin-3 prohibited EMT through blocking the phosphorylation
of Smad2 in human cancer cells (30). Therefore, we determined whether
Nutlin-3 inhibits GR-mediated EMT in HCC cells via targeting Smad2.
Consistent with the previous report (29), our western blot analysis revealed
that Nutlin-3 inhibited the expression of Smad2 in both the GR
HepG2 and GR SMMC-7721 cells (Fig. 4C
and D), indicating that Nutlin-3 suppressed GR-induced EMT
partly through inhibition of Smad2 in HCC cells.
Depletion of Smad2 reverses EMT to MET in
GR cells
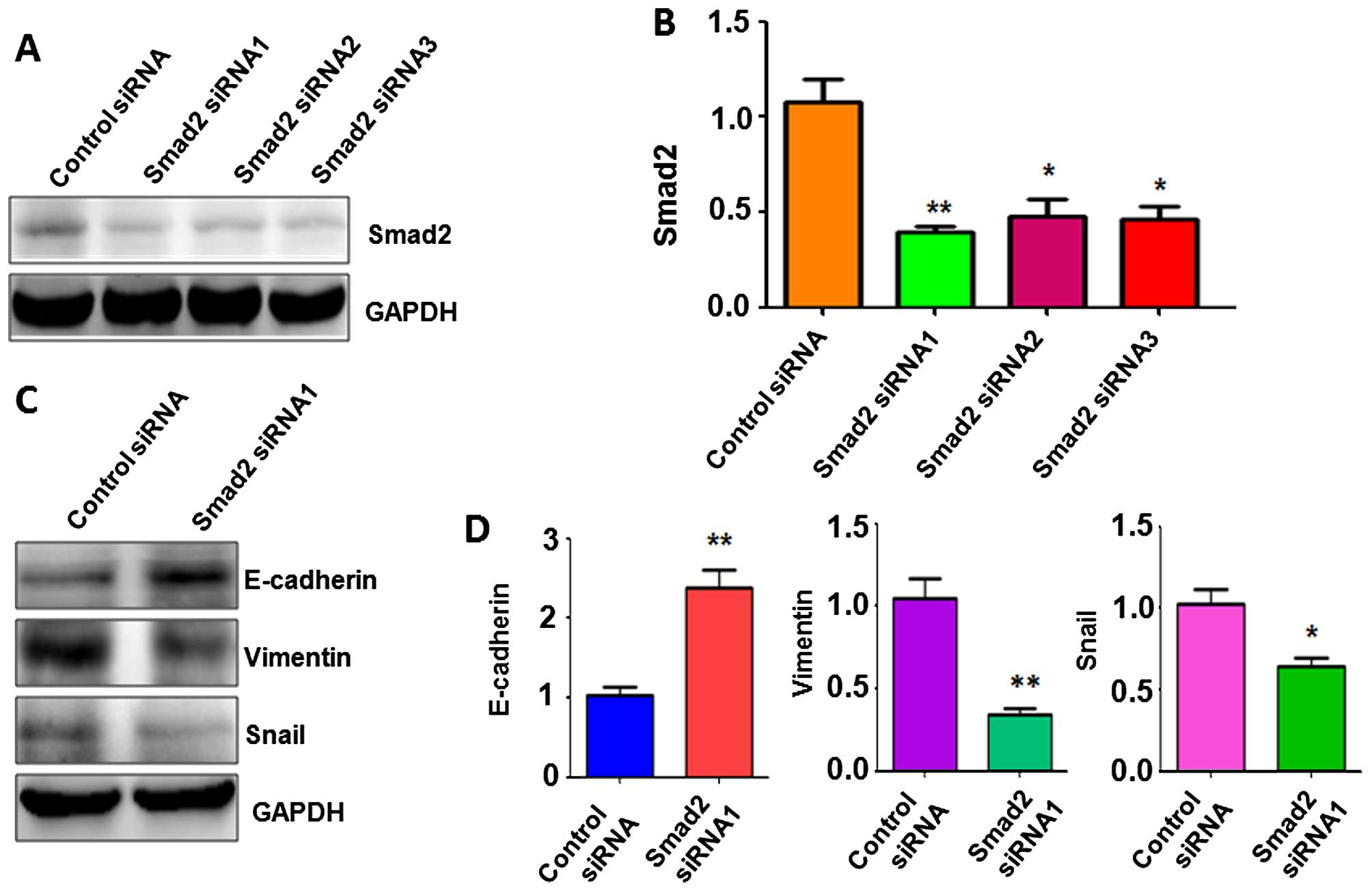
To determine whether Smad2 plays a key role in
GR-mediated EMT, we depleted Smad2 using specific siRNAs in the GR
HepG2 cells. Western blot analysis was used to detect the efficacy
of multiple Smad2 siRNAs on the downregulation of Smad2 in the GR
HCC cells. We found that all three Smad2 siRNAs significantly
depleted Smad2 expression (Fig. 5A and
B). Smad2 siRNA1 was then used for our subsequent studies. To
confirm whether depletion of Smad2 regulates GR-induced EMT, we
measured the expression of EMT markers by western blot analysis in
the GR HCC cells transfected with Smad2 siRNA1. Our results
demonstrated that depletion of Smad2 upregulated E-cadherin protein
levels, but downregulated the protein levels of vimentin and Snail
in the GR HepG2 cells (Fig. 5C and
D). These findings suggest that depletion of Smad2 resulted in
the reversal of EMT to MET in GR HCC cells.
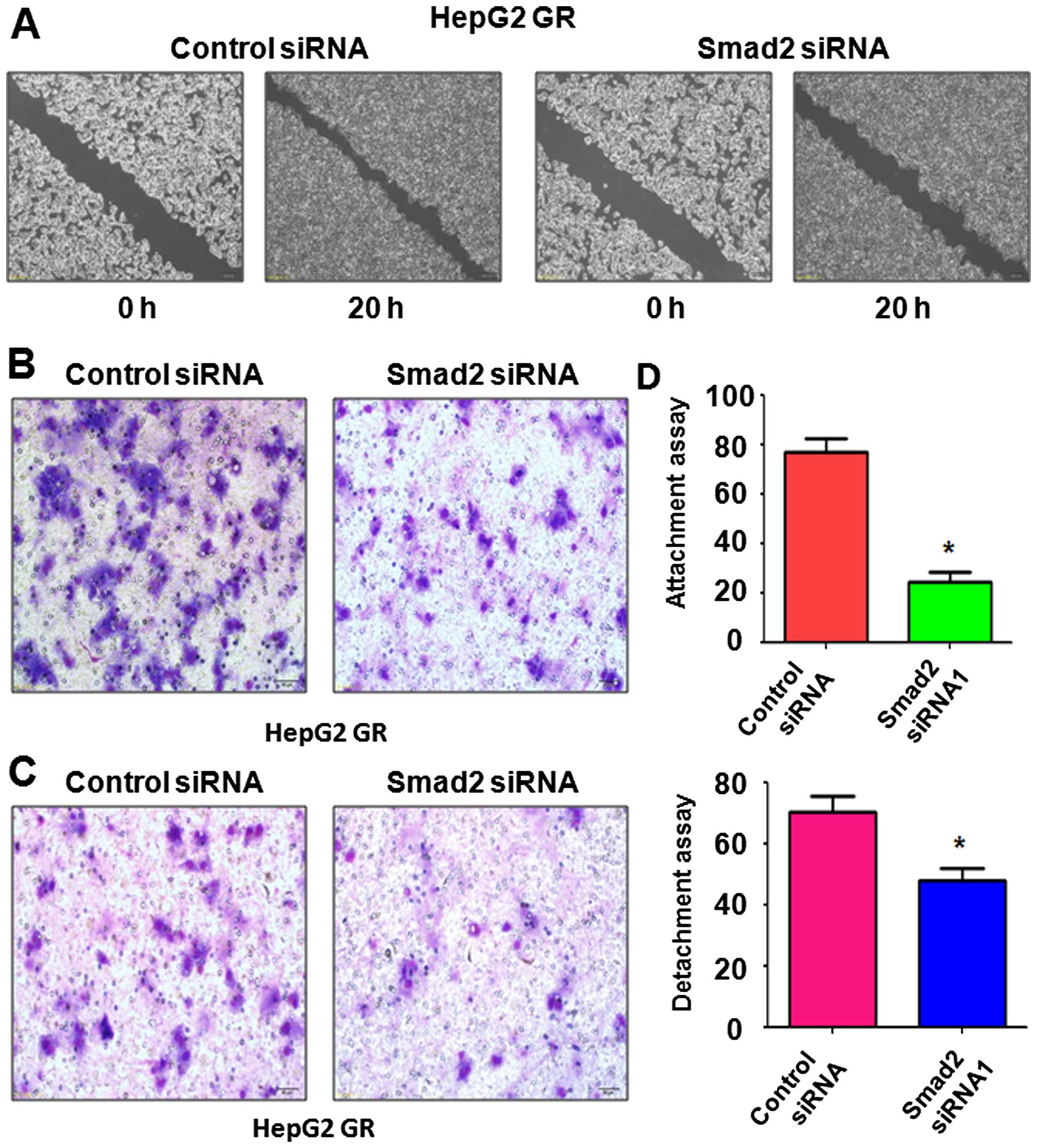
Depletion of Smad2 inhibits migration and
invasion in GR cells
To further characterize the reversal of EMT by
depletion of Smad2, we measured cell motility capacities by wound
healing, migration and invasion assays. Our wound healing assay
results showed that Smad2 depletion led to a decrease in the number
of cells migrating across the wound (Fig. 6A). In further support of this
finding, depletion of Smad2 suppressed cell migration and invasion
in the GR HCC cells (Fig. 6B and
C). In addition, we also performed the cell detachment and
attachment assays following Smad2 depletion. We observed that GR
HepG2 cells depleted of Smad2 displayed reduced detachment and
attachment capacity (Fig. 6D).
Taken together, our data demonstrated that downregulation of Smad2
retarded cell motility capacities in GR HCC cells, and provides a
potential mechanism by which Nutlin-3 suppresses tumor cell growth
and metastasis.
Discussion
In this study, we identified that Nutlin-3 reversed
EMT to MET in GR HCC cells in part through downregulation of Smad2.
Previous results demonstrated that Nutlin-3 selectively inhibited
cell proliferation and enhanced apoptosis by activating the p53
pathway in human cancers including HCC (24). For example, it has been reported
that Nutlin-3 enhanced the growth inhibition by doxorubicin and
potentiated the apoptotic effects of doxorubicin in HepG2 cells via
the disruption of p53-MDM2 binding (31). Moreover, Nutlin-3 was found to
inhibit cell proliferation and induce G0/G1
phase-arrest through downregulation of cyclin D1, cyclin E, CDK2,
CDK4, PCNA and E2F-1 and upregulation of p21 and p27 in human HCC
cells (32). Furthermore, Shi et
al discovered that Nutlin-3 induced apoptosis through
downregulation of phospho-Ser392-p53 in HCC cells (33). Recently, they further identified
that Nutlin-3 significantly increased the expression levels of
interferon-γ-inducible protein 16 (IFI16) and partially
redistributed IFI16 protein to the cytoplasm in SMMC-7721 cells
(wild-type p53), but not in Huh-7 (p53 mutant) and Hep3B (p53-null)
cells, suggesting that Nutlin-3 controlled the subcellular
localization of IFI16 in a p53-dependent manner (34). Nutlin-3 was further reported to
potentiate the antiproliferative activity of gefitinib and
lapatinib in cancer cells (30). In
line with these findings, we found that Nutlin-3 inhibited cell
proliferation in GR HepG2 and GR SMMC-7721 cells.
Emerging evidence has revealed that Nutlin-3 could
be a useful tool to overcome chemoresistance in human cancer. For
example, it has been demonstrated that Nutlin-3 could reverse
P-glycoprotein-mediated multidrug resistance in neuroblastoma and
rhabdomyosarcoma cell lines (35).
Moreover, one study showed that Nutlin-3 sensitized neuroblastoma
cells to doxorubicin through upregulation of E2F1 and p73 (36). Similarly, Nutlin-3 activated the p53
pathway and inhibited tumor growth in chemoresistant neuroblastoma
cells (37). Notably, Nutlin-3 was
considered as a potent enhancer of DR5-selective TRAIL variant
D269H/E195R-induced apoptosis in cancer cells (38). Additionally, cisplatin further
enhanced Nutlin-3 and DR5-mediated apoptosis (38). Nutlin-3 was also identified to
overcome arsenic trioxide resistance and inhibit tumor metastasis
via activation of p73 and enhancing mutant p53 degradation mediated
by arsenic trioxide in HCC cells (26). These reports indicate that Nutlin-3
is involved in regulating drug resistance of cancer cells. A number
of studies havev validated that EMT is associated with drug
resistance (10). Indeed, in this
study, we observed that GR HCC cells acquired EMT features.
Importantly, Nutlin-3 reversed EMT, leading to inhibition of
migration and invasion in GR HCC cells. Mechanistically, Nutlin-3
treatment increased E-cadherin expression and downregulated the
expression of vimentin, Snail, and Slug in GR HCC cells.
It has been previously demonstrated that TGF-β
triggers EMT via governing Smad family expression. One study showed
that Nutlin-3 could abolish the decreased in E-cadherin levels
induced by TGF-β1 in cancer cells (30). Furthermore, this same group found
that Nutlin-3 suppressed EMT by inhibition of phosphorylation of
Smad2/3, leading to decreased motility of cancer cells (30). Since Smad2 has previously been shown
to play an oncogenic role in HCC cells, inhibition of Smad2 could
be a potential strategy for treating HCC (39,40).
Consistent with this notion, we also identified that Nutlin-3
inhibited the expression of Smad2 in GR HCC cells. Strikingly,
depletion of Smad2 inhibited cell migration and invasion in GR
cells and regulated EMT marker expression, indicating that Nutlin-3
may reverse GR-mediated EMT partly via downregulation of the Smad2
pathway.
Nutlin-3 exerts its antitumor activity in a
p53-dependent manner. In tumors carrying a wild-type p53, Nutlin-3
prevented p53 protein degradation leading to maintained activation
of p53, and subsequent induction of p21 and Bax expression, which
are regulated by p53. However, multiple studies also found that
Nutlin-3 inhibited cell growth and enhanced apoptosis in a
p53-independent manner. For instance, Zheng et al reported
that Nutlin-3 disrupted the p73-MDM2 interaction in Huh-7 (p53
mutant) and Hep3B (p53-null) HCC cells and subsequently activated
the apoptotic pathway, leading to the increased chemosensitivity to
doxorubin (31). This group further
found that Nutlin-3 suppressed cell growth and induced
G0/G1 cell cycle arrest in p53-mutant and
p53-null HCC cells (32).
Similarly, Nutlin-3 was reported to induce apoptosis in Huh-7 cells
(33). These reports argued that
Nutlin-3 could have anticancer effects against human cancer cells
regardless of p53 status. Collectively, Nutlin-3 could be an
attractive agent for the treatment of HCC and offers new prospects
to overcome chemoresistance. Without a doubt, further in-depth
investigation is required to explore the molecular mechanism of
Nutlin-3-mediated tumor growth inhibition.
Acknowledgments
This study was supported by funding from the Natural
Science Program of the Education Office of Anhui Province
(KJ2013B147), the Natural Science Research Key Project of Education
Office of Anhui Province (KJ2014A152), and the Natural Science
Foundation of Zhejiang Province (LY14H160045).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Trovato FM, Tognarelli JM, Crossey MM,
Catalano D, Taylor-Robinson SD and Trovato GM: Challenges of liver
cancer: Future emerging tools in imaging and urinary biomarkers.
World J Hepatol. 7:2664–2675. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al SHARP Investigators Study Group: Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S,
Kim JS, Luo R, Feng J, Ye S, Yang TS, et al: Efficacy and safety of
sorafenib in patients in the Asia-Pacific region with advanced
hepatocellular carcinoma: A phase III randomised, double-blind,
placebo-controlled trial. Lancet Oncol. 10:25–34. 2009. View Article : Google Scholar
|
|
6
|
Qin S, Bai Y, Lim HY, Thongprasert S, Chao
Y, Fan J, Yang TS, Bhudhisawasdi V, Kang WK, Zhou Y, et al:
Randomized, multi-center, open-label study of oxaliplatin plus
fluorouracil/leucovorin versus doxorubicin as palliative
chemotherapy in patients with advanced hepatocellular carcinoma
from Asia. J Clin Oncol. 31:3501–3508. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zaanan A, Williet N, Hebbar M, Dabakuyo
TS, Fartoux L, Mansourbakht T, Dubreuil O, Rosmorduc O, Cattan S,
Bonnetain F, et al: Gemcitabine plus oxaliplatin in advanced
hepatocellular carcinoma: A large multicenter AGEO study. J
Hepatol. 58:81–88. 2013. View Article : Google Scholar
|
|
8
|
Llovet JM, Villanueva A, Lachenmayer A and
Finn RS: Advances in targeted therapies for hepatocellular
carcinoma in the genomic era. Nat Rev Clin Oncol. 12:408–424. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wörns MA and Galle PR: HCC
therapies-lessons learned. Nat Rev Gastroenterol Hepatol.
11:447–452. 2014. View Article : Google Scholar
|
|
10
|
Wang Z, Li Y, Ahmad A, Azmi AS, Kong D,
Banerjee S and Sarkar FH: Targeting miRNAs involved in cancer stem
cell and EMT regulation: An emerging concept in overcoming drug
resistance. Drug Resist Updat. 13:109–118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Puisieux A, Brabletz T and Caramel J:
Oncogenic roles of EMT-inducing transcription factors. Nat Cell
Biol. 16:488–494. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mitra A, Mishra L and Li S: EMT, CTCs and
CSCs in tumor relapse and drug-resistance. Oncotarget.
6:10697–10711. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu Q, Wang R, Yang Q, Hou X, Chen S, Hou
Y, Chen C, Yang Y, Miele L, Sarkar FH, et al: Chemoresistance to
gemcitabine in hepatoma cells induces epithelial-mesenchymal
transition and involves activation of PDGF-D pathway. Oncotarget.
4:1999–2009. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang R, Cheng L, Xia J and Wang Z, Wu Q
and Wang Z: Gemcitabine resistance is associated with
epithelial-mesenchymal transition and induction of HIF-1α in
pancreatic cancer cells. Curr Cancer Drug Targets. 14:407–417.
2014. View Article : Google Scholar
|
|
16
|
Güngör C, Zander H, Effenberger KE,
Vashist YK, Kalinina T, Izbicki JR, Yekebas E and Bockhorn M: Notch
signaling activated by replication stress-induced expression of
midkine drives epithelial-mesenchymal transition and
chemoresistance in pancreatic cancer. Cancer Res. 71:5009–5019.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A,
Azmi AS, Ali S, Abbruzzese JL, Gallick GE and Sarkar FH:
Acquisition of epithelial-mesenchymal transition phenotype of
gemcitabine-resistant pancreatic cancer cells is linked with
activation of the notch signaling pathway. Cancer Res.
69:2400–2407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rieber M and Strasberg-Rieber M: p53
inactivation decreases dependence on estrogen/ERK signalling for
proliferation but promotes EMT and susceptility to 3-bromopyruvate
in ERα+ breast cancer MCF-7 cells. Biochem Pharmacol.
88:169–177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin Y, Mallen-St Clair J, Luo J, Sharma S,
Dubinett S and St John M: p53 modulates NF-κB mediated
epithelial-to-mesenchymal transition in head and neck squamous cell
carcinoma. Oral Oncol. 51:921–928. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alam SK, Yadav VK, Bajaj S, Datta A, Dutta
SK, Bhattacharyya M, Bhattacharya S, Debnath S, Roy S, Boardman LA,
et al: DNA damage-induced ephrin-B2 reverse signaling promotes
chemoresistance and drives EMT in colorectal carcinoma harboring
mutant p53. Cell Death Differ. 23:707–722. 2016. View Article : Google Scholar
|
|
21
|
Wade M, Li YC and Wahl GM: MDM2, MDMX and
p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 13:83–96.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Khoo KH, Verma CS and Lane DP: Drugging
the p53 pathway: Understanding the route to clinical efficacy. Nat
Rev Drug Discov. 13:217–236. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Van Maerken T, Rihani A, Van Goethem A, De
Paepe A, Speleman F and Vandesompele J: Pharmacologic activation of
wild-type p53 by nutlin therapy in childhood cancer. Cancer Lett.
344:157–165. 2014. View Article : Google Scholar
|
|
24
|
Secchiero P, Bosco R, Celeghini C and
Zauli G: Recent advances in the therapeutic perspectives of
Nutlin-3. Curr Pharm Des. 17:569–577. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barone G, Tweddle DA, Shohet JM, Chesler
L, Moreno L, Pearson AD and Van Maerken T: MDM2-p53 interaction in
paediatric solid tumours: Preclinical rationale, biomarkers and
resistance. Curr Drug Targets. 15:114–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zheng T, Yin D, Lu Z, Wang J, Li Y, Chen
X, Liang Y, Song X, Qi S, Sun B, et al: Nutlin-3 overcomes arsenic
trioxide resistance and tumor metastasis mediated by mutant p53 in
hepatocellular carcinoma. Mol Cancer. 13:1332014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moran DM and Maki CG: Nutlin-3a induces
cytoskeletal rearrangement and inhibits the migration and invasion
capacity of p53 wild-type cancer cells. Mol Cancer Ther. 9:895–905.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang R, Li Y, Hou Y, Yang Q, Chen S, Wang
X, Wang Z, Yang Y, Chen C, Wang Z, et al: The
PDGF-D/miR-106a/Twist1 pathway orchestrates epithelial-mesenchymal
transition in gemcitabine resistance hepatoma cells. Oncotarget.
6:7000–7010. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Saitoh M: Epithelial-mesenchymal
transition is regulated at post-transcriptional levels by
transforming growth factor-β signaling during tumor progression.
Cancer Sci. 106:481–488. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu Y, Fu Y, Zheng L, Lin G, Ma J, Lou J,
Zhu H, He Q and Yang B: Nutlin-3 inhibits epithelial-mesenchymal
transition by interfering with canonical transforming growth
factor-β1-Smad-Snail/Slug axis. Cancer Lett. 342:82–91. 2014.
View Article : Google Scholar
|
|
31
|
Zheng T, Wang J, Song X, Meng X, Pan S,
Jiang H and Liu L: Nutlin-3 cooperates with doxorubicin to induce
apoptosis of human hepatocellular carcinoma cells through p53 or
p73 signaling pathways. J Cancer Res Clin Oncol. 136:1597–1604.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang J, Zheng T, Chen X, Song X, Meng X,
Bhatta N, Pan S, Jiang H and Liu L: MDM2 antagonist can inhibit
tumor growth in hepatocellular carcinoma with different types of
p53 in vitro. J Gastroenterol Hepatol. 26:371–377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shi X, Liu J, Ren L, Mao N, Tan F, Ding N,
Yang J and Li M: Nutlin-3 downregulates p53 phosphorylation on
serine392 and induces apoptosis in hepatocellular carcinoma cells.
BMB Rep. 47:221–226. 2014. View Article : Google Scholar :
|
|
34
|
Shi XL, Yang J, Mao N, Wu JH, Ren LF, Yang
Y, Yin XL, Wei L, Li MY and Wang BN: Nutlin-3-induced
redistribution of chromatin-bound IFI16 in human hepatocellular
carcinoma cells in vitro is associated with p53 activation. Acta
Pharmacol Sin. 36:252–258. 2015. View Article : Google Scholar :
|
|
35
|
Michaelis M, Rothweiler F, Klassert D, von
Deimling A, Weber K, Fehse B, Kammerer B, Doerr HW and Cinatl J Jr:
Reversal of P-glycoprotein-mediated multidrug resistance by the
murine double minute 2 antagonist nutlin-3. Cancer Res. 69:416–421.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peirce SK and Findley HW: The MDM2
antagonist nutlin-3 sensitizes p53-null neuroblastoma cells to
doxorubicin via E2F1 and TAp73. Int J Oncol. 34:1395–1402.
2009.PubMed/NCBI
|
|
37
|
Van Maerken T, Ferdinande L, Taildeman J,
Lambertz I, Yigit N, Vercruysse L, Rihani A, Michaelis M, Cinatl J
Jr, Cuvelier CA, et al: Antitumor activity of the selective MDM2
antagonist nutlin-3 against chemoresistant neuroblastoma with
wild-type p53. J Natl Cancer Inst. 101:1562–1574. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Meijer A, Kruyt FA, van der Zee AG,
Hollema H, Le P, ten Hoor KA, Groothuis GM, Quax WJ, de Vries EG
and de Jong S: Nutlin-3 preferentially sensitises wild-type
p53-expressing cancer cells to DR5-selective TRAIL over rhTRAIL. Br
J Cancer. 109:2685–2695. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zheng X, Gai X, Han S, Moser CD, Hu C,
Shire AM, Floyd RA and Roberts LR: The human sulfatase 2 inhibitor
2,4-disul-fonylphenyl-tert-butylnitrone (OKN-007) has an antitumor
effect in hepatocellular carcinoma mediated via suppression of
TGFB1/SMAD2 and Hedgehog/GLI1 signaling. Genes Chromosomes Cancer.
52:225–236. 2013. View Article : Google Scholar
|
|
40
|
Wang J, Liu G, Li Q, Wang F, Xie F, Zhai
R, Guo Y, Chen T, Zhang N, Ni W, et al: Mucin1 promotes the
migration and invasion of hepatocellular carcinoma cells via
JNK-mediated phosphorylation of Smad2 at the C-terminal and linker
regions. Oncotarget. 6:19264–19278. 2015. View Article : Google Scholar : PubMed/NCBI
|