Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignancies worldwide, which tends to metastasize within
the liver and to the lung, brain and kidney (1–3). The
incidence of HCC is increasing, especially in China, due to the
high prevalence of hepatitis virus infection, which conveys a high
risk of HCC (4,5). Despite advances in diagnosis and
treatment, HCC still ranks in the top three in cancer-associated
mortality worldwide. Surgical resection is the first choice for
most of HCC patients, but the overall survival rate 5 years after
surgery remains <12% (6–8). With the development of interventional
treatment, chemotherapy drugs can directly act on hepatic carcinoma
cells, so as to prolong the life of patients with liver cancer
(1,9). However, some of the HCC patients are
not sensitive to chemotherapy and prone to resistance. For these
patients, there is a lack of effective treatments. Therefore,
related research focus on seeking effective drugs which can improve
the effect of cancer chemotherapy.
Many studies have been focusing on utilizing ginseng
as an option for HCC treatment (10,11).
The ginsenoside chemoprevention and anticancer effects are achieved
through mechanisms such as DNA damage mitigation, apoptosis
induction, proliferation inhibition positive and immunomodulation
(12,13). 20(S)-ginsenoside Rh2 [(S)Rh2]
increases the proportion of SMMC-7721 cells in G1 phase, and
decreases those in S and G2/M phases (14). Moreover, (S)Rh2 downregulates
the expression of positive regulating factors (cyclin D1 and E) and
upregulates the expression of negative regulating factors (p16
protein, p21 mRNA) in SMMC-7721 cells (15). Many studies have shown that
(S)Rh2 acts upstream of glycogen synthase kinase-3β
(GSK-3β), on EGFR and Akt (16,17).
Inaction of Akt could result in decreased phosphorylation of
GSK-3β, leading to its activation (17). (S)Rh2 is one of the most
widely investigated ginsenosides and exerts potent anticancer
activity in vitro and in vivo (12).
GSK-3β is a serine/threonine kinase that was first
isolated and purified as an enzyme capable of phosphorylating and
inactivating the enzyme glycogen synthase (18). We now know that, beyond its role in
glycogen metabolism, GSK-3β acts as a downstream regulatory switch
that determines the output of numerous signaling pathways initiated
by diverse stimuli (19). GSK-3β
has a high basal activity within the cell, and both insulin and Wnt
stimulation lead to a decrease in kinase activity. In the case of
insulin this is by activation of protein kinase B (PKB), which
phosphorylates a serine residue in the N terminus, residues Ser-9
in GSK-3 and Ser-21 in GSK-3α, and inhibits GSK-3 activity
(20). Wnt stimulation, on the
other hand, acts on GSK-3β in a multiprotein complex that also
includes axin, adenomatous polyposis coli (APC)-associated protein,
and β-catenin. GSK-3β phosphorylates all three of these proteins;
however, phosphorylation of β-catenin leads to its degradation. Wnt
stimulation inhibits the activity of GSK-3β within this complex,
through mechanisms that may involve axin binding to the proteins
dishevelled and LRP-5 (21). This
allows unphosphorylated β-catenin to accumulate in the cytoplasm
and nucleus. By binding to TCF family transcription factors,
nuclear β-catenin regulates transcription of target mRNAs such as
c-Myc and cyclin D1 (22–24). Mutations, which perturb the function
of the axin-APC complex, such as truncation of APC or deletion of
the GSK-3 sites of β-catenin, are present in 90% of colon cancers.
The pathways in which GSK-3 acts as a key regulator, when
dysregulated, have been implicated in the development of human
diseases such as diabetes, Alzheimer's disease, bipolar disorder
and cancer (18).
It has been proven by substantial evidence that
(S)Rh2 displays dramatic inhibitory effect on HepG2 cells,
although its specific molecular mechanism has not been well
understood. GSK-3 is apparently related to tumor progression.
Therefore, we hypothesize that there may be some correlation
between (S)Rh2 and GSK-3. Mechanism in such phenomena is
that (S)Rh2 exerts its anticancer effects via
GSK-3/β-catenin pathway.
Materials and methods
Cell culture
HepG2 cells (Bogoo, Shanghai, China) cryopreserved
in our laboratory were cultured in DMEM-F12 containing 10% fetal
bovine serum (HyClone, Waltham, MA, USA), which does not contain
antibiotics, in incubators with 5% CO2, at 37°C and
constant humidity.
Establishment and identification of
HepG2-β-catenin cells
The lentivirus carrying β-catenin cells were
constructed by Neuron Biotechnology Co., Ltd, which provided that
multiplicity of infection was 20 and puromycin working
concentration was 0.5 µg/µl. The cells were seeded at
a concentration of 2×105 cells/ml and incubated in two
disposable sterile bottles, at 37°C, for 24 h. The culture medium
was discarded, and β-catenin lentiviral particles were added in a
bottle with 50 µl and 4 mg/ml of polybrene 1 µl,
while in another bottle was added DMEM-F12. Laser scanning confocal
microscopy, flow cytometry (FCM), PCR were used in observation.
Stably expressed single clones were established by puromycin
selection (0.5 µg/µl).
HepG2-β-catenin cells were established by our
laboratory, which were cultured in DMEM-F12 containing 10% fetal
bovine serum and puromycin (0.5 µg/µl).
Nude mouse xenograft model
Mice (BALB/c, 6–8 weeks old) were purchased from the
Laboratory Animal Center of Chongqing Medical University. HepG2 and
HepG2-β-catenin cells were injected into the mice. When the
diameter of the new neoplasm was 0.5 cm, the mice were given by
gavage (S)Rh2 (20 mg/kg) once a day for 20 days. The
diameter of the new neoplasms and the weight were recorded every
day.
Antibodies and chemicals
(S)Rh2 was purchased from the National
Standard Network, with a purity of 98%. Dimethylsulphoxide (DMSO),
Cell Counting Kit-8 (Takara Bio, Inc., Shiga, Japan), and Annexin
V-FITC notation apoptosis detection kit (KeyGen Biotech Co., Ltd.,
Nanjing, China) were used. The primary antibodies, GSK-3β
(1:1,000), β-catenin (1:1,000), MMP3 (1:1,000), TCF4 (1:100), were
purchased from AB Antibody Technology. Bax (1:1,000), cyclin D1
(1:1,000) antibodies were purchased from Sigma Co. GSK-3β ELISA kit
was purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA). The chromatin immunoprecipitation (ChIP) and IHC kits were
purchased from Cell Signaling Technology, Inc. The secondary
antibodies were horseradish peroxidase (HRP)-conjugated goat
anti-rabbit IgG and anti-mouse IgG antibodies (Beyotime Institute
of Biotechnology, Shanghai, China).
CCK-8 assay
For detecting cell proliferation, a CCK-8 assay was
performed. Briefly, 1×104 cells/well were plated in
96-well plates and cultured at different times. At the end of each
time, 20 µl CCK-8 was added to each well and then incubated
at 37°C for 3 h. Then plates were detected by 450 nm on a
spectrophotometric plate reader (Shanghai Precision &
Scientific Instrument Co., Ltd., Shanghai, China). Cells were
treated with 0.1% DMSO which served as a solvent control. Complete
culture medium without cells served as a blank control. All
experiments were performed in triplicate. The drug concentration
resulting in inhibition of growth (IC50) to 50% was
determined.
Cell cycle assay
The cells were seeded at a concentration of
2×105 cells/ml and incubated for 24–72 h with
(S)Rh2 at various concentrations. (S)Rh2 dissolved in
DMSO was added to the medium in serial dilution. The cells were
collected by centrifugation at 2,500 × g for 5 min, fixed in 70%
ethanol then washed once with PBS and resuspended in 1 ml of PBS
containing 2.5 µg/ml ribonuclease and 50 µg/ml
propidium iodide (Beyotime Institute of Biotechnology), incubated
in the dark for 30 min at room temperature and analyzed using
FCM.
Apoptosis
Briefly, for the cell death assay, HepG2 and
HepG2-β-catenin cells were seeded at a concentration of
2×105 cells/ml and maintained for logarithmic growth by
passaging them every 48–96 h and incubated for 48 h with
(S)Rh2. Samples were prepared according to the instructions
provided together with Annexin V apoptosis kit. Briefly, after
treatment for the indicated time, cells were collected and washed
twice with binding buffer containing 10 mM HEPES, pH 7.4, 140 mM
NaCl, 2.5 mM CaCl2. Then 1×105 cells were
resuspended in 100 µl of binding buffer. Last, 5 µl
of Annexin V-FITC and 10 µl of propidium iodide (50
µg/ml, stocking concentration) were added to the cell
suspension. Gently mixed, the cells were incubated for 15 min at
room temperature, then 400 µl of binding buffer was added to
get the sample ready. Quantification of cell death was analyzed
with a BD FACScan.
H&E coloration
First tissue blocks were fixed with formalin for 16
h, after which they were embedded with paraffin and dewaxed with
xylene. Then they were dehydrated with ethanol, after which they
were stained with hematoxylin for 5 min and differentiated with
ethanol of hydrochloric acid, then they were immersed in warm water
for 15 min, after which they were put in eosin solution for 2 min
and dehydrated with ethanol again. Finally they were sealed with
neutral resin.
ELISA analysis
The protein content of cell extracts was determined
by the Bradford assay (Bio-Rad). A total of 10 ng of protein was
added in each well. The ELISA kit was used to assay activities of
protein (Cell Signaling Technology, Inc.) according to the
manufacturer's instructions.
Immunocytochemistry
Immunocytochemistry was performed to identify the
subcellular localization of β-catenin, GSK-3β, and MMP3. The
tissues were permeabilized with 0.5% Triton X-100 in PBS for 5 min,
and incubated with 3% H2O2 to inactivate
endogenous peroxidase. Blocking was carried out with goat serum for
1 h to minimize non-specific binding of the primary antibody. The
β-catenin (1:100), GSK-3β (1:200), and MMP3 (1:100) antibodies were
applied overnight, and washed three times with PBS. As a specific
control, PBS was used instead of the primary antibodies to exclude
non-specific binding of the secondary antibodies. After incubation
with goat anti-rabbit secondary antibody for 1 h, slides were
rinsed for 5 min in PBS three times before application of the
HRP-conjugated anti-goat antibody. Finally, immunocytochemical
staining was visualized with 3,3′-diaminobenzidine (DAB) reagent.
Images were acquired using an Olympus DX-51 microscope. The mean
density values of the immunocytochemical stains were quantified by
Image-Pro Plus (IPP) software.
ChIP-PCR
ChIP was performed by the Cell Signaling Technology
ChIP assay kit with minor modifications. A total of
1.1×108 cells were incubated with 1% formaldehyde for 15
min and 0.125 M glycine was added for 5 min. Washed cells were
incubated in NP-40 lysis buffer and nuclei pelleted, resuspended in
sodium dodecyl sulfate lysis buffer and sonicated five times on
ice. Precleared soluble chromatin was incubated with anti-TCF4, an
isotype control, or no antibody followed by Protein G
agarose/salmon sperm DNA. qPCR was performed on immune
complex-associated DNA by primers spanning rs1876453
(5-GGAAAGTTTCTGTGCCGCGA-3, 5-GACAATCAGGACCAGGCGGT-3),
SYBR-Green-based detection, and the Illumina Eco Real-Time PCR
System. A standard curve was constructed by a chromatin input
control.
Western blotting
The protein content of cell extracts was determined
by the Bradford assay (Bio-Rad). A total of 20–30 µg of
protein was electrophoresed on 10–15% SDS-PAGE and transferred to
PDVF membranes. Membranes were blocked and incubated with primary
Abs at the appropriate concentration. Subsequently they were
incubated with HRP-conjugated goat anti-rabbit IgG or goat
anti-mouse IgG (1:2,000 dilutions; Bio-Rad). Labeled bands were
detected by Immun-Star™ HRP Chemiluminescent Kit, and images were
captured and the intensity of the bands was quantified by the
Bio-Rad VersaDoc™ Imaging System (Bio-Rad, Regents Park, NSW,
Australia).
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
Total RNA was extracted by TRIzol reagent
(Invitrogen). RT-PCR was carried out with M-MLV transcrip-tase and
oligod(T) and the resulting cDNA products were used as templates
for real-time PCR assays. Real-time RT-PCR was performed by the ABI
PRISM 7700 Sequence Detection System (Applied Biosystems). A total
of 25 µl mixture was used for reaction. Fold change in mRNA
expression was determined with the 2−ΔΔCT method with
β-actin as endogenous control. Primer sequence was as follows:
β-catenin forward, CGCCAGGGCGCCAGGGTTTTCCCAGTC and reverse,
TAATACGACTCACTAGAGGG; GSK-3β forward, GGATTCGTCAGGAACAGGACA and
reverse, TTAGCATCTGACGCTGCTGT; Bcl-2 forward, GGTGAACTGGGGGAGGATTG
and reverse, GGCAGGCATGTTGACTTCAC; Cyclin D1 forward,
CATGGAGAGACAGACAGAGCA and reverse, TATCCACGGGGCTGTTCCTA; Bax
forward, TTCATCCAGGATCGAGCAGG and reverse, CTTGGTGGACGCATCCTGAG;
MMP3 forward, TAATGGAGATGCCCACTTTGATG and reverse,
GAGTGAAAGAGACCCAGGGAGTG. After incubation at 5°C for 2 min, then
95°C for 10 min, the reaction was carried out for 40 cycles as
follows: 95°C for 15 sec and 60°C for 1 min.
Statistical analysis
The intensity of the immunoreactive bands was
determined by a densitometer (Bio-Rad, Hercules, CA, USA). The
statistical significance of the differences between the control and
treated samples was calculated by Student's t-test (SSPS 17.0).
P<0.05 was considered significant. All experiments were repeated
at least three times, for each time with three or more independent
observations.
Results
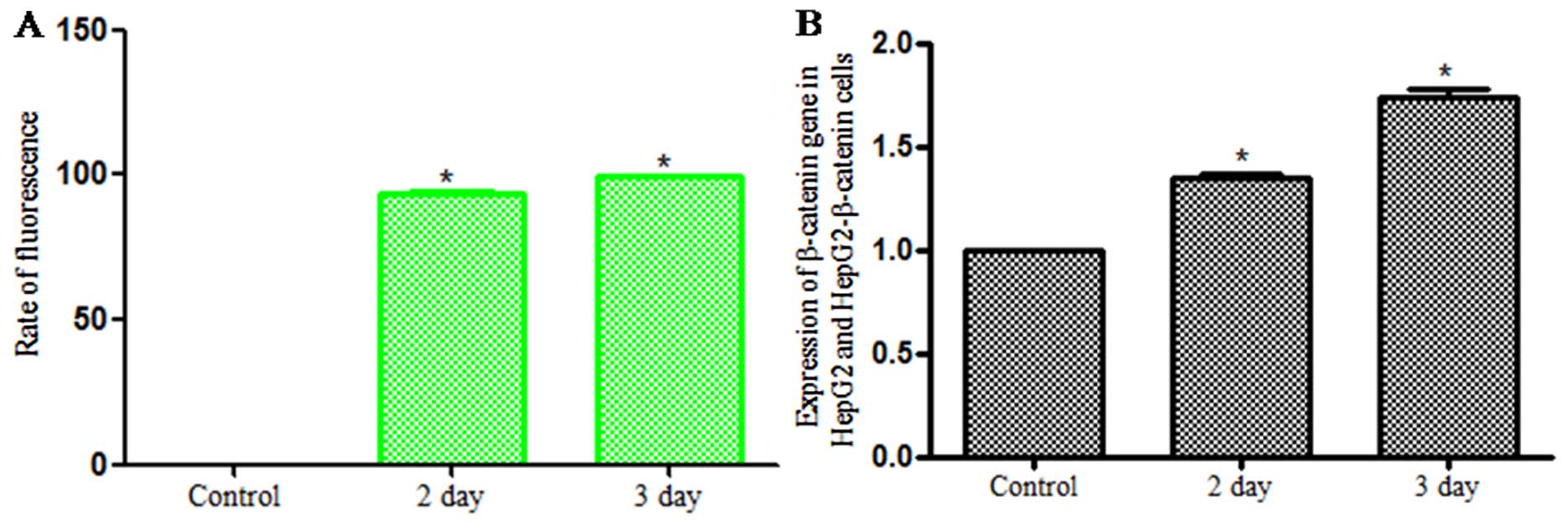
Identification of HepG2-β-catenin
cells
Lentivirus carrying β-catenin was used to infect
HepG2 cells for 2 days. Laser scanning confocal microscopy was used
for observation and it showed that cells emitted green
fluorescence, which indicated that the HepG2 cells were
successfully infected by lentivirus. Infection rate was analyzed by
FCM which increased as time went on (Fig. 1A). The PCR was used for checking the
expression of β-catenin and the results demonstrated that the
expression of β-catenin in infected group was higher than that of
the control group (Fig. 1B). So it
was identified that HepG2 cells were infected by lentivirus
carrying β-catenin, and were named HepG2-β-catenin cells.
Overexpression of β-catenin decreases the
pharmacological effect of (S)Rh2
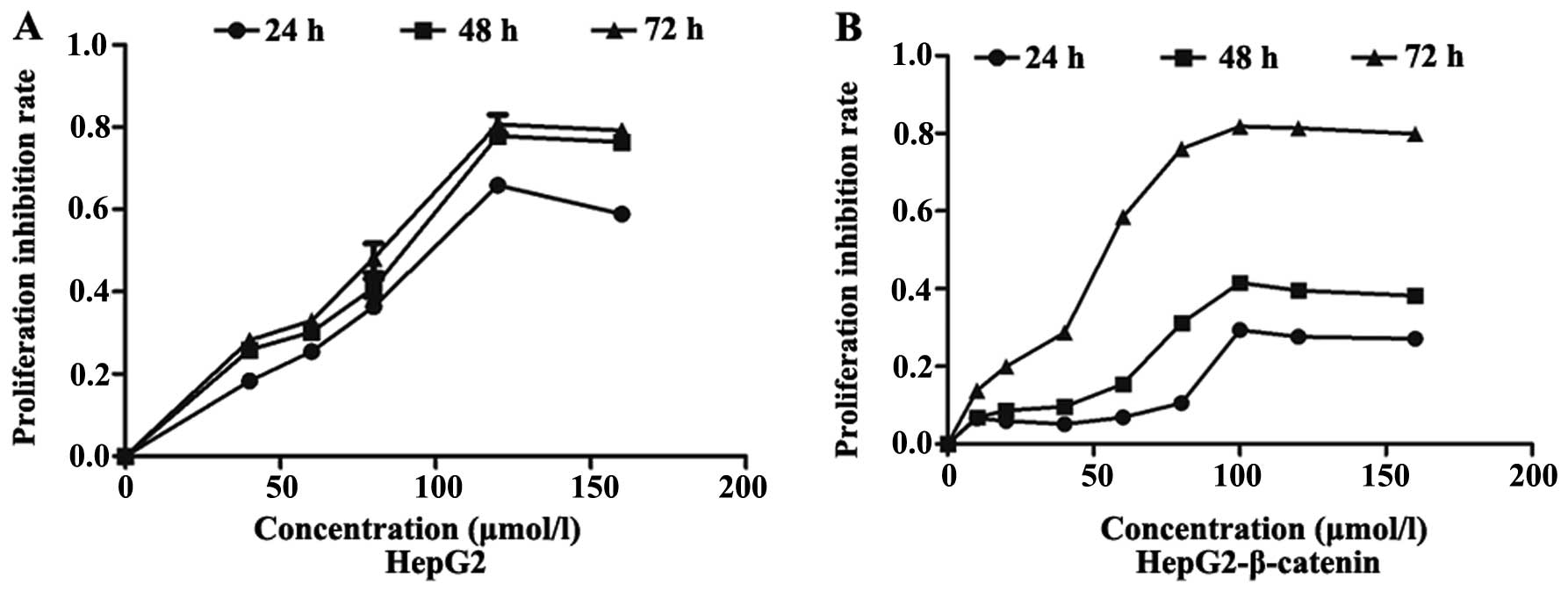
CCK-8 showed that ginsenoside (S)Rh2 can
effectively inhibit the survival of HepG2 and HepG2-β-catenin cells
in vitro, which exhibited a dose-dependent manner at a range
of 10–160 µmol/l (S)Rh2. The IC50 of
(S)Rh2 exposure on HepG2 cells for 48 and 72 h was 100 and
58.12 µmol/l, respectively, and the IC50 of
(S)Rh2 exposure on HepG2-β-catenin for 48 and 72 h was 129.2
and 83.33 µmol/l, respectively. The IC50 of
(S)Rh2 exposure on HepG2-β-catenin cells was higher than
that on HepG2 cells. The effect of overexpression of β-catenin on
cell cycle and apoptosis was detected by FCM which indicated that
(S)Rh2 could arrest HepG2 and HepG2-β-catenin cells in G0/G1
phase. The cell population in G0/G1 phase of the HepG2 group was
61.02±1.48%, of the HepG2 + (S)Rh2 group 64.57±0.65%, of the
HepG2-β-catenin group 52.86±1.46%, and of the HepG2-β-catenin +
(S)Rh2 group 58.61±2.01%. HepG2-β-catenin cells in G0/G1
phase (52.86±1.46%) was significantly lower than that in the HepG2
group (61.02±1.48%), which illustrated that the percentage of
overexpression of β-catenin in G0/G1 phase decreased so as to
accelerate proliferation. The apoptotic rate of the HepG2 group was
10.01±2.02%, of the HepG2 + (S)Rh2 group 17.27±2.77%, of the
HepG2-β-catenin group 5.26±0.50%, and of the HepG2-β-catenin +
(S)Rh2 group 9.02±1.76%. The apoptotic rate of the
HepG2-β-catenin + (S)Rh2 group was lower than that of the
HepG2 + (S)Rh2 group, and the difference between them was
statistically significant (p<0.01). The results of FCM showed
that overexpression of β-catenin could decrease the pharmacological
effect on apoptosis of HepG2 (Fig.
3B).
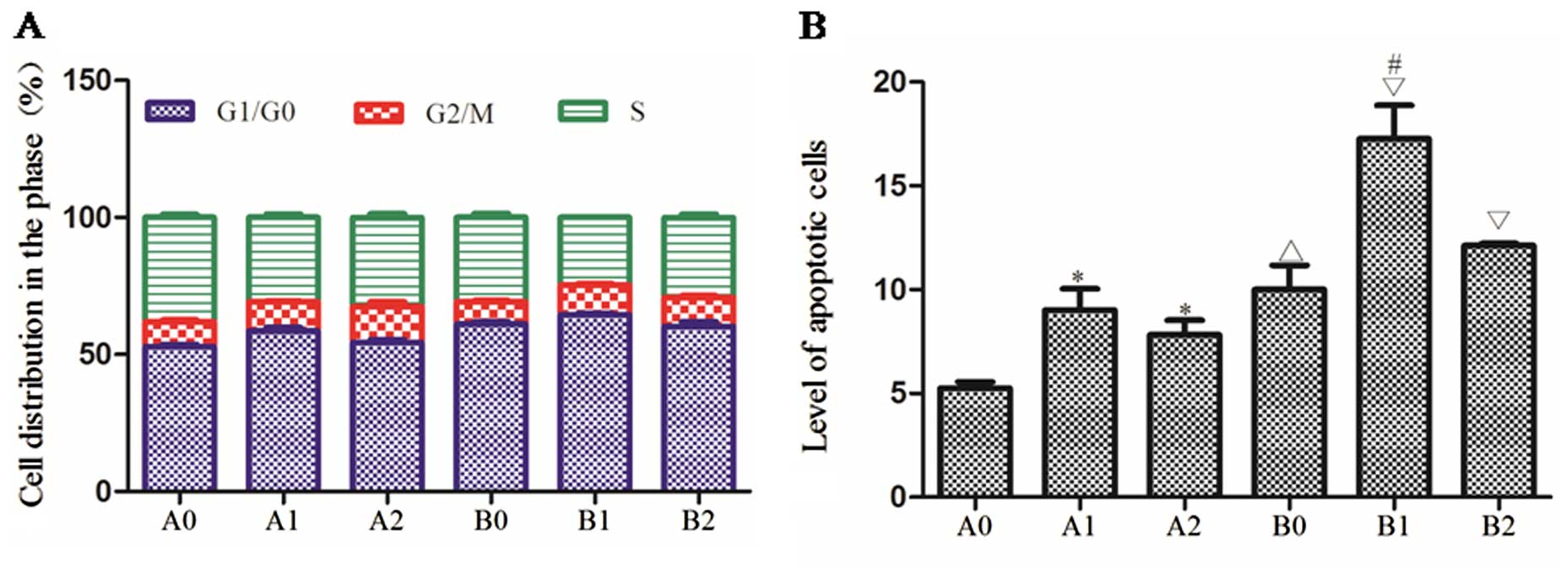 | Figure 3Cell cycle and apoptosis. (A)
HepG2-β-catenin and HepG2 cell cycle arrest was induced for 48 h
with (S)Rh2 or (S)Rh2 + BIO. Cell cycle distribution
was analyzed by FCM. Green areas, S phase; red areas, G2/M phase;
blue areas, G0/G1 phase of the cell cycle. Proportion of the cells
is expressed as the mean ± SD of three independent experiments. (B)
Apoptosis in HepG2-β-catenin and HepG2 cells treated for 48 h with
(S)Rh2 or (S)Rh2 + BIO was measured by Annexin
V-FITC/PI. Controls were treated with the appropriate vehicle.
Duplicate samples were measured and representative experimental
results are shown. A0, HepG2-β-catenin; A1, HepG2-β-catenin +
(S)Rh2; A2, HepG2-β-catenin + (S)Rh2 + BIO; B0,
HepG2; B1, HepG2 + (S)Rh2; B2, HepG2 + (S)Rh2 + BIO.
(S)Rh2, 20(S)-ginsenoside Rh2; FCM, flow
cytometry. |
(S)Rh2 activated GSK-3β to degraded
β-catenin in HepG2-β-catenin and HepG2 cells
The stability of β-catenin depends on GSK-3β. ELISA
kit was used to analyze the activity of GSK-3β, and the results
indicated that the activity of GSK-3β gradually increased when
HepG2 cells were induced by (S)Rh2 for 12, 24, 48 and 72 h,
reaching the peak in 48 h and then decreased (Fig. 4A). Next, GSK-3β inhibitor BIO was
used to verify whether (S)Rh2 activated GSK-3β. The activity
of GSK-3β in HepG2-β-catenin + (S)Rh2 and HepG2 +
(S)Rh2 groups reduced after BIO was added (Fig. 4B). These results suggested that
(S)Rh2 activates the activity of GSK-3β. Furthermore, GSK-3β
and β-catenin mRNA and protein expression treated with
(S)Rh2 or (S)Rh2 + BIO was analyzed. The PCR and
western blotting results showed that the expression of GSK-3β mRNA
and proteins increased in HepG2 and HepG2-β-catenin cells induced
by (S)Rh2, but the expression of GSK-3β did not show
significant difference between HepG2-β-catenin + (S)Rh2 and
HepG2 + (S)Rh2 groups (Fig.
5A–C). In order to further validate that upregulated GSK-3β
would adjust to the effect of downstream mRNA and protein, PCR and
western blotting were used to determine the expression of β-catenin
mRNA and protein. The PCR and western blotting results showed that
the expression of β-catenin mRNA and proteins was downregulated in
HepG2 and HepG2-β-catenin cells induced by (S)Rh2. So we
presumed that ginsenoside (S)Rh2 degraded β-catenin through
activating GSK-3β.
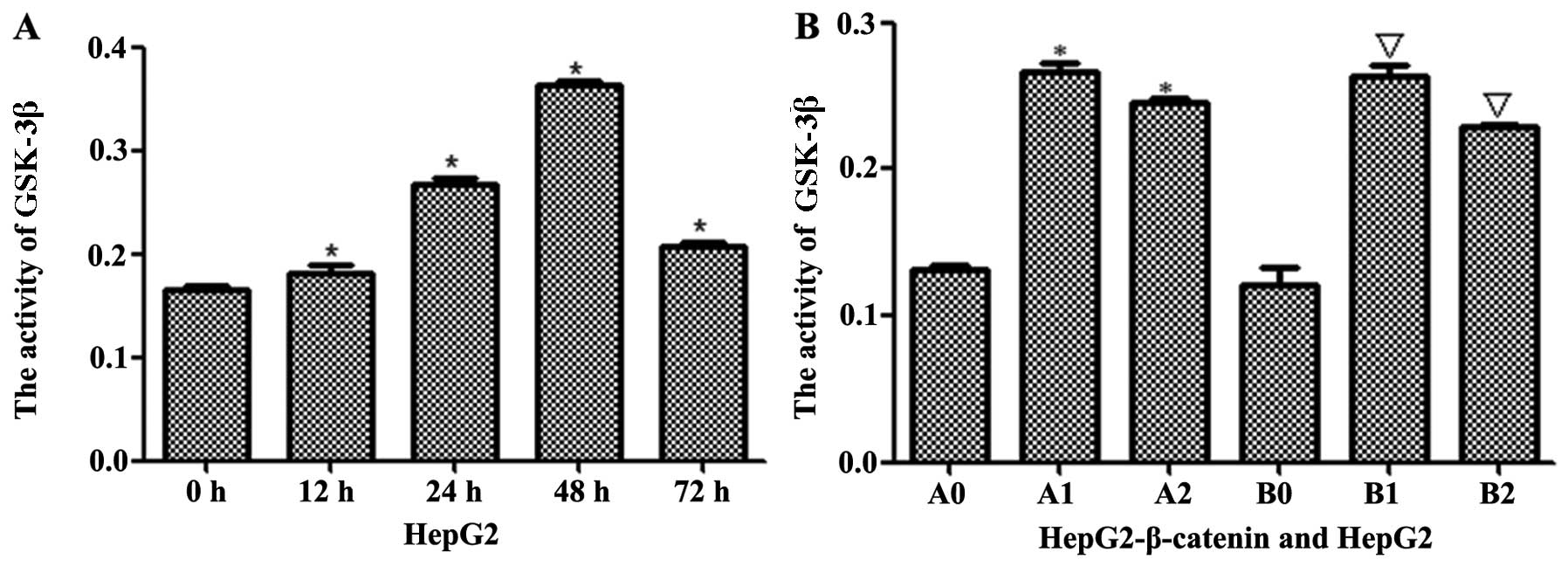 | Figure 4GSK-3β was activated by
(S)Rh2. (A) The activity of GSK-3β was checked by ELISA kit.
In treatment group, the activity of GSK-3β increased in a
time-dependent manner. (B) GSK-3β was activated by (S)Rh2 in
HepG2-β-catenin and HepG2 cells. Results shown are representative
of at least three independent experiments. *P<0.01,
HepG2-β-catenin + (S)Rh2, shRNA-β-catenin-HepG2 +
(S)Rh2 + BIO vs. HepG2-β-catenin group;
△p<0.01, HepG2 vs. HepG2-β-catenin;
∇p<0.01, HepG2 + (S)Rh2, HepG2 + (S)Rh2
+ BIO vs. HepG2; #p<0.01, HepG2 + (S)Rh2 vs.
HepG2-β-catenin + (S)Rh2. A0, HepG2-β-catenin; A1,
HepG2-β-catenin + (S)Rh2; A2, HepG2-β-catenin +
(S)Rh2 + BIO; B0, HepG2; B1, HepG2 + (S)Rh2; B2,
HepG2 + (S)Rh2 + BIO. GSK-3β, glycogen synthase kinase-3β;
(S)Rh2, 20(S)-ginsenoside Rh2. |
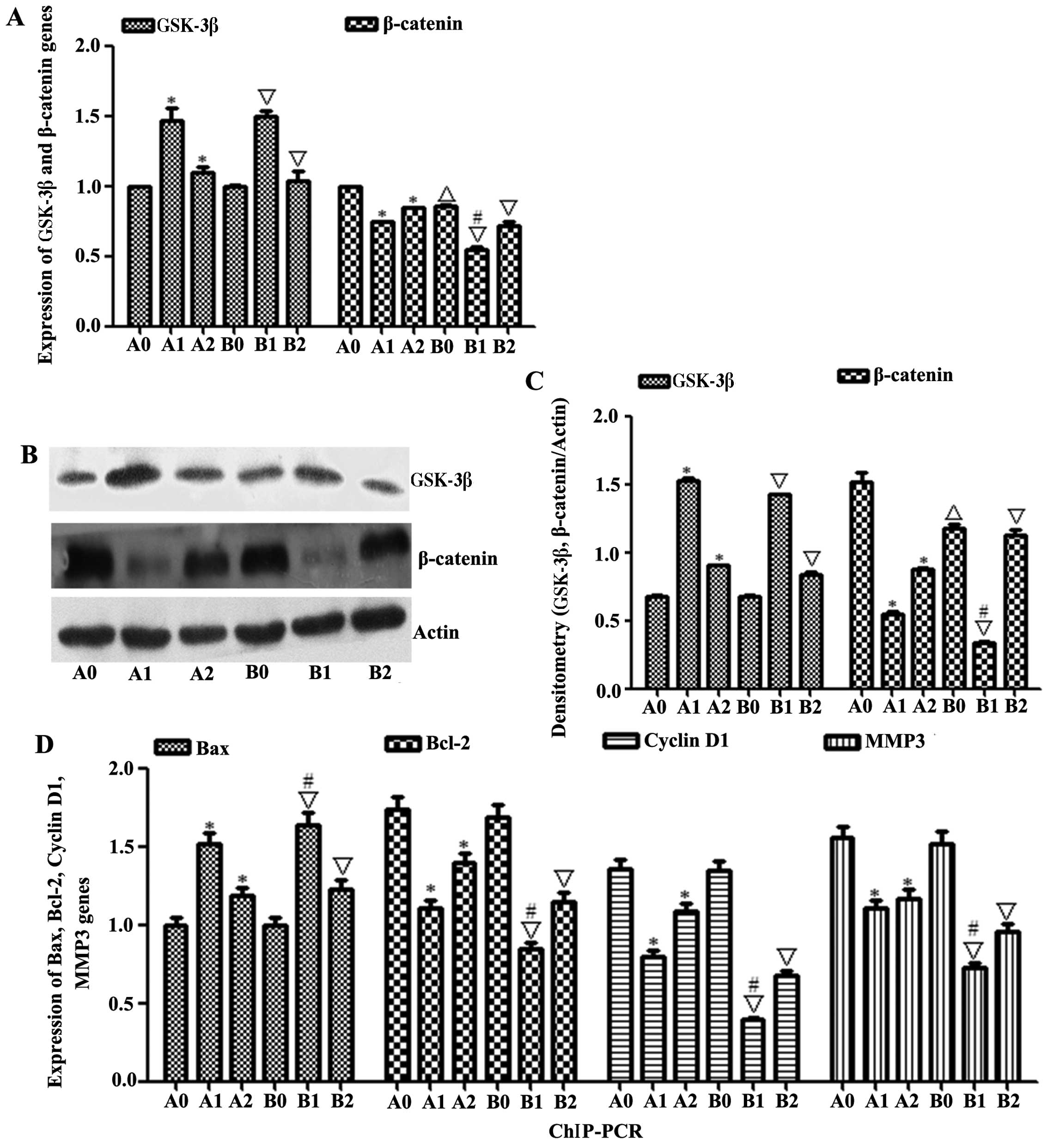 | Figure 5The expression of β-catenin was
degraded through activating GSK-3β and ChIP-PCR. (A) The
expressions of GSK-3β and β-catenin mRNAs were measured by qRT-PCR.
(B and C) GSK-3β and β-catenin expression levels were determined by
western blotting; β-actin served as a protein loading control. (D)
The expressions of Bax, Bcl-2, cyclin D1 and MMP3 mRNAs were
measured by PCR. Results shown are representative of at least three
independent experiments. *P<0.01, HepG2-β-catenin +
(S)Rh2, shRNA-β-catenin-HepG2 + (S)Rh2 + BIO vs.
HepG2-β-catenin group; △p<0.01, HepG2 vs.
HepG2-β-catenin; ∇p<0.01, HepG2 + (S)Rh2,
HepG2 + (S)Rh2 + BIO vs. HepG2; #p<0.01, HepG2
+ (S)Rh2 vs. HepG2-β-catenin + (S)Rh2. A0,
HepG2-β-catenin; A1, HepG2-β-catenin + (S)Rh2; A2,
HepG2-β-catenin + (S)Rh2 + BIO; B0, HepG2; B1, HepG2 +
(S)Rh2; B2, HepG2 + (S)Rh2 + BIO. GSK-3β, glycogen
synthase kinase-3β; ChIP, chromatin immunoprecipitation;
(S)Rh2, 20(S)-ginsenoside Rh2. |
Changes of downstream mRNAs
To check the relationship between β-catenin and
TCF4, ChIP assay kit was used. The expression of downstream genes,
including Bax, Bcl-2, cyclin D1 and MMP3, was measured by PCR. The
ChIP results showed that the expression of Bax mRNA increased,
while the cyclin D1, Bcl-2 and MMP3 mRNA expressions were
downregulated in HepG2 and HepG2-β-catenin cells induced by
(S)Rh2. Compared with HepG2-β-catenin + (S)Rh2 group,
the expression of Bax mRNA in HepG2 + (S)Rh2 group increased
significantly, and the expressions of Bcl-2, cyclin D1 and MMP3
mRNA were also significantly low, between which the difference was
statistically significant (Fig.
5D). The PCR and western blotting results showed that the
expression of Bax mRNA and proteins increased, while the expression
of cyclin D1, Bcl-2 and MMP3 mRNA and proteins were down-regulated
in HepG2 and HepG2-β-catenin cells induced by (S)Rh2
(Fig. 6A–C).
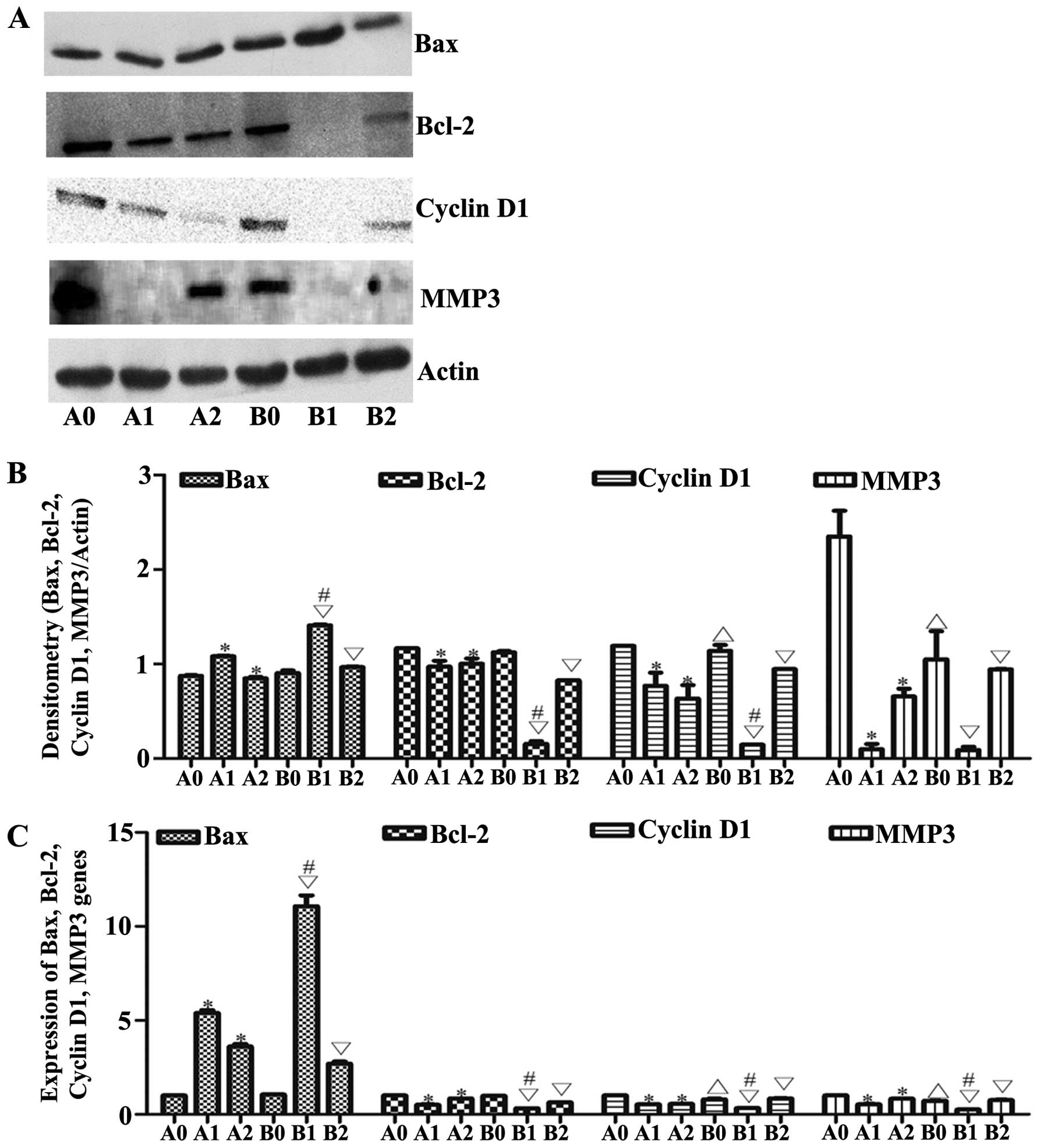 | Figure 6Changes of downstream mRNAs. (A and
B) Bax, Bcl-2, cyclin D1, MMP3 expression levels were determined by
western blotting; β-actin served as a protein loading control. (C)
The expressions of Bax, Bcl-2, cyclin D1 and MMP3 mRNAs were
measured by qRT-PCR. Results shown are representative of at least
three independent experiments. *P<0.05 vs. control.
*P<0.01, HepG2-β-catenin + (S)Rh2,
shRNA-β-catenin-HepG2 + (S)Rh2 + BIO vs. HepG2-β-catenin
group; △p<0.0, HepG2 vs. HepG2-β-catenin;
∇p<0.01, HepG2 + (S)Rh2, HepG2 + (S)Rh2
+ BIO vs. HepG2; #p<0.01, HepG2 + (S)Rh2 vs.
HepG2-β-catenin + (S)Rh2. A0, HepG2-β-catenin; A1,
HepG2-β-catenin + (S)Rh2; A2, HepG2-β-catenin +
(S)Rh2 + BIO; B0, HepG2; B1, HepG2 + (S)Rh2; B2,
HepG2 + (S)Rh2 + BIO; (S)Rh2,
20(S)-ginsenoside Rh2. |
Effect of (S)Rh2 on nude mouse xenograft
model
To evaluate the effect of (S)Rh2 on tumor
growth, nude mice inoculated with HepG2-β-catenin and HepG2 cells
were treated with (S)Rh2 (20 mg/kg). (S)Rh2 was
administered every day for 20 days consecutively. The mice grew
well, and the weight of the mice increased every day. The weight of
tumor in HepG2-β-catenin group (1.7±0.19 g) was greater than that
in the HepG2 group (1.6±0.16 g); the weight of tumor in
HepG2-β-catenin + (S)Rh2 group (1.10±0.12 g) was greater
than that in the HepG2 + (S)Rh2 group (1.0±0.13 g), and the
difference between them was statistically significant (p<0.01).
The alterations in tumor volume as well as diameter changed
similarly to weight (Fig. 7A–D).
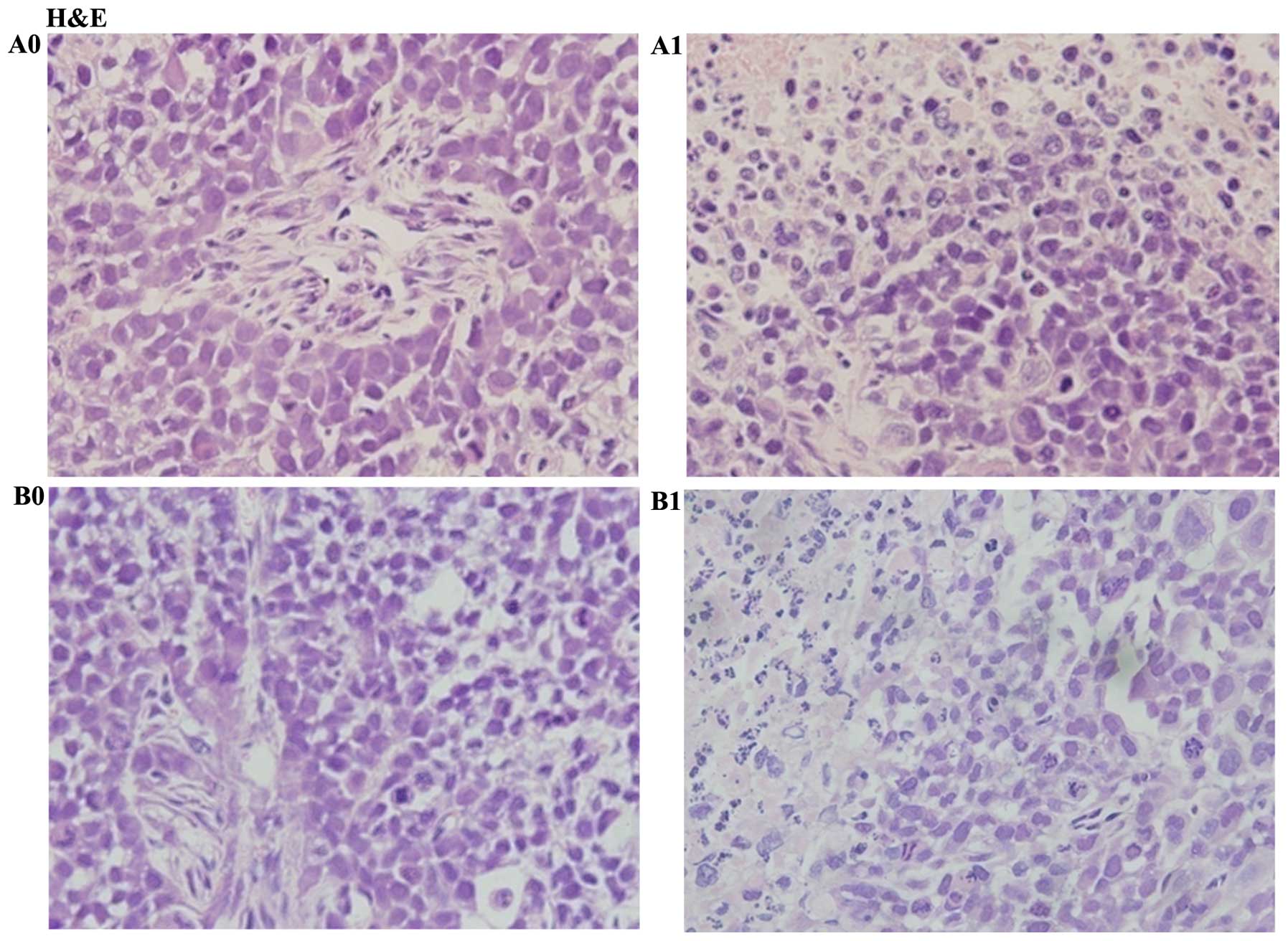
Tumor sections were paraffin-embedded and H&E stain was used to
observe the cell morphology. The results showed nucleus atypia and
accounted for a large proportion of the cells in HepG2 and
HepG2-β-catenin cells. But atypia in the nucleus in HepG2-β-catenin
group was more obvious. Condensation in nuclei and many broken
cells were observed in HepG2-β-catenin + (S)Rh2 and HepG2 +
(S)Rh2 groups. However, condensation in nuclei and broken
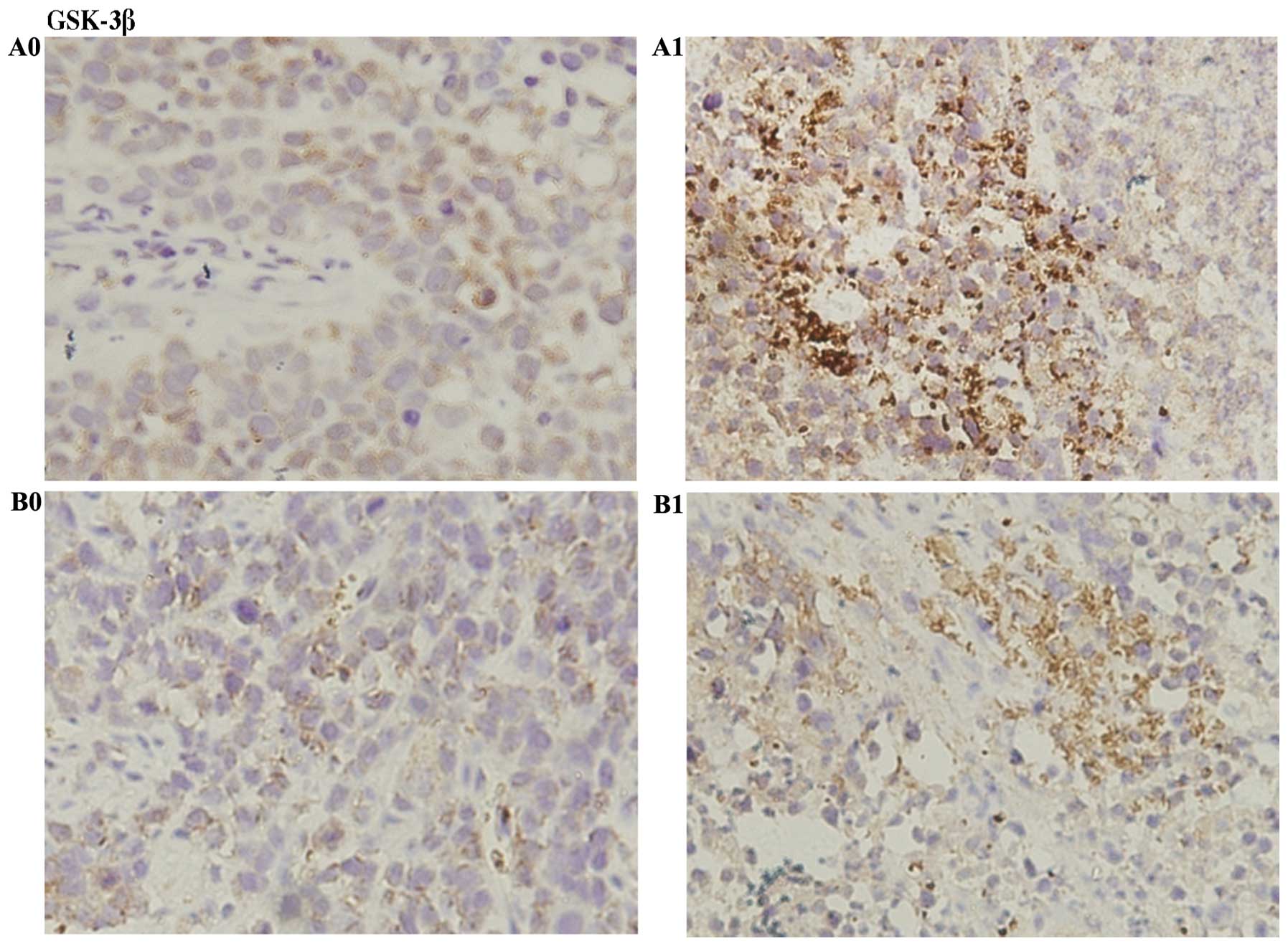
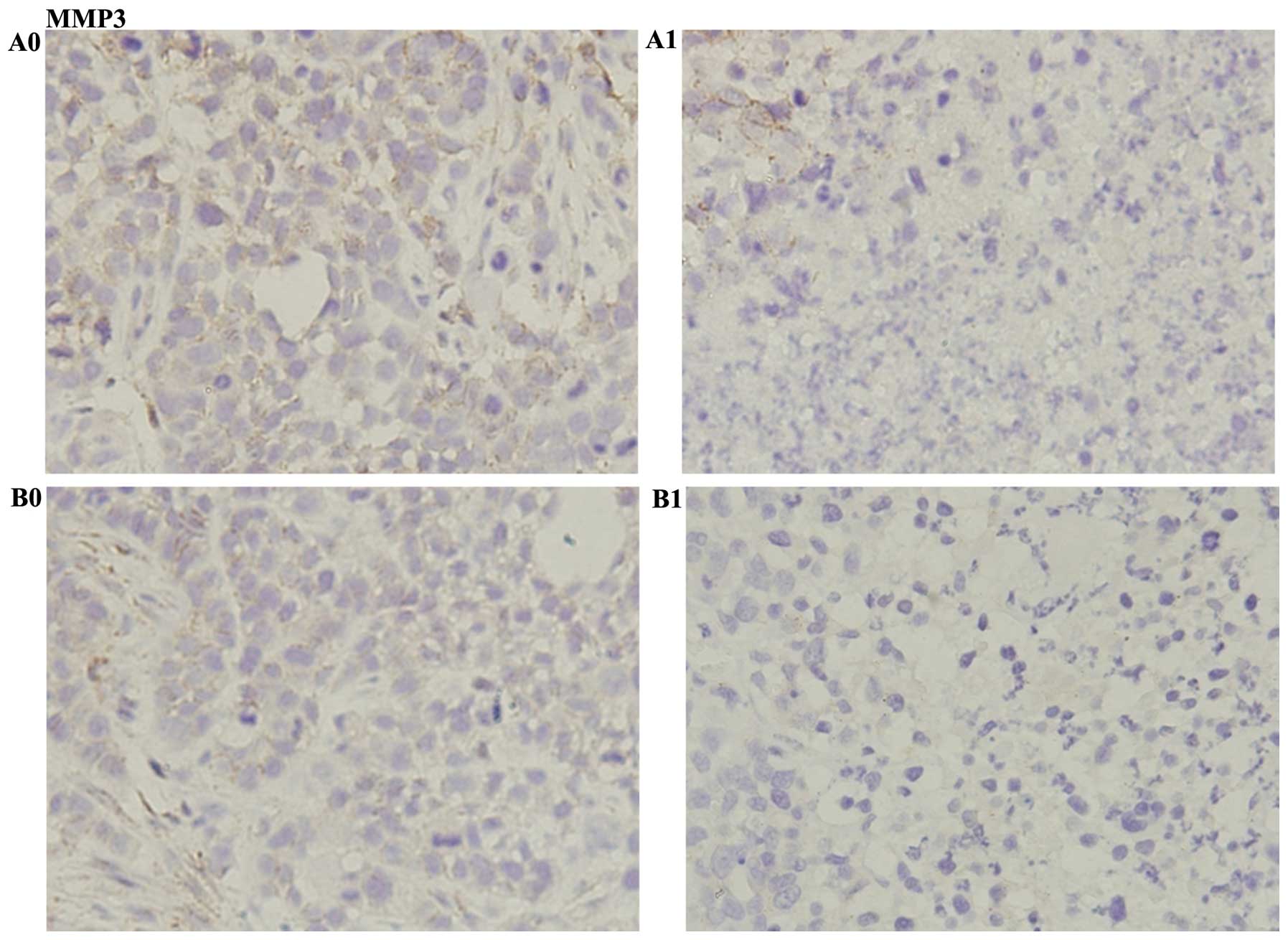
cells in HepG2 + (S)Rh2 group was greater (Fig. 8). Immunohistochemical results
indicated that the expression of GSK-3β increased, and β-catenin
and MMP3 expressions decreased in HepG2-β-catenin + (S)Rh2
group, compared with HepG2-β-catenin group. The expression of
β-catenin and MMP3 in HepG2 + (S)Rh2 group was weaker than
that of the HepG2-β-catenin + (S)Rh2 group, while GSK-3β was
not significantly different (Figs.
9Figure 10–11). ELISA kit was used to analyze the
activity of GSK-3β and it was found that the activity of GSK-3β
increased in HepG2 + (S)Rh2 and HepG2-β-catenin +
(S)Rh2 groups (Fig. 12A).
The PCR and western blotting results showed that the expression of
GSK-3β mRNA and protein increased, while the expression of
β-catenin mRNA and protein was downregulated in HepG2 and
HepG2-β-catenin cells induced by (S)Rh2. Compared with the
HepG2-β-catenin + (S)Rh2 group, the expression of β-catenin
protein in HepG2 + (S)Rh2 group was also significantly low,
and the difference between them was statistically significant.
In vivo experiment, the expression of Bax gene increased,
while the expression of cyclin D1, Bcl-2 and MMP3 genes was
downregulated in HepG2 and HepG2-β-catenin cells induced by
(S)Rh2. Compared with HepG2-β-catenin + (S)Rh2 group,
the expression of Bax gene in HepG2 + (S)Rh2 group increased
significantly, and the expression of Bcl-2, cyclin D1 and MMP3 mRNA
was also significantly low, and the difference between them was
statistically significant (p<0.01). These results supported that
activating GSK-3β in vivo could contribute to inhibiting the
tumor growth in HepG2-β-catenin + (S)Rh2 and HepG2 +
(S)Rh2 groups.
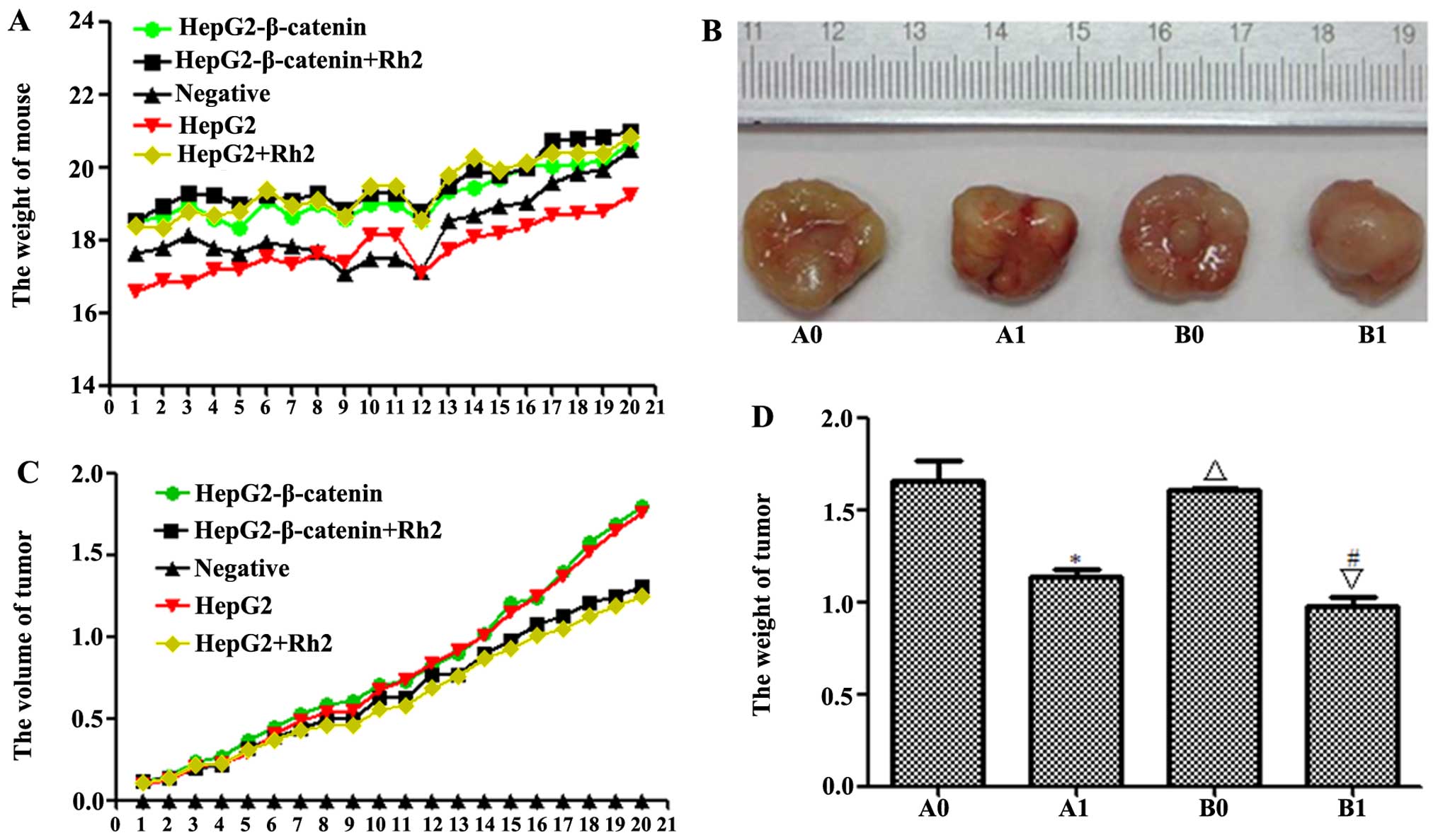 | Figure 7Effect of (S)Rh2 on the growth
of established HepG2-β-catenin and HepG2 tumors in nude mice. Mice
were randomized into five groups. Each mouse was inoculated
subcutaneously in the left flank with 5×107
HepG2-β-catenin and HepG2 cells or normal saline in a total volume
of 0.1 ml. (S)Rh2 was given to the tumor-bearing mice. There
was no statistically significant difference in changes in the body
weight of mice between treatment groups, and no signs of other
toxic effects were observed during the period. (A) Mean weight of
mice for each group is indicated. (B) Each bar represents the mean
± SEM of tumor volume of five animals per group. (C) Mean diameter
of tumor for each group is indicated. (D) Mean weight of tumor for
each group is indicated. Results are represented as the mean ± SEM
of five animals per group. *P<0.01, HepG2-β-catenin +
(S)Rh2 vs. HepG2-β-catenin group; △p<0.01,
HepG2 vs. HepG2-β-catenin; ∇p<0.01, HepG2 +
(S)Rh2 vs. HepG2; #p<0.01, HepG2 +
(S)Rh2 vs. HepG2-β-catenin + (S)Rh2. A0,
HepG2-β-catenin; A1, HepG2-β-catenin + (S)Rh2; B0, HepG2;
B1, HepG2 + (S)Rh2. (S)Rh2, 20(S)-ginsenoside
Rh2. |
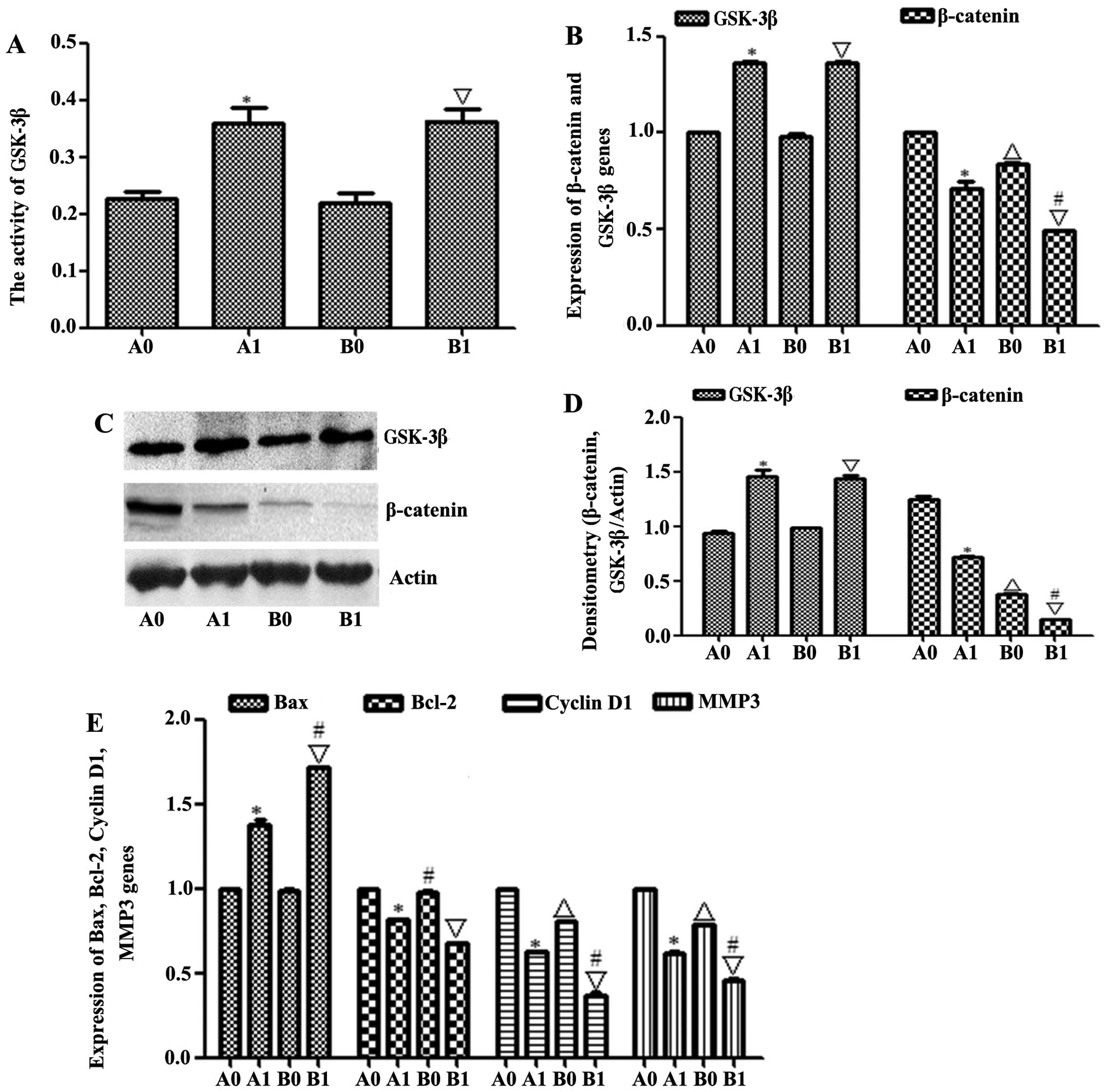 | Figure 12GSK-3β was activated and β-catenin
was degraded in a nude mouse xenograft model. (A) The activity of
GSK-3β was checked by ELISA kit. (B and C) GSK-3β and β-catenin
expression levels were determined by western blotting; β-actin
served as a protein loading control. (D) The expression of GSK-3β
and β-catenin mRNAs was measured by qRT-PCR. (E) The expressions of
Bax, Bcl-2, cyclin D1 and MMP3 mRNAs was measured by qRT-PCR.
Results shown are representative of at least three independent
experiments. *P<0.01, HepG2-β-catenin + (S)Rh2
vs. HepG2-β-catenin group; △p<0.01, HepG2 vs.
HepG2-β-catenin; ∇p<0.01, HepG2 + (S)Rh2 vs.
HepG2; #p<0.01, HepG2 + (S)Rh2 vs.
HepG2-β-catenin + (S)Rh2. A0, HepG2-β-catenin; A1,
HepG2-β-catenin + (S)Rh2; B0, HepG2; B1, HepG2 +
(S)Rh2. GSK-3β, glycogen synthase kinase-3β; (S)Rh2,
20(S)-ginsenoside Rh2. |
Discussion
Panax ginseng has been used for treatment of
disease in Chinese Traditional Medicine for thousands of years, and
it has also been employed in recent years for adjuvant therapy in
various types of cancers (25). It
has been found that (S)Rh2 could help induce apoptosis in
pancreatic cancer, hepatoma and A549 lung cancer cells (11). It was found in this experiment that
the proliferation of HepG2 cells at various concentrations of
(S)Rh2 were inhibited to some degree, compared with the
control group, and the difference between them is statistically
significant. The inhibition effect of (S)Rh2 on HepG2 liver
cancer cell growth was dose- and time-dependent, and the
IC50 of (S)Rh2 exposure on HepG2 cells for 48 and
72 h was 100 and 58.12 µmol/l, respectively (Fig. 2A). It has been found through other
studies in the laboratory that the effect of TSPG and (S)Rh2
inhibiting cell proliferation and inducing apoptosis in KG1α and
β-catenin protein significantly reduced in the nucleus, and
expression in the nucleus transferred to the cell membrane.
Clinical studies showed that overexpression of β-catenin could
resist to chemotherapy or radiotherapy and lead to poor prognosis
(18). Lentivirus carrying
β-catenin was used to infect HepG2 cells so as to establish
HepG2-β-catenin cells. (S)Rh2 inhibited HepG2-β-catenin cell
proliferation dose- and time-dependently, and the IC50
of (S)Rh2 exposure on HepG2-β-catenin cells for 48 and 72 h
are 129.2 and 83.33 µmol/l (Fig.
2B), respectively. The results showed that overexpression of
β-catenin decreased the pharmacological effects of ginsenoside
(S)Rh2 on hepatoma HepG2 cells.
According to previous reports, the constitutive
activation of Wnt/β-catenin signaling would promote cell
proliferation and tumorigenesis in tissues as well as in the colon
and the pancreas (21). In order to
explore the effect of (S)Rh2 and over-expression of
β-catenin on cell cycle and apoptosis of HepG2 hepatoma cells, FCM
was used to detect the cycle proportion in each group of cells,
including HepG2, HepG2 + (S)Rh2, HepG2-β-catenin and
HepG2-β-catenin + (S)Rh2 group. Compared with HepG2 cells,
the proportion of HepG2-β-catenin cells in G0/G1 phase
(52.86±1.46%) was significantly lower than that in HepG2 group
(61.02±1.48%). This result indicated that overexpression of
β-catenin could reduce the cycle proportion in G0/G1 phase, which
could accelerate cell proliferation (Fig. 3A). It was found that cycle
percentage of G0/G1 phase was increased in HepG2 and
HepG2-β-catenin cells induced by (S)Rh2. The result showed
that the anticancer effect of (S)Rh2 was achieved by
arresting the cell cycle in G0/G1 phase (Fig. 3A). Cyclin D1 played a critical role
in controlling the proliferation of malignant tumors, which could
transit G1 to S phase (26),
detected by PCR, ChIP and western blotting. The expression of
cyclin D1 mRNA and protein reduced in HepG2 and HepG2-β-catenin
cells induced by (S)Rh2 (Fig.
6A–C). Downregulating the cyclin D1 expression could suppress
uncontrolled proliferation of tumor cells. The expressions of
cyclin D1 mRNA and protein levels in HepG2 + (S)Rh2 group
were significantly lower than that in HepG2-β-catenin +
(S)Rh2 group, and the difference between them was
statistically significant (p<0.01). The result showed that
overexpression of β-catenin might weaken the effect of
(S)Rh2 downregulating the expression of cyclin D1 mRNA and
protein levels. FCM was used to detect apoptosis of cells. The
experimental results indicated that the rate of cell apoptosis
increased in HepG2 + (S)Rh2 and HepG2-β-catenin +
(S)Rh2 groups, among which the rate of cell apoptosis in
HepG2-β-catenin + (S)Rh2 was lower than that in HepG2 +
(S)Rh2 (Fig. 3B) group. The
above results indicated that overexpression of β-catenins might
weaken the effect of survival inhibition and reduce the rate of
apoptosis inhibition on HepG2 liver cancer cells. From analysis of
80 cases, it was found that the expression level of GSK-3β protein
in HCC was significantly lower than that of normal liver tissue and
cancerous tissue, and indicated a poor prognosis. GSK-3β expression
was also correlated with vascular invasion, TNM classification.
Therefore it could be involved in the process of HCC metastasis
(19). GSK-3β has carcinogenic
potential and is also a favorable target for anticancer therapy
(20). An ELISA assay kit was used
to detect the activity of GSK-3β in HepG2 and HepG2-β-catenin
cells, and GSK-3β was able to be activated by (S)Rh2 in a
time-dependent manner (Fig. 4A).
Next, GSK-3β inhibitor BIO was used to verify whether (S)Rh2
activated GSK-3β. The activity of GSK-3β was decreased both in
HepG2 and HepG2-β-catenin cells induced by (S)Rh2 and BIO,
significantly lower than HepG2 and HepG2-β-catenin cell group,
which proved that (S)Rh2 activated GSK-3β and the
pharmacologic action of (S)Rh2 could be antagonized by BIO
(Fig. 4B). PCR and western blotting
were used to detect the expression of GSK-3β mRNA and protein in
HepG2 and HepG2-β-catenin cells induced by (S)Rh2 for 48 h.
The expression of GSK-3β mRNA and protein increased. These results
suggested that (S)Rh2 could activate GSK-3β and enhance the
expression of GSK-3β (Fig. 5A and
B).
β-catenin both in cancer and normal tissues is an
important molecule, whose stability depends on the degradation
complex, including Axin, APC and GSK-3β, and is regulated mainly by
GSK-3β in the cells (21,27,28).
The activity of GSK-3β increased in HepG2 and HepG2-β-catenin cells
induced by (S)Rh2 for 48 h. The stability of β-catenin
depended on the regulation of GSK-3β. PCR and western blotting were
used to detect the expression of β-catenin mRNA and protein in
HepG2. As a result the expression of β-catenin mRNA and protein in
HepG2 + (S)Rh2 group significantly decreased, in contrast to
HepG2. HepG2-β-catenin cells were also detected. The results showed
that β-catenin mRNA and protein in HepG2-β-catenin + (S)Rh2
group were lower than that in HepG2-β-catenin cells, and the
β-catenin mRNA and protein reducing rate was lower than that in
HepG2 + (S)Rh2 group (Fig. 5A
and B). The results showed that overexpression of β-catenin
could weaken the effect of (S)Rh2 degrading β-catenin.
Furthermore, it was confirmed that the GSK-3β was increased by
ginsenoside (S)Rh2 so as to degrade β-catenin.
In Wnt stimulation, GSK-3β transferred to the cell
membrane and bound with dishevelled and LRP-5 receptor, thereby
preventing the GSK-3β to phosphorylate β-catenin (20,27).
As a result, the β-catenin escaped the proteasome to degradation,
which accumulated in the cytoplasm and then shifted to the nucleus
(28). When chromatin was in a
loose state, β-catenin shifting into the nucleus could combine with
TCF and HDACs/Groucho complex. After combination, N- and C-terminus
of β-catenin exposure, HAT protein CBP and p300 interacted with
β-catenin R10-C area. Next, transcription factors, transcription
complexes and RNA polymerase were recruited on β-catenin platform
to activate the downstream genes, including Bax, Bcl-2, cyclin D1
and MMP3 (22,23). PCR was used to detect the changes of
Bax, Bcl-2, cyclin D1 and MMP3 mRNA in HepG2 cells induced by
(S)Rh2. The results showed that the expression of Bax mRNA
in HepG2 + (S)Rh2 group increased, while the expressions of
Bcl-2 and MMP3 mRNA downregulated, in contrast to the HepG2 group.
Previous experimental results showed that β-catenin expression was
significantly decreased in HepG2 cells induced by (S)Rh2
(Fig. 6C). In order to further
explore the decreasing effect of β-catenin in cell nuclear on
downstream mRNAs, ChIP method was used to detect downstream mRNAs.
The results showed that the expression of Bax mRNA in HepG2 +
(S)Rh2 group increased, while the expression of cyclin D1,
Bcl-2 and MMP3 mRNA downregulated, compared with HepG2 group
(Fig. 5C). It was important that
the western blotting results agreed with ChIP results (Fig. 6A and B). The Bax, Bcl-2, cyclin D1,
MMP3 mRNA and protein in HepG2-β-catenin + (S)Rh2 and HepG2
+ (S)Rh2 groups were also examined. It was found that the
degree of Bax mRNA and protein was increased and the extent of
Bcl-2, cyclin D1, MMP3 mRNA and protein was reduced in
HepG2-β-catenin + (S)Rh2 group, which was weaker than that
of HepG2 + (S)Rh2 group, and the difference between them was
statistically significant (p<0.01) (Fig. 6A–C). The results showed that
(S)Rh2 activated GSK-3β to degrade β-catenin, reduced the
number of β-catenin shifting into the nucleus, thereby inhibiting
the expression of the cycle, proliferation and migration-related
protein, then promoted the expression of apoptosis-related
proteins, and ultimately inhibited the proliferation of HepG2 cells
and promoted their apoptosis. Re-expression of β-catenin could
weaken apoptosis in HepG2 cells induced by (S)Rh2, which was
also consistent with the results of apoptosis by FCM.
In vivo, (S)Rh2 was administered for
mice by oral gavage with 20 mg/kg, which equates to 1.6 mg/kg for
humans. In addition, the dosage used in this study was based on
several reports by other researchers. The weight of tumor in
HepG2-β-catenin and HepG2 groups induced by (S)Rh2 was
significantly reduced. Earlier experiments illustrated that the
overexpression of β-catenin accelerated the proliferation of HepG2
cells in vitro. In vivo experiments, it was found
that the weight of tumor in HepG2-β-catenin group (1.7±0.19 g) was
greater than that of HepG2 group (1.6±0.16 g), and the difference
between them was statistically significant (p<0.01). The results
indicated that overexpression of β-catenin increased the weight of
tumor. It was also observed that the weight of tumor in
HepG2-β-catenin + (S)Rh2 group (1.7±0.19 g) was greater than
that of HepG2 + (S)Rh2 group (1.0±0.13 g), and the
difference between them was statistically significant (p<0.01).
The results elucidated that overexpression of β-catenin could
weaken the pharmacological effect of (S)Rh2 on HepG2 cells
(Fig. 7A–D). Tumor sections were
paraffin-embedded. H&E staining was used to observe the
morphology of cells in the tumor tissue. It showed nucleus atypia
and accounted for a large proportion of the whole cells in HepG2
and HepG2-β-catenin cells. But the nuclear atypia in
HepG2-β-catenin group was more obvious. Condensation in nuclei and
abundant broken cells were observed in HepG2-β-catenin +
(S)Rh2 and HepG2 + (S)Rh2 groups. However,
condensation in nuclei and broken cells in HepG2 + (S)Rh2
group was more significant (Fig.
8). The results indicated that overexpression of β-catenin
could cause abnormal proliferation of the tumor, and weaken the
pharmacological effect of (S)Rh2 on HepG2 cells.
In vitro, it was found that anticancer
effect of (S)Rh2 on HepG2 cells was achieved by the
GSK-3β/β-catenin pathway. In order to further detect the role of
the signal pathway in vivo, immunohistochemistry was used to
detect the distribution of GSK-3β. Brown granules of GSK-3β in
HepG2-β-catenin and HepG2 tumor tissues mainly located in the
cytoplasm. When they were induced by (S)Rh2, the number of
brown granules increased significantly (Fig. 9). ELISA results also showed that the
activity of GSK-3β significantly increased in HepG2-β-catenin and
HepG2 tumor tissues induced by (S)Rh2. The results showed
that (S)Rh2 also activated GSK-3β in tumor tissues (Fig. 12A). In vitro, it was found
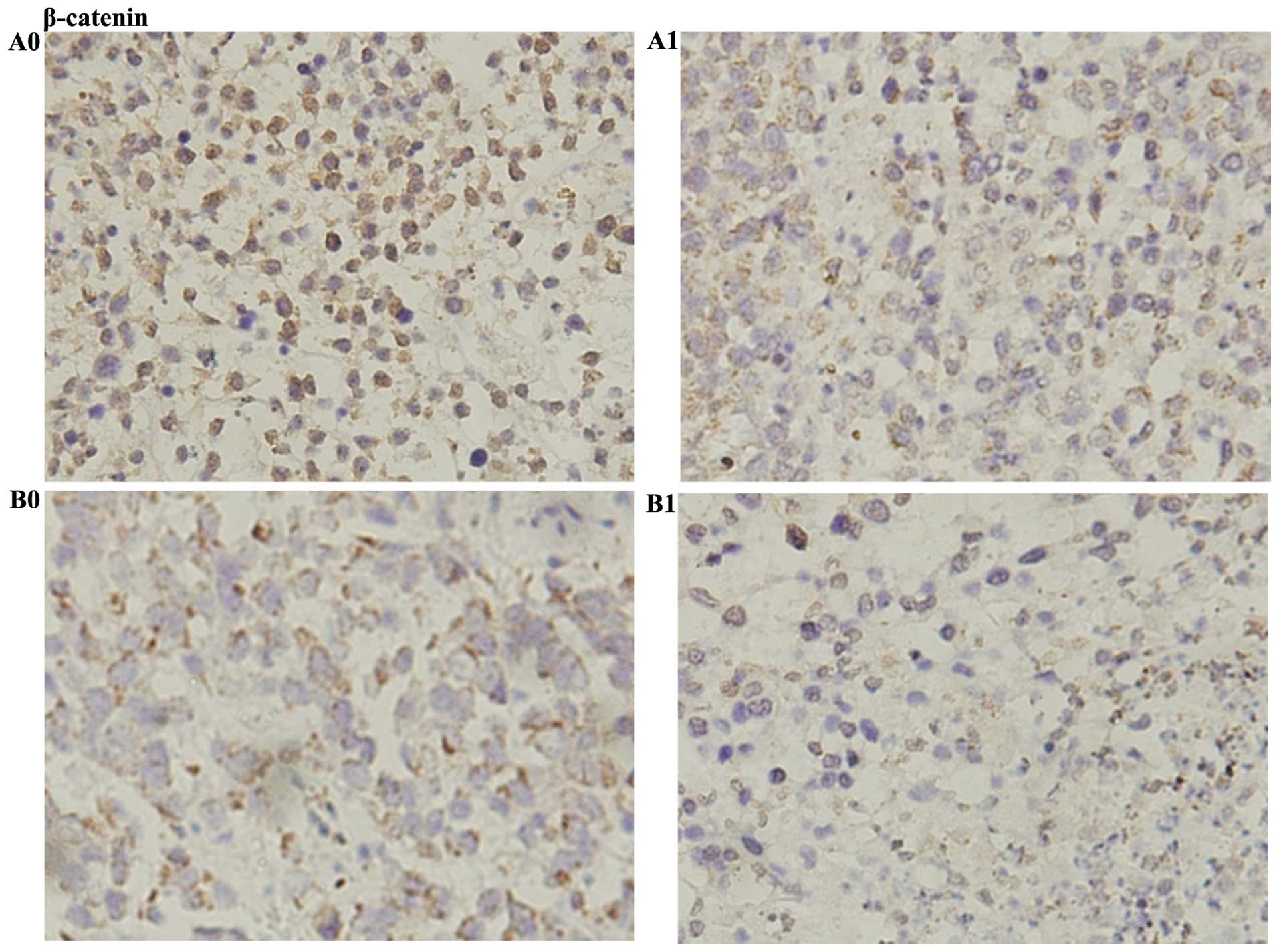
that stability of β-catenin depended on the regulation of GSK-3β.
Immunohistochemistry was used to detect the position of β-catenin,
and it was found that brown granules of β-catenin stored in
cytoplasm and nucleus in HepG2-β-catenin and HepG2 tumor tissues,
but brown granules of β-catenin in the HepG2-β-catenin group was
more abundant than that in HepG2 group. When they were induced by
(S)Rh2, the volume of β-catenin brown granules in the
cytoplasm and the nucleus was reduced, which further confirmed that
(S)Rh2 degraded β-catenin through activating GSK-3β in
vivo (Fig. 11).
The effect of ginsenoside (S)Rh2 on cancer
was evident, but its ability to inhibit tumor migration had not
been previously elucidated. β-catenin is also an adhesion molecule,
but there has been only a handful of studies reporting its role in
metastasis. MMP3 is a significant proteolytic enzyme to degrade the
extracellular matrix, which degraded the main component of the
extracellular matrix and basement membrane collagen IV to
participate in the process of tumor invasion and metastasis,
therefore it contributed to metastasis (29–31).
In order to investigate this, western blotting and
immunohistochemical staining was used to check the β-catenin and
MMP3. The results indicated that (S)Rh2 reduced the
expression levels of β-catenin, MMP3 mRNA and protein (Fig. 12B–E). The results also suggested
that (S)Rh2 could inhibit tumor metastasis.
In general, (S)Rh2 suppressed proliferation,
promoted apoptosis and inhibited metastasis of HepG2, decreased the
weight of tumors by downregulating β-catenin through activating
GSK-3β, and the pharmacological effect of (S)Rh2 on HepG2
cells might be weakened by overexpression of β-catenin.
Acknowledgments
We would like to express our great gratitude to the
National Natural Science Foundation of China for grant support for
this research (nos. KJ130312 and 81171929). We also wish to thank
the College of Life Sciences for its technical support.
References
|
1
|
Tsai WL, Lai KH, Liang HL, Hsu PI, Chan
HH, Chen WC, Yu HC, Tsay FW, Wang HM, Tsai HC, et al: Hepatic
arterial infusion chemotherapy for patients with huge unresectable
hepatocellular carcinoma. PLoS One. 9:e927842014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Page AJ, Cosgrove DC, Philosophe B and
Pawlik TM: Hepatocellular carcinoma: Diagnosis, management, and
prognosis. Surg Oncol Clin N Am. 23:289–311. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Scaggiante B, Kazemi M, Pozzato G, Dapas
B, Farra R, Grassi M, Zanconati F and Grassi G: Novel
hepatocellular carcinoma molecules with prognostic and therapeutic
potentials. World J Gastroenterol. 20:1268–1288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tanase A-M, Dumitrascu T, Dima S, Grigorie
R, Marchio A, Pineau P and Popescu I: Influence of hepatitis
viruses on clinicopathological profiles and long-term outcome in
patients undergoing surgery for hepatocellular carcinoma.
Hepatobiliary Pancreat Dis Int. 13:162–172. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ji Z, Wang T, Shao Z, Huang D, Wang A, Guo
Z, Long Y, Zhang L, Su H, Zhang Q, et al: A population-based study
examining hepatitis B virus infection and immunization rates in
Northwest China. PLoS One. 9:e974742014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu L-B, Wang J, Liu C, Pang HW, Chen YJ,
Ou QJ and Chen JS: Staging systems for predicting survival of
patients with hepatocellular carcinoma after surgery. World J
Gastroenterol. 16:5257–5262. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Osaki A, Suda T, Kamimura K, Tsuchiya A,
Tamura Y, Takamura M, Igarashi M, Kawai H, Yamagiwa S and Aoyagi Y:
A safe and effective dose of cisplatin in hepatic arterial infusion
chemotherapy for hepatocellular carcinoma. Cancer Med. 2:86–98.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ker CG, Chen JS, Kuo KK, Chuang SC, Wang
SJ, Chang WC, Lee KT, Chen HY and Juan CC: Liver surgery for
hepatocellular carcinoma: Laparoscopic versus open approach. Int J
Hepatol. 2011:5967922011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miyahara K, Nouso K and Yamamoto K:
Chemotherapy for advanced hepatocellular carcinoma in the sorafenib
age. World J Gastroenterol. 20:4151–4159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song SB, Tung NH, Quang TH, Ngan NT, Kim
KE and Kim YH: Inhibition of TNF-α-mediated NF-κB transcriptional
activity in HepG2 cells by dammarane-type saponins from Panax
ginseng leaves. J Ginseng Res. 36:146–152. 2012. View Article : Google Scholar
|
|
11
|
Guo XX, Guo Q, Li Y, Lee SK, Wei XN and
Jin YH: Ginsenoside Rh2 induces human hepatoma cell apoptosisvia
bax/bak triggered cytochrome C release and caspase-9/caspase-8
activation. Int J Mol Sci. 13:15523–15535. 2012. View Article : Google Scholar
|
|
12
|
Wang Z, Zheng Q, Liu K, Li G and Zheng R:
Ginsenoside Rh(2) enhances antitumour activity and decreases
genotoxic effect of cyclophosphamide. Basic Clin Pharmacol Toxicol.
98:411–415. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ho YL, Li KC, Chao W, Chang YS and Huang
GJ: Korean red ginseng suppresses metastasis of human hepatoma
SK-Hep1 cells by inhibiting matrix metalloproteinase-2/-9 and
urokinase plasminogen activator. Evid Based Complement Alternat
Med. 2012:9658462012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chung KS, Cho SH, Shin JS, Kim DH, Choi
JH, Choi SY, Rhee YK, Hong HD and Lee KT: Ginsenoside Rh2 induces
cell cycle arrest and differentiation in human leukemia cells by
upregulating TGF-β expression. Carcinogenesis. 34:331–340. 2013.
View Article : Google Scholar
|
|
15
|
Yan XJ, Gong LH, Zheng FY, Cheng KJ, Chen
ZS and Shi Z: Triterpenoids as reversal agents for anticancer drug
resistance treatment. Drug Discov Today. 19:482–488. 2014.
View Article : Google Scholar
|
|
16
|
Li S, Gao Y, Ma W, Guo W, Zhou G, Cheng T
and Liu Y: EGFR signaling-dependent inhibition of glioblastoma
growth by ginsenoside Rh2. Tumour Biol. 35:5593–5598. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park EK, Lee EJ, Lee SH, Koo KH, Sung JY,
Hwang EH, Park JH, Kim CW, Jeong KC, Park BK, et al: Induction of
apoptosis by the ginsenoside Rh2 by internalization of lipid rafts
and caveolae and inactivation of Akt. Br J Pharmacol.
160:1212–1223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maurer U, Preiss F, Brauns-Schubert P,
Schlicher L and Charvet C: GSK-3 - at the crossroads of cell death
and survival. J Cell Sci. 127:1369–1378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang JS, Herreros-Villanueva M, Koenig A,
Deng Z, de Narvajas AA, Gomez TS, Meng X, Bujanda L, Ellenrieder V,
Li XK, et al: Differential activity of GSK-3 isoforms regulates
NF-κB and TRAIL- or TNFα induced apoptosis in pancreatic cancer
cells. Cell Death Dis. 5:e11422014. View Article : Google Scholar
|
|
20
|
Gao Y, Liu Z, Zhang X, He J, Pan Y, Hao F,
Xie L, Li Q, Qiu X and Wang E: Inhibition of cytoplasmic GSK-3β
increases cisplatin resistance through activation of Wnt/β-catenin
signaling in A549/DDP cells. Cancer Lett. 336:231–239. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee H, Bae S, Kim YS and Yoon Y:
WNT/β-catenin pathway mediates the anti-adipogenic effect of
platycodin D, a natural compound found in Platycodon grandiflorum.
Life Sci. 89:388–394. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thompson MD and Monga SP: WNT/beta-catenin
signaling in liver health and disease. Hepatology. 45:1298–1305.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mosimann C, Hausmann G and Basler K:
Beta-catenin hits chromatin: Regulation of Wnt target gene
activation. Nat Rev Mol Cell Biol. 10:276–286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ying Y and Tao Q: Epigenetic disruption of
the WNT/beta-catenin signaling pathway in human cancers.
Epigenetics. 4:307–312. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen L, Dai J, Wang Z, Zhang H, Huang Y
and Zhao Y: Ginseng total saponins reverse corticosterone-induced
changes in depression-like behavior and hippocampal
plasticity-related proteins by interfering with GSK-3 β-CREB
signaling pathway. Evid Based Complement Alternat Med.
2014:5067352014. View Article : Google Scholar
|
|
26
|
Fu M, Wang C, Rao M, Wu X, Bouras T, Zhang
X, Li Z, Jiao X, Yang J, Li A, et al: Cyclin D1 represses p300
transactivation through a cyclin-dependent kinase-independent
mechanism. J Biol Chem. 280:29728–29742. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dahmani R, Just PA and Perret C: The
Wnt/β-catenin pathway as a therapeutic target in human
hepatocellular carcinoma. Clin Res Hepatol Gastroenterol.
35:709–713. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Debeb BG, Lacerda L, Xu W, Larson R,
Solley T, Atkinson R, Sulman EP, Ueno NT, Krishnamurthy S, Reuben
JM, et al: Histone deacetylase inhibitors stimulate
dedifferentiation of human breast cancer cells through
WNT/β-catenin signaling. Stem Cells. 30:2366–2377. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Correia AL, Mori H, Chen EI, Schmitt FC
and Bissell MJ: The hemopexin domain of MMP3 is responsible for
mammary epithelial invasion and morphogenesis through extracellular
interaction with HSP90β. Genes Dev. 27:805–817. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang X, Li F, Fan C, Wang C and Ruan H:
Effects and relationship of ERK1 and ERK2 in interleukin-1β-induced
alterations in MMP3, MMP13, type II collagen and aggrecan
expression in human chondrocytes. Int J Mol Med. 27:583–589.
2011.PubMed/NCBI
|
|
31
|
Konstantinopoulos PA, Karamouzis MV,
Papatsoris AG and Papavassiliou AG: Matrix metalloproteinase
inhibitors as anticancer agents. Int J Biochem Cell Biol.
40:1156–1168. 2008. View Article : Google Scholar : PubMed/NCBI
|