Introduction
Extracellular sulfatases, especially heparan
endosulfatases (Sulfs), play important roles in cancer progression
by modifying the sulfate patterns of heparan sulfate proteoglycans
(HSPGs) located on the surface of most animal cells (1–3). HSPGs
can be released into the extracellular matrix and can also be
detected in serum. HSPGs carry out many structural and signaling
functions through binding to protein ligands (4,5). The
Sulf family includes two structurally similar endogenous sulfatases
(Sulf1 and Sulf2) with 64% homology in highly conserved
heparin-binding domains, but with different functions (2–4).
Sulfatase 2 (Sulf2) is an extracellular endoglucosamine-6-sulfatase
and considered as a bona fide cancer-causing agent in multiple
types of cancer (6,7). Sulf2 is overexpressed in many tumor
cells and was shown to promote tumorigenesis in many human cancers
such as hepatocellular (8),
pancreatic (9), ovarian (10), breast (6,10), and
non-small cell lung carcinoma (11). Sulf2 also increased the activities
of growth factors such as vascular endothelial growth factor (VEGF)
and fibroblast growth factor-1 (FGF-1), and certain chemokines such
as stromal cell-derived factor-1 (SDF-1) and secondary
lymphoid-tissue chemokine (SLC), stimulating the biological
functions of endothelial cells to promote angiogenesis (5,12).
Although Sulf2 was confirmed to facilitate angiogenesis, the effect
of Sulf2 on lymphangiogenesis in tumors is still unknown.
Lymphangiogenesis, which is the formation of new
lymphatic vessels, is a common process in normal tissue
development, inflammation, wound healing and lymphatic edema
(13,14). Recently, more and more research has
found lymphangiogenesis to play an important role in tumor
progression and metastasis (15,16).
New lymphatic vessels are composed of one single layer of lymphatic
endothelial cells. The basement membranes of new lymphatic vessels
are incomplete, and the endothelial cells do not connect tightly.
These factors allow tumor cells to easily invade new lymphatic
vessels and metastasize to regional lymph nodes or distant organs
(15–18). In recent years more research
oncologists are becoming interested in the mechanisms of
tumor-induced lymphangiogenesis in various tumors (19,20).
Breast cancer is one of the most common types of cancer among women
worldwide (21,22). Over 50% of early-stage breast cancer
patients have local lymph node metastasis (18). Moreover, regional lymph node
metastasis in breast cancer is also one of the main factors that
leads to breast cancer metastasis and poor prognosis (23,24).
Tumor size and regional lymph node metastasis are used as
biological indicators for breast cancer classification and
selection markers for treatment strategies. Vascular endothelial
growth factor-C (VEGF-C) and VEGF-D could combine with vascular
endothelial growth factor receptor-3 (VEGFR-3) to induce
lymphangiogenesis (20,25,26).
Our previous studies also suggested that breast cancer patients
with high VEGF-D expression would have more regional lymph node
metastasis, poor disease-free survival (DFS) and poor overall
survival (OS) (20,25). Karpanen et al (24) demonstrated that when
VEGF-D-overexpressing cells were implanted into transgenic mice,
tumor-associated lymphangiogenesis was induced in several
orthotopic mouse models.
Previously, we demonstrated that VEGF-D/FIGF, a
member of the VEGF family, was upregulated by Sulf2 (6). In this study, we hypothesized that
Sulf2 facilitates lymphangiogenesis in breast cancer by regulating
VEGF-D. To evaluate the functions of Sulf2 in lymphangiogenesis in
breast cancer, we examined the proliferation, apoptosis, cell
cycle, mobility and tube-like structure formation of LECs in
vitro, as well as lymphangiogenesis in mouse ears and
xenografts in vivo. The expression of related signaling
pathway genes was also screened and verified in LECs.
Materials and methods
Cell lines
Human breast cancer cell lines (MCF-7, MDA-MB-231)
were purchased from the The Cell Bank of the Chinese Academy of
Sciences (Shanghai, China). HEK293T cells used for lentivirus
packaging were stocked in our own laboratory. All cells were
cultured in Dulbecco's modified Eagle's medium (DMEM; HyClone
Laboratories, Inc., Logan, UT, USA) with 10% fetal bovine serum
(FBS) and penicillin-streptomycin (all from Gibco, Grand Island,
NY, USA). LECs were cultured in Endothelial Cell Growth Medium
(ECGM) (both from PromoCell, Heidelberg, Germany). All cells were
cultured at 37°C in 5% CO2.
Conditioned medium (CM)
collection
CM was collected from the supernatant of MCF-7
cells. MCF-7 cells release high levels of Sulf2 protein in the
supernatant which was confirmed in our previous study (6). The MCF-7 cells were cultured in DMEM
until 80% confluence and were subsequently cultured in OptiMEM
(HyClone Laboratories, Inc.) for another 72 h. The supernatant was
collected and concentrated using Amicon Ultra filters 30 D
(Millipore, Billerica, MA, USA) and then was kept in 50 mM HEPES
buffer (pH 8.0; Biochrom GmbH, Berlin, Germany) for further
study.
Flag-Sulf2 vector construct
The signal peptide sequence of Sulf2 was removed.
The new peptide sequences of signal Flag and Ig-k were added with
three rounds of PCR using three forward primers (Table I) and reverse primer
5′-CGGGATCCTTAACCTTCCCAGCCTTCCC-3′. The PCR conditions were 95°C
for 5 min, followed by 35 cycles of amplification, 95°C for 15 sec,
55°C for 15 sec and 72°C for 1 min. The PCR sequence structures of
flag-Sulf2 were signal peptide, signal peptide cleavage site, Flag,
the linker portion of GSG and the Sulf2 cDNA sequence (Table II). The amplified fragment was
cloned into the pCDH (System Biosciences, Mountain View, CA, USA)
to form the pCDH-Flag-Sulf2 vector construct. The sequences of the
positive clone were identified using enzyme digestion and gene
sequencing detection (Shanghai Meiji, Shanghai, China).
 | Table I.Upstream primers of flag-Sulf2. |
Table I.
Upstream primers of flag-Sulf2.
| No. | Primer sequences |
|---|
| Flag-Sulf2 F1 |
5-GACGATTACAAGGATGACGACGATAAGGGTTCTGGCTTCCTGTCGCACCACCGC-3 |
| Flag-Sulf2 F2 |
5-TGGGTACTGCTGCTCTGGGTTCCAGGTTCCACTGGTGACGATTACAAGGATGACGACG-3 |
| Flag-Sulf2
F3(Nhe I) |
5-CTAGCTAGCATGGAGACAGACACACTCCTGCTATGGGTACTGCTGCTCTGG-3 |
| Table II.DNA sequences and amino acid sequences
of flag-Sulf2. |
Table II.
DNA sequences and amino acid sequences
of flag-Sulf2.
| Genes | DNA sequence | Amino acid
sequence |
|---|
| Signal peptide |
ATGGAGACAGACACACTCCTGCTATGGGTACTGCTGCTCTGGGTTCCAGGTTCCACTGGT |
METDTLLLWVLLLWVPGST |
| Cleavage site | GAC | D |
| Flag |
GATTACAAGGATGACGACGATAAG | DYKDDDDK |
| Linker | GGTTCTGGC | GSG |
| Sulf2 |
TTCCTGTCGCACCACCGCCTGAAA…(Sulf2 full
length…) | FLSHHRLK…(Sulf2
full length amino acid chain…) |
rSulf2 combination and
purification
The pCDH-Flag-Sulf2 lentivirus was packaged in
HEK293F cells using Lipofectamine 2000 (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturers
instructions. The supernatant from the lentivirus-transfected
HEK293F cells was collected and incubated with anti-Flag/M2 agarose
beads (Sigma-Aldrich, St. Louis, MO, USA) at 4°C overnight. The
beads were washed three times with washing buffer. The bound
proteins were eluted with 0.1 mg/ml Flag peptide (DYKDDDDK). The
eluate was concentrated though Amicon Ultra filters 30 D and kept
in 50 mM HEPES buffer (pH 8.0).
qRT-PCR analysis
Total RNA was isolated from cells using TRIzol
reagent and reverse transcribed into cDNA using SuperScript III
Reverse Transcriptase (both from Invitrogen Madison, WI, USA). The
mRNA level was determined using the 7900HT qRT-PCR system (Applied
Biosystems, Foster City, CA, USA) using SYBR® Green
Real-time PCR Master Mix (Takara, Shiga, Japan). Primers for
qRT-PCR are listed in Table III.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an
internal control. Relative mRNA levels were calculated using the
ΔΔCt method.
| Table III.Real-time PCR primers. |
Table III.
Real-time PCR primers.
| Gene | Primer
sequences | Length (bp) |
|---|
| GAPDH (HUMAN) | F
5′-GGGAAACTGTGGCGTGAT-3′ | 299 |
|
| R
5′-GAGTGGGTGTCGCTGTTGA-3 |
|
| PLA2G1B | F
5′-TGTGGCAGTTCCGCAAAAT-3′ | 77 |
|
| R
5′-GCAGCCGTAGTTGTTGTATTCC-3 |
|
| PLA2G5 | F
5′-AACCCCAGAGATGAAAGGC-3′ | 134 |
|
| R
5′-CGTAGTTTGTCAGGGCGTTC-3 |
|
| PLA2G6 | F
5′-CCACATCATCCCTTCTCCCT-3′ | 181 |
|
| R
5′-CTTTCACTCCTCCTCCATCCA-3 |
|
| PLA2G2D | F
5′-GGCCTAGAGTGGCAAATGG-3′ | 104 |
|
| R
5′-GGGAAAACAGGGGAAACAGA-3 |
|
| AKT1 | F
5′-GCCCTGCTACCTGTTCTTGG-3′ | 266 |
|
| R
5′-AAGCAAATGGCAAAGTGTGAG-3 |
|
| PIK3R1 | F
5′-TTGGAAGCAGCAACCGAAAC-3′ | 123 |
|
| R
5′-CTTCGCCGTCCACCACTACA-3 |
|
| PIK3R3 | F
5′-TGGTTCAGCACAACGACTCC-3′ | 99 |
|
| R
5′-CACCTCTCTTCCCACTTCCT-3 |
|
Western blot (WB) analysis
Cells were harvested in the presence of a protease
inhibitor cocktail (Sigma-Aldrich) in RIPA lysis buffer (Beyotime,
Jiangsu, China). Equal amounts of proteins from the cells were
resolved on SDS-PAGE and then transferred onto PVDF membranes as
previously described (6). The
membranes were separately probed with rabbit anti-FIGF (1:1,000,
PAB4879; Abnova, Atlanta, GA, USA), rabbit anti-AKT1 (1:1,000,
ab32505), rabbit anti-pAKT1 (s473) (1:1,000, ab66138; both from
Abcam, Cambridge MA, USA), rabbit anti-mouse LYVE-1 (1:1,000,
ab36993; AngioΒio, San Diego, CA, USA), rabbit anti-GAPDH (1:500;
Sigma-Aldrich) for 1 h. Subsequently, the membranes were washed
with TBST, and then incubated with goat anti-rabbit HRP (1:500;
Sigma-Aldrich) for 1 h. Bound antibody chemiluminescence was
detected using chemiluminescence kits (Thermo Fisher Scientific,
Darmstadt, Germany). The optical density was determined using a
scanning densitometer and analyzed using Quantity One software
(Bio-Rad, Hercules, CA, USA).
Proliferation assay
LECs were divided into four groups (control-1,
rSulf2, VEGF-D, rSulf2+VEGF-D). Control-1 was cultured with only
DMEM media. The other groups were separately cultured with rSulf2,
VEGF-D and rSulf2+VEGF-D. The final concentration of rSulf2 and
VEGF-D in each group was 50 ng/ml. The proliferation of LECs was
assessed by the MTT method. Cells were dissociated from cell flasks
by trypsin (Sigma-Aldrich) digestion and were seeded into 96-well
plates (1×105 cells/ml). The proliferation of LECs in
the four groups was detected at different time-points (0, 12, 24,
36 and 48 h). All cells were incubated at 37°C for 4 h followed by
the addition of 10 µl MTT (5 mg/ml) and 100 µl DMSO. The absorbance
value of each well was measured using a microplate reader (Bio-Tek,
Winooski, VT, USA) at a wavelength of 570 nm.
Apoptosis assay
The aforementioned four groups were used. Apoptosis
was determined by dual staining using Annexin V/FITC and propidium
iodide (Invitrogen). Briefly, the log phase of LECs was seeded into
24-well cell culture plates (1×105 cells/well).
Subsequently, LECs were treated with 10 µg/ml cisplatin (Qilu
Pharmaceutical Co., Shanghai, China) for 24 h and then dissociated
from the wells with 0.25% trypsin, spun at 1,500 rpm for 5 min,
resuspended in Annexin V binding buffer, stained with 1 µl Annexin
V/FITC for 15 min and 1 µl propidium iodide for 1 min. The cells
were analyzed using the FACSCalibur System (BD Biosciences, San
Jose, CA, USA). The relative proportion of Annexin V-positive
cells, representing apoptotic cells, was determined using FlowJo
software (FlowJo, LLC, Ashland, OR, USA).
Cell cycle assay
The aforementioned four groups were used. The cell
cycle distribution of LECs was determined by propidium iodide
staining and flow cytometric analysis. After LECs were treated with
10 µg/ml cisplatin for 24 h, the cells were dissociated from the
wells with 0.25% trypsin. Subsequently, they were fixed in 70%
ethanol overnight at −20°C and incubated in RNase A at 37°C for 30
min. Propidium iodide was then added and the cells were incubated
in a dark room for 30 min. Flow cytometry was used to detect the
cell cycle distribution. The proliferation index (PI) was
calculated using the formula PI = (S + G2)/(S + G1 + G2) ×
100%.
LEC mobility assay
The aforementioned four groups were used. The
mobility of LECs was determined in 12-well Boyden chamber plates
and polycarbonate membrane filter inserts (CoStar Group, Inc.,
Washington, DC, USA) with 8-µm pores. For the cell mobility assay,
the interior of the Transwell insert was coated with Matrigel (BD
Pharmingen, San Diego, CA, USA), which mimics the basement
membrane. In all, 1×105 cells were seeded into the upper
chamber. The cell suspension was also seeded onto the membrane in
the upper chamber and the lower chamber was filled with 1 ml medium
with 10% FBS. After 48 h, the non-migrating cells on the surface of
the upper chamber were removed with cotton swabs. The migrating
cells at the bottom of the membrane were fixed in formaldehyde for
1 min and then stained with crystal violet. The stained membranes
were cut and placed onto a glass slide and the number of invading
cells at the bottom surface of the membrane was counted three times
under a bright-field light microscope.
Lymphatic tube-like structure
formation assay
The aforementioned four groups were used. The
lymphangiogenic capacities of LECs on Matrigel were determined
according to the manufacturers instructions. The Matrigel was
melted in 4°C and was diluted to half its concentration by media
and then was added into 24-well plates for cooling down. The
24-well plates were placed in an incubator for 30 min to solidify
the glue. LECs (5×105) were digested and added into each
well before this solidification process. The numbers of new
lymphatic tubes were detected using an inverted phase contrast
microscope (AMG, Bovenden, Germany) after 24 h of culture.
Lymphangiogenesis in mouse ears
Four-week-old, BALB/c-nu mice (weight, 15 g) were
purchased from the Shanghai Experimental Animal Center of the
Chinese Academy of Sciences (Shanghai, China). The mice were
divided into three groups (control-2, CM-nu, rSulf2-nu). A total of
0.1 ml 0.9% saline, CM (50 mM) and rSulf2 (50 mM) were separately
injected into the root of the mouse ears every day for six weeks.
Excised mouse ears were fixed in formalin buffer and embedded in
paraffin for advanced testing. All experimental protocols followed
the instructions of the Chinese Council on Animal Care and were
approved by the Animal Experimental Ethical Inspection of Shanghai
Ninth People's Hospital affiliated to Shanghai Jiaotong University,
School of Medicine [permit (no. 20015) 25].
Lymphangiogenesis in breast cancer
xenografts
Six-week-old, 18-g female Nod/scid mice were
purchased from Shanghai Experimental Animal Center of the Chinese
Academy of Sciences. MDA-MB-231 breast cancer cells were detached
with 0.25% trypsin and resuspended in HBSS/Matrigel (1:1 volume) to
107 cells/ml. Xenografts were generated by injecting 0.2
ml cell suspension into the area of the mammary fat pad. Mice were
divided into three groups (control-3, CM-scid, rSulf2-scid). A
total of 0.1 ml 0.9% saline, CM (50 mM) and rSulf2 (50 mM) were
separately injected into xenografts every day until 6 weeks.
Excised xenografts were fixed in formalin buffer and embedded in
paraffin for advanced testing. All experimental protocols followed
the instructions of the Chinese Council on Animal Care and were
approved by the Animal Experimental Ethical Inspection of Shanghai
Ninth People's Hospital affiliated to Shanghai Jiaotong University,
School of Medicine [permit (no. 20015) 25].
Immunohistochemistry (IHC)
Five-micron-thick sections of the paraffin-embedded
tissues were deparaffinized in xylenes and rehydrated through a
graded alcohol series. Heat-induced epitope retrieval (HIER) was
performed by immersion of the tissue sections in 8 mM EDTA buffer
(pH 8.0) for 20 min at 98°C. IHC staining was performed using a
horseradish peroxidase-labeled polymer K4001 (Dako, Zagreb,
Croatia) according to the manufacturers instructions. Briefly, the
slides were incubated with 3% hydrogen peroxide and 1% bovine serum
albumin (BSA; Sigma-Aldrich) for 10 min each. To visualize
lymphatic vessels, the sections were exposed to the primary
antibody, rabbit anti-mouse LYVE-1 which was diluted as recommended
in 3% BSA, for 1 h at room temperature. The slides were then
incubated with goat anti-rabbit HRP for 30 min followed by
incubation with the DAB chromogen (Dako) for 5 min. Finally, the
slides were counterstained with hematoxylin (Thermo Fisher
Scientific, Waltham, MA, USA), blued in 1% ammonium hydroxide,
dehydrated, and mounted with Acrymount. Consecutive sections where
the primary antibody was omitted were used as negative controls.
The washing buffer used was 1X TBS with 0.05% Tween-20 (Thermo
Fisher Scientific).
Statistical analysis
Data are presented as the mean ± standard deviation
(SD) of at least three independent experiments with three or more
replicates. Continuous data were analyzed using a two-tailed
Student's t-test. P<0.05 was considered to indicate a
significant difference.
Results
Sulf2 with VEGF-D promotes LEC
proliferation
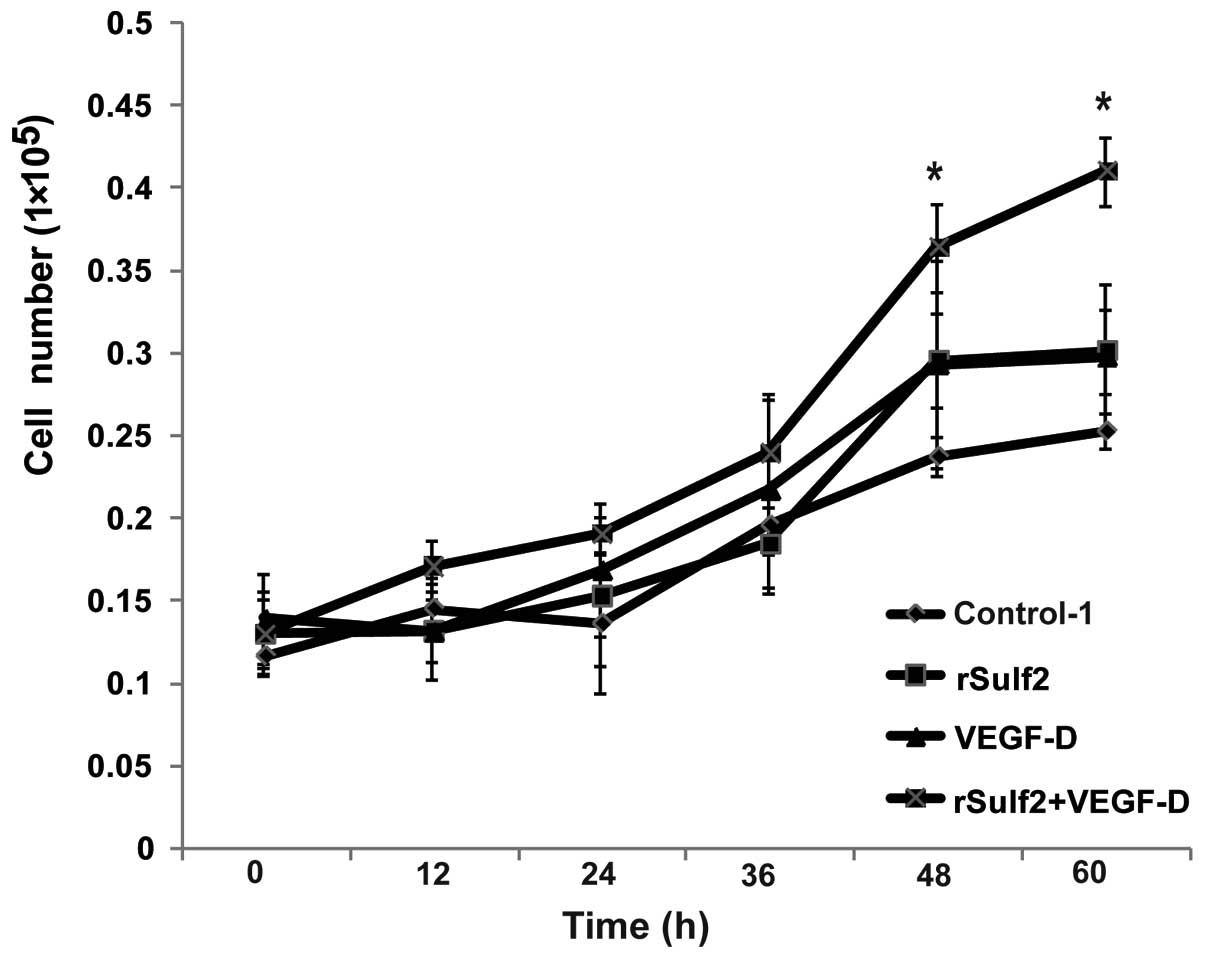
To evaluate the role of Sulf2 in LEC proliferation,
an MTT assay was used to detect the proliferation of LECs at
different time-points. Cell growth curves were drawn based on the
absorbance value of live cells at different time-points. Compared
with the control-1, the rSulf2 and VEGF-D groups showed higher cell
growth after 36 h, but the difference was not significant. However,
the group treated with rSulf2+VEGF-D showed a significant
difference in the absorbance of live cells at 48 and 60 h
(0.36±0.03 vs. 0.24±0.01, 0.41±0.02 vs. 0.25±0.01 respectively,
P<0.05, Fig. 1). The results
indicated that Sulf2 or VEGF-D could enhance LEC proliferation, but
their effects were not significant. Furthermore, LECs treated with
rSulf2 and VEGF-D showed a significantly higher growth rate than
the cells treated with control-1. Collectively, these data
indicated that Sulf2 could promote breast cancer proliferation
through the activation of VEGF-D.
Sulf2 inhibits cisplatin-induced LEC
apoptosis
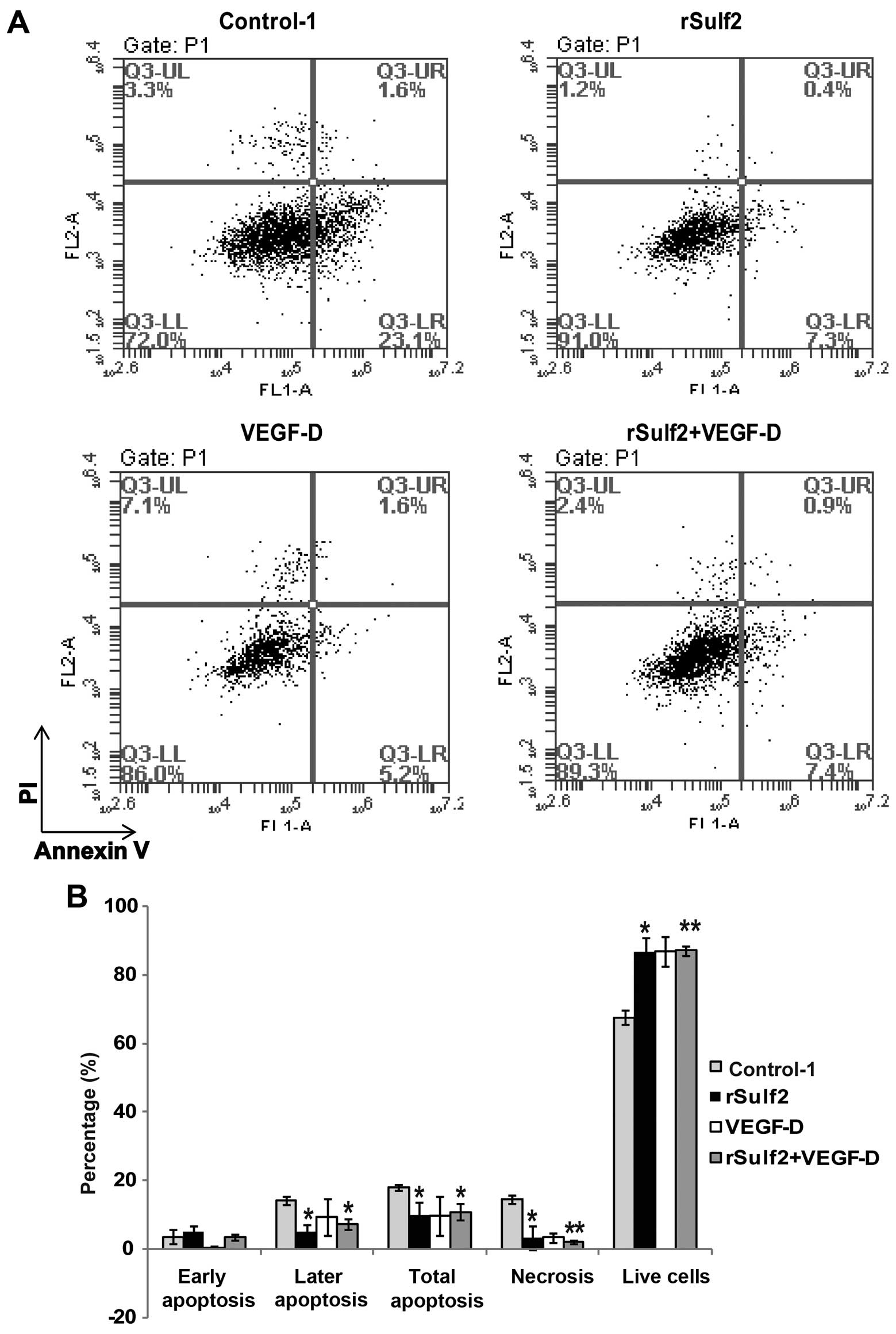
To evaluate the role of Sulf2 in LEC apoptosis, we
measured the cisplatin-induced apoptosis and necrosis of LECs by
flow cytometry. Compared with the control-1, treatment with rSulf2
resulted in a significant increase in the percentage of live cells
(86.98±3.84 vs. 67.60±2.12, P<0.05) and a significant decrease
in total apoptosis (9.75±4.03 vs. 17.95±0.78, P<0.05). A closer
look at the different stages in apoptosis revealed that the most
significant difference occured in the late stage (4.95±2.19 vs.
14.3±1.27, P<0.05) instead of the early stage (4.80±1.83 vs.
3.65±2.05, P>0.05). Treatment with VEGF-D caused a significant
decrease in the percentage of dead cells (3.35±1.48 vs. 14.5±1.27,
P<0.05), but had no significant effects on the percentage of
live cells (86.75±4.31 vs. 67.6±2.12, P>0.05) and total
apoptosis (9.85±5.72 vs. 17.95±0.78, P>0.05). Treatment with
rSulf2+VEGF-D resulted in a significant increase in the percentage
of live cells (87.11±1.27 vs. 67.60±2.12, P<0.01) and a more
significant decrease in total apoptosis (10.81±2.40 vs. 17.95±0.78,
P<0.05) and percentage of dead cells (2.05±0.64 vs. 14.50±1.27,
P<0.01), especially in the late stage (7.31±1.56 vs. 14.31±1.27,
P<0.05) (Fig. 2A and B). The
results showed that VEGF-D had no direct effect on
cisplatin-induced LEC apoptosis. The rSulf2- and
rSulf2+VEGF-D-treated groups showed a significant increase in the
percentage of live cells, decreased cell necrosis and inhibited
cisplatin-induced LEC apoptosis, particularly in the late stage of
apoptosis. However, rSulf2+VEGF-D treatment had a greater effect on
apoptosis. Based on these results, rSulf2 inhibited the apoptosis
of LECs by activating VEGF-D.
Sulf2 with VEGF-D improves cell cycle
distibution of cisplatin-pretreated LECs
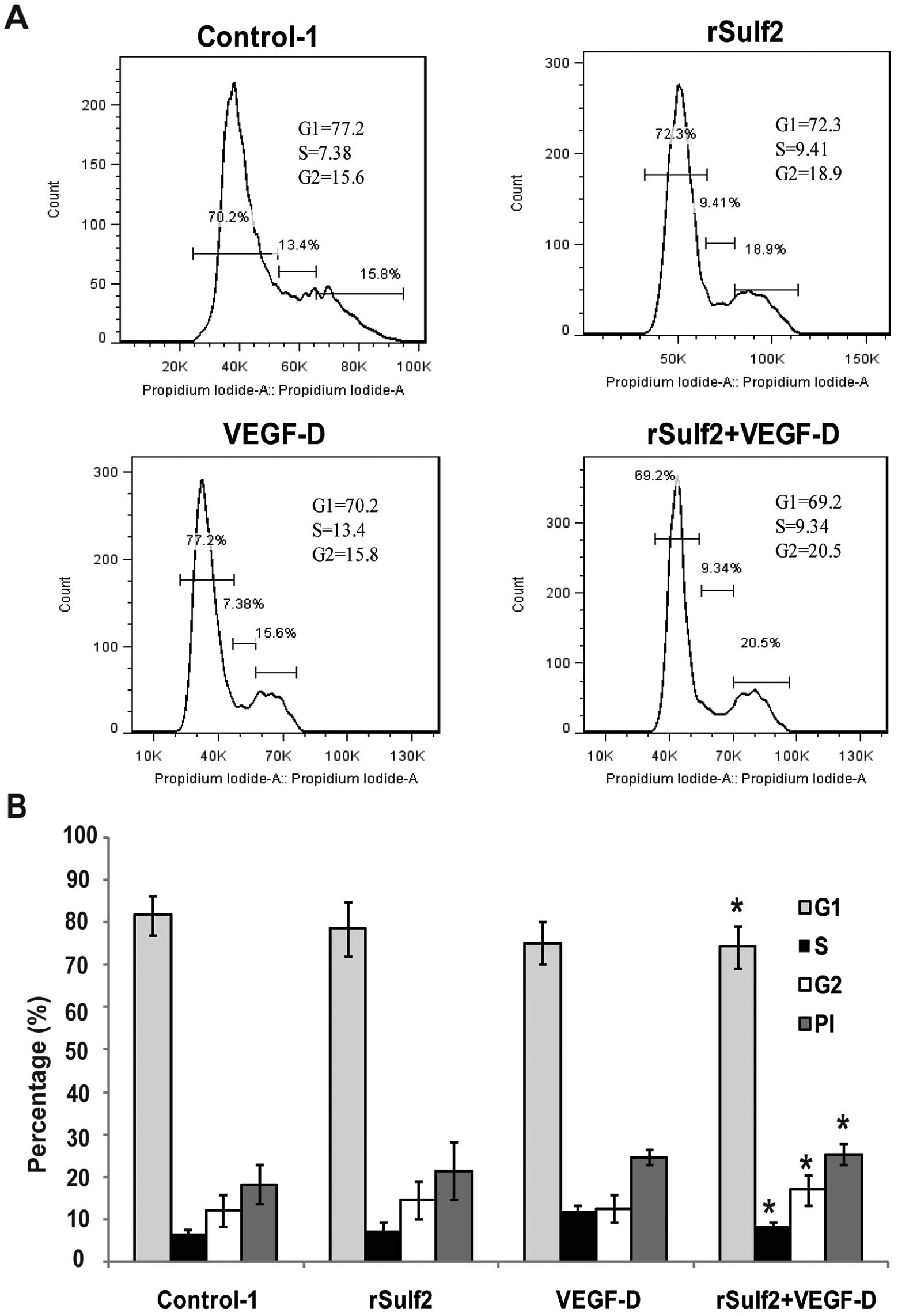
To ascertain the role of Sulf2 in the cell cycle
control of LECs, the cell cycle distribution of LECs was assessed
by flow cytometry. Compared with the control-1, rSulf2 treatment
caused no difference in the number of cells in the G1 phase
(78.70±6.40 vs. 81.75±4.55, P>0.05), the S phase (6.99±2.42 vs.
6.31±1.08, P>0.05) and the G2/M phase (14.45±4.45 vs.
11.99±3.61, P>0.05). Moreover, it had no effect on the PI index
(21.41±6.76 vs. 18.29±4.65, P>0.05). Furthermore, VEGF-D
treatment had no significant effect on the PI index (24.55±3.23 vs.
18.29±4.65, P>0.05), the number of cells in the G1 phase
(75.23±5.03 vs. 81.75±4.55, P>0.05), the S phase (11.9±1.5 vs.
6.31±1.08, P>0.05) and the G2/M phase (12.56±3.26 vs.
11.99±3.61, P>0.05). rSulf2+VEGF-D treatment caused a
significant decrease in the number of cells in the G1 phase
(74.3±5.10 vs. 81.75±4.55, P<0.05), the S phase (8.32±1.02 vs.
6.31±1.08, P<0.05) and the G2/M phase (16.95±3.55 vs.
11.99±3.61, P<0.05) as well as a significantly higher PI index
(25.37±2.50 vs. 18.29±4.65, P<0.05). We concluded that Sulf2
together with VEGF-D significantly promoted cell cycle progression
from the G1 phase to the G2/M phase and increased the PI index in
the LECs (Fig. 3A and B), while
Sulf2 or VEGF-D alone had no significant effect on the cell cycle
of the LECs.
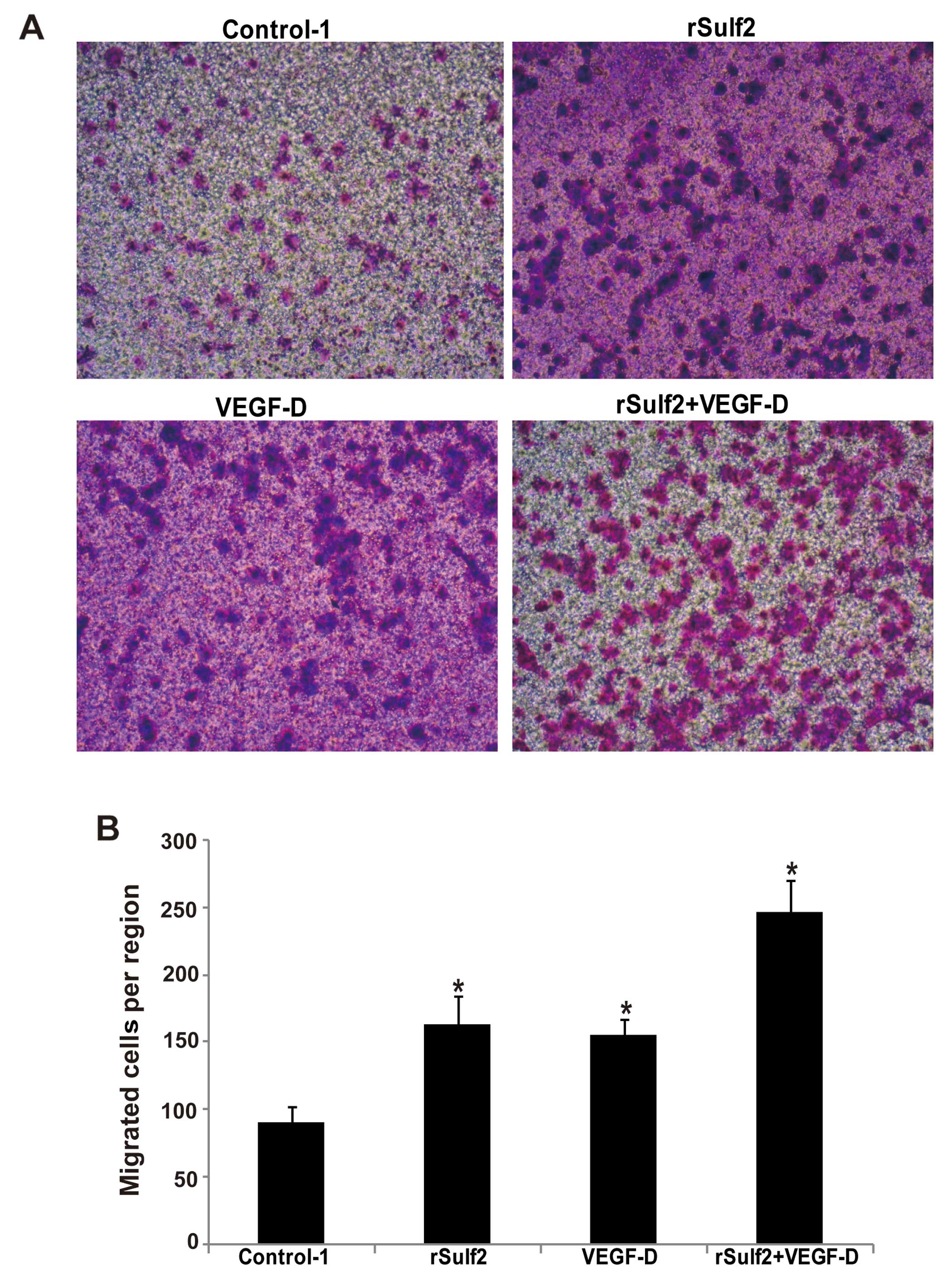
Sulf2 promotes breast cancer
migration
Compared with the control-1, the rSulf2- or
VEGF-D-treated LECs showed higher migration through the membrane of
the Boyden chamber (163.33±20.98, 155.67±10.96 vs. 90.0±12.52,
P<0.05). Moreover, the rSulf2+VEGF-D treated cells showed the
highest migration rate (247.33±23.07 vs. 90.0±12.52, P<0.05)
(Fig. 4A and B). These observations
clearly suggested that rSulf2 or VEGF-D enhanced LEC migration, but
rSulf2 with VEGF-D might work synergistically.
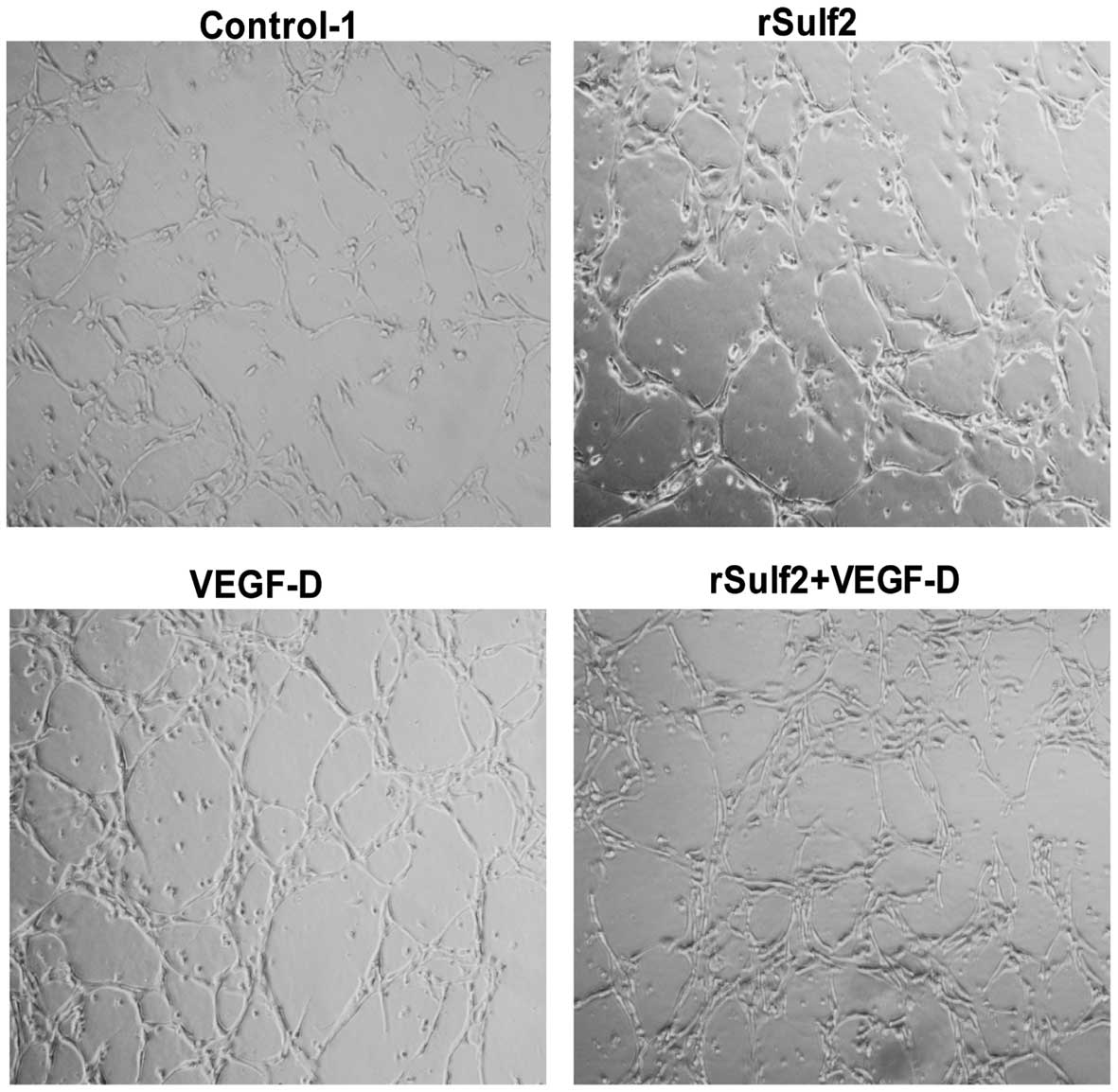
Sulf2 promotes lymphatic tube-like
structure formation in vitro
To examine the effect of Sulf2 on lymphatic
tube-like structure formation of LECs in vitro, LECs were
seeded on Matrigel substrate. Compared with the control-1, more
lymphatic tube-like structures were formed by LECs treated with
rSulf2, VEGF-D, and rSulf2+VEGF-D, after 24 h (Fig. 5). The results showed that rSulf2 or
VEGF-D increased lymphatic tube-like structure formation of the
LECs, however, the effect of Sulf2 with VEGF-D was more
significant, suggesting that Sulf2 could promote lymphangiogenesis
in vitro through the activation of VEGF-D.
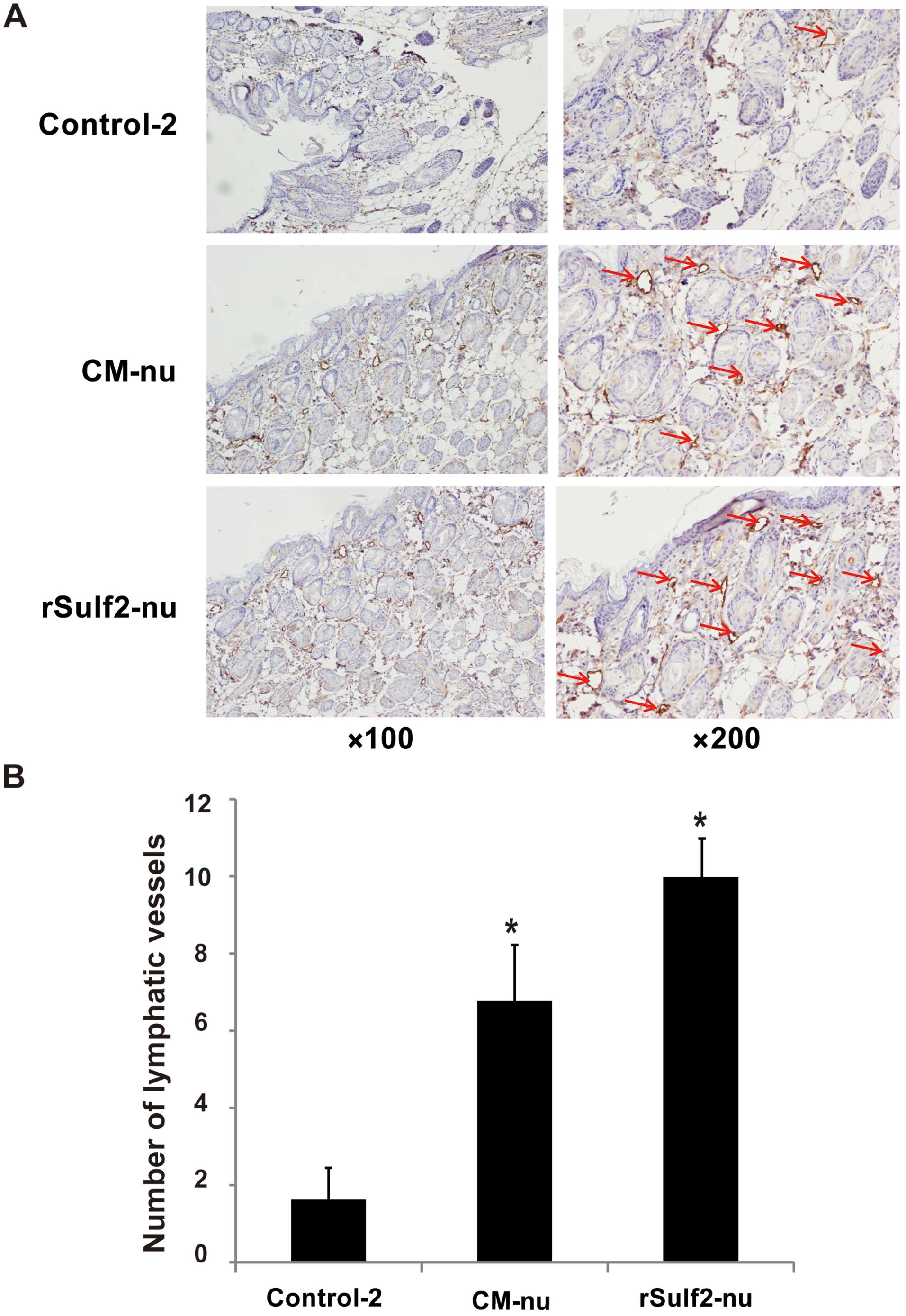
Sulf2 improves lymphangiogenesis in
nude mouse ears
The nude mouse ears were examined by pathological
sections. The lymphatic vessels were detected using IHC. Compared
with the control-2, the CM-nu and rSulf2-nu groups showed
significantly more lymphatic vessels (6.8±1.48 vs. 1.6±0.89,
P<0.01, 10±1.00 vs. 1.6±0.89, P<0.05). Furthermore, rSulf2-nu
also showed more lymphatic vessels compared with the CM-nu group
(10±1.00 vs. 6.8±1.48, P<0.05). The results demonstrated that
both exogenous and endogenous Sulf2 from breast cancer xenografts
promoted lymphangiogenesis in nude mouse ears. Moreover, the
effects of purified exogenous Sulf2 on lymphangiogenesis were more
pronounced than endogenous Sulf2 (Fig.
6).
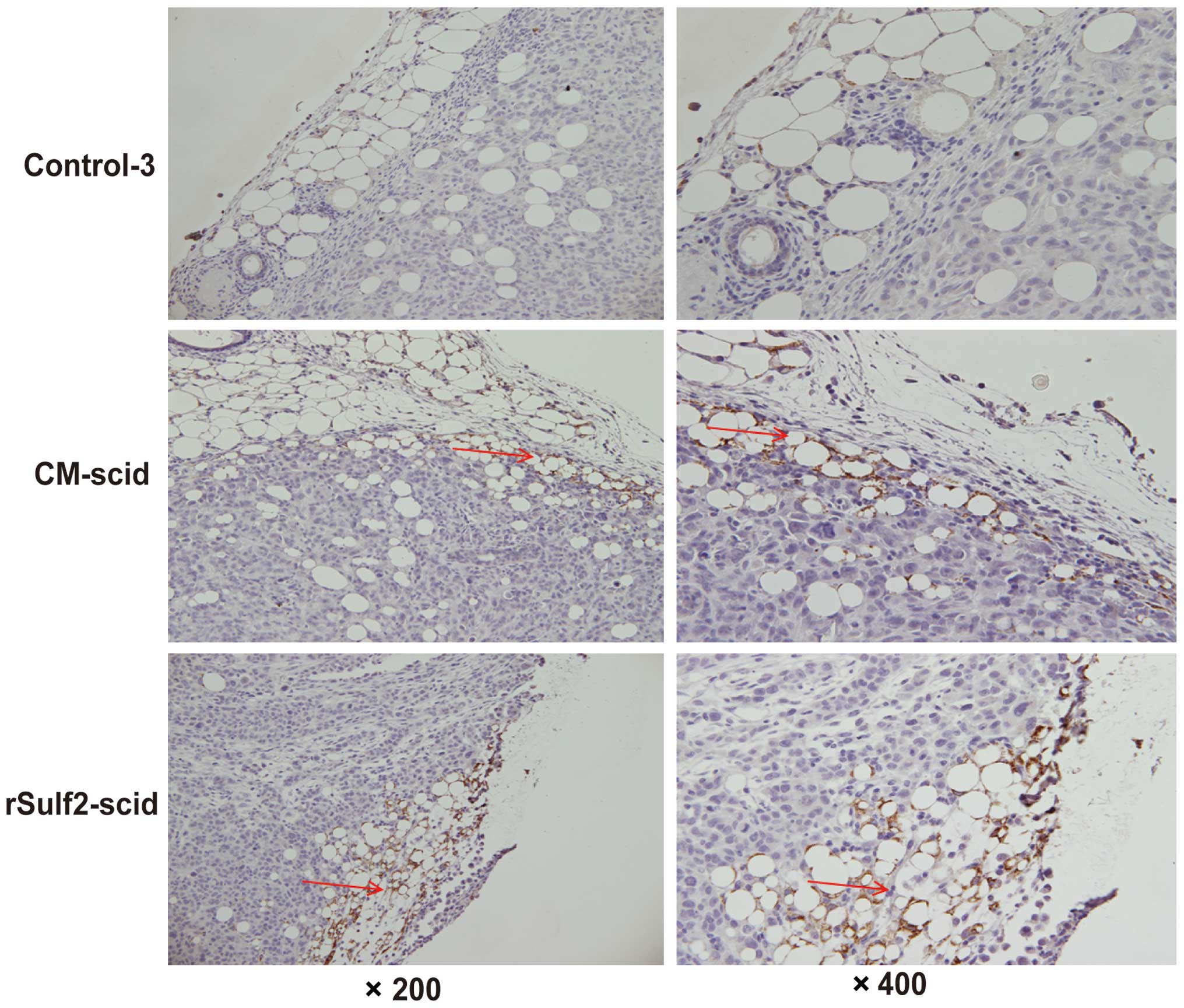
Sulf2 promotes lymphangiogenesis in
the breast cancer xenografts
To detect the effect of Sulf2 on lymphangiogenesis
in the breast cancer xenografts, we detected the density of
lymphatic vessels in MDA-MB-231 breast cancer xenografts, which did
not express Sulf2. No significant lymphatic vessels were detected
inside or around the xenografts in control-3. More lymphatic
vessels around the xenografts were detected in the CM-scid and
rSulf2-scid groups (Fig. 7). The
results further certified that Sulf2 increased lymphangiogenesis in
breast cancer xenografts and that breast cancer cells secreted
Sulf2 to promote lymphangiogenesis.
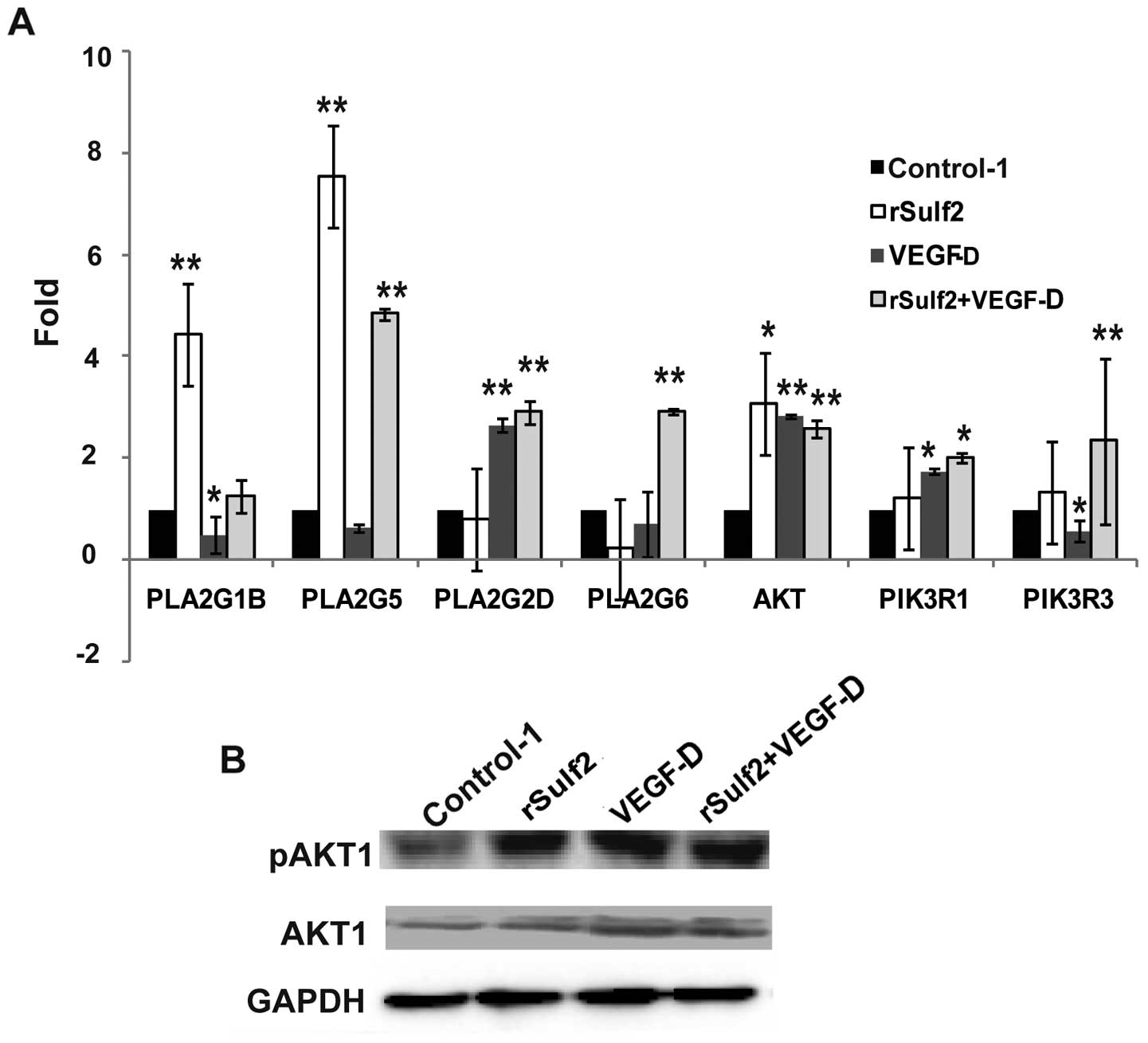
Sulf2 regulates signaling pathway
molecular interactions in LECs
Messenger RNA levels of a panel of VEGF signaling
pathway genes were first analyzed by PCR microarray, followed by
qRT-PCR and WB analysis verification. Compared with the control-1,
the genes significantly upregulated following treatment with rSulf2
were PLA2G1B (4.44±0.84 vs. 1.00, P<0.01), PLA2G5 (7.54±1.21 vs.
1.00, P<0.01) and AKT1 (3.09±0.62 vs. 1.00, P<0.05). The
genes significantly upregulated by VEGF-D were PLA2G2D (2.67±0.14
vs. 1.00, P<0.01), AKT1 (2.85±0.04 vs. 1.00, P<0.01) and
PI3KR1 (1.76±0.06 vs. 1.00, P<0.05). The genes downregulated
were PLA2G1B (0.49±0.36 vs. 1.00, P<0.05) and PI3KR3 (0.57±0.21
vs. 1.00, P<0.05). The genes significantly upregulated in the
rSulf2+VEGF-D group were PLA2G5 (4.84±0.12 vs. 1.00, P<0.01),
PLA2G2D (2.91±0.21 vs. 1.00, P<0.01), PLA2G6 (2.93±0.04 vs.
1.00, P<0.01), AKT1 (2.59±0.16 vs. 1.00, P<0.01) and PI3KR1
(2.01±0.1 vs. 1.00, P<0.01) (Fig.
8A). Only AKT1 mRNA showed the same significant trends in the
three groups and was chosen for further WB analysis verification.
Furthermore, we tested the AKT1 and the phosphorylated AKT1 protein
by WB analysis in the four groups. Compared with the control-1, the
expression and phosphorylation of AKT1 in the LECs revealed a
significant increase in the other groups (Fig. 8B). The results revealed that Sulf2
and/or VEGF-D could promote AKT1 expression and activation in the
LECs.
Discussion
Sulf2 has been reported to modify the activities of
heparan-binding growth factors (VEGF and FGF-1) and influence the
signaling pathways of the corresponding receptors to facilitate
angiogenesis. Uchimura et al (5) validated Sulf2 as a new molecule
involved in angiogenesis through the activation of VEGF and FGF-1.
Skobe et al (19) and Cherng
et al (26) certified that
the VEGF family is comprised of different monomeric forms including
VEGF145, VEGF165 and VEGF189 and VEGF206. These different monomeric
forms had similar heparan-binding regions, which could be regulated
by Sulf2. VEGF-D is one member of the VEGF family and also shares
similar structures. Harris et al (27) reported that VEGF-D is an angiogenic
and lymphangiogenic glycoprotein. Heparan-binding regions in VEGF-D
were found within the N- and C-terminal propeptides, which
suggested that VEGF-D could also bind to heparan. The C-terminal
propeptide significantly enhanced this interaction through the
removal of this propeptide from full-length VEGF-D. The removal of
either the N- or C-terminal propeptide was required for VEGF-D
binding to VEGFR-2/VEGFR-3 and formation of heterodimers, which
have recently been shown to positively regulate angiogenic and
lymphangiogenic sprouting (28,29).
In contrast, the removal of both propeptides was required for high
rates of lymph node metastasis. It was also reported that the
propeptides profoundly influenced the molecular interactions of
VEGF-D with VEGFR-3, and these propeptide structures also promoted
the effects of VEGF-D on tumor development.
In our previous study, we demonstrated that Sulf2
was upstream of VEGF-D and upregulated VEGF-D expression in breast
cancer cells (6). In this study, we
studied the role of Sulf2 in lymphangiogenesis and the mechanism
involved in its function. MCF-7 breast cancer cells released a high
level of Sulf2 protein into the culture medium, which was
demonstrated in our previous study (6). In this study, we collected the CM from
the supernatant of MCF-7 cells to study the effect of endogenous
Sulf2 on lymphangiogenesis in breast cancer cells. We also combined
and purified exogenous rSulf2 to study the direct function and
mechanism of Sulf2 in lymphangiogenesis. We found that Sulf2
significantly increased LEC mobility and lymphatic tube-like
structure formation, inhibited cisplatin-induced LEC apoptosis
in vitro, but had no direct effect on cell proliferation and
the cell cycle. Moreover, rSulf2 together with VEGF-D, further
promoted the proliferation, cell cycle progression, mobility and
tube formation in LECs, while at the same time inhibited
cisplatin-induced apoptosis, especially in the late stage. Sulf2
also significantly improved the densities of lymphatic vessels in
mouse ears and breast cancer xenografts in vivo. These
results showed that Sulf2 not only enhanced VEGF-D expression, but
also enhanced the activity of VEGF-D. Furthermore, we found that
the signaling pathway gene AKT1 was upregulated and activated by
Sulf2.
In summary, Sulf2 markedly promoted
lymphangiogenesis in breast cancer, possibly by promoting VEGF-D
expression and by activating the AKT1-related signaling pathway.
This finding confirmed the role of Sulf2 as a biomarker of breast
cancer progression. More importantly, new therapeutic approaches
targeting Sulf2 could improve the clinical outcomes in patients
with lymph node-positive breast cancer.
References
|
1
|
Bishop JR, Schuksz M and Esko JD: Heparan
sulphate proteoglycans fine-tune mammalian physiology. Nature.
446:1030–1037. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosen SD and Lemjabbar-Alaoui H: Sulf-2:
An extracellular modulator of cell signaling and a cancer target
candidate. Expert Opin Ther Targets. 14:935–949. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maltseva I, Chan M, Kalus I, Dierks T and
Rosen SD: The SULFs, extracellular sulfatases for heparan sulfate,
promote the migration of corneal epithelial cells during wound
repair. PLoS One. 8:e696422013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lamanna WC, Baldwin RJ, Padva M, Kalus I,
Ten Dam G, van Kuppevelt TH, Gallagher JT, von Figura K, Dierks T
and Merry CL: Heparan sulfate 6-O-endosulfatases: Discrete in vivo
activities and functional co-operativity. Biochem J. 400:63–73.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Uchimura K, Morimoto-Tomita M, Bistrup A,
Li J, Lyon M, Gallagher J, Werb Z and Rosen SD: HSulf-2, an
extracellular endoglucosamine-6-sulfatase, selectively mobilizes
heparin-bound growth factors and chemokines: Effects on VEGF,
FGF-1, and SDF-1. BMC Biochem. 7:22006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu C, He L, Zhou X, Nie X and Gu Y:
Sulfatase 2 promotes breast cancer progression through regulating
some tumor-related factors. Oncol Rep. 35:1318–1328.
2016.PubMed/NCBI
|
|
7
|
Hammond E, Khurana A, Shridhar V and
Dredge K: The role of heparanase and sulfatases in the modification
of heparan sulfate proteoglycans within the tumor microenvironment
and opportunities for novel cancer therapeutics. Front Oncol.
4:1952014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Al-Gayyar MM, Abbas A and Hamdan AM:
Chemopreventive and hepatoprotective roles of adiponectin (SULF2
inhibitor) in hepatocelluar carcinoma. Biol Chem. 397:257–267.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gill RM, Michael A, Westley L, Kocher HM,
Murphy JI and Dhoot GK: SULF1/SULF2 splice variants differentially
regulate pancreatic tumour growth progression. Exp Cell Res.
324:157–171. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Khurana A, Beleford D, He X, Chien J and
Shridhar V: Role of heparan sulfatases in ovarian and breast
cancer. Am J Cancer Res. 3:34–45. 2013.PubMed/NCBI
|
|
11
|
Lui NS, Yang YW, van Zante A, Buchanan P,
Jablons DM and Lemjabbar-Alaoui H: SULF2 expression is a potential
diagnostic and prognostic marker in lung cancer. PLoS One.
11:e01489112016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fujita K, Takechi E, Sakamoto N, Sumiyoshi
N, Izumi S, Miyamoto T, Matsuura S, Tsurugaya T, Akasaka K and
Yamamoto T: HpSulf, a heparan sulfate 6-O-endosulfatase, is
involved in the regulation of VEGF signaling during sea urchin
development. Mech Dev. 127:235–245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Plate K: From angiogenesis to
lymphangiogenesis. Nat Med. 7:151–152. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Karkkainen MJ and Petrova TV: Vascular
endothelial growth factor receptors in the regulation of
angiogenesis and lymphangiogenesis. Oncogene. 19:5598–5605. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sleeman JP and Thiele W: Tumor metastasis
and the lymphatic vasculature. Int J Cancer. 125:2747–2756. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reis-Filho JS and Schmitt FC:
Lymphangiogenesis in tumors: What do we know? Microsc Res Tech.
60:171–180. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tammela T, Petrova TV and Alitalo K:
Molecular lymphangiogenesis: New players. Trends Cell Biol.
15:434–441. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Acs G, Paragh G, Rakosy Z, Laronga C and
Zhang PJ: The extent of retraction clefts correlates with lymphatic
vessel density and VEGF-C expression and predicts nodal metastasis
and poor prognosis in early-stage breast carcinoma. Mod Pathol.
25:163–177. 2012.PubMed/NCBI
|
|
19
|
Skobe M, Hawighorst T, Jackson DG, Prevo
R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K and Detmar
M: Induction of tumor lymphangiogenesis by VEGF-C promotes breast
cancer metastasis. Nat Med. 7:192–198. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu C, Qi X, Chen Y, Sun B, Dai Y and Gu
Y: PI3K/Akt and MAPK/ERK1/2 signaling pathways are involved in
IGF-1-induced VEGF-C upregulation in breast cancer. J Cancer Res
Clin Oncol. 137:1587–1594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Joy AA, Ghosh M, Fernandes R and Clemons
MJ: Systemic treatment approaches in Ηer2-negative advanced breast
cancer-guidance on the guidelines. Curr Oncol. 22:(Suppl 1).
S29–S42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fina E, Callari M, Reduzzi C, DAiuto F,
Mariani G, Generali D, Pierotti MA, Daidone MG and Cappelletti V:
Gene expression profiling of circulating tumor cells in breast
cancer. Clin Chem. 61:278–289. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takei H, Kurosumi M, Yoshida T, Ninomiya
J, Hagiwara Y, Kamimura M, Hayashi Y, Tozuka K, Suemasu K, Inoue K,
et al: Current trends of sentinel lymph node biopsy for breast
cancer - a surgeons perspective. Breast Cancer. 14:362–370. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Karpanen T, Wirzenius M, Mäkinen T,
Veikkola T, Haisma HJ, Achen MG, Stacker SA, Pytowski B,
Ylä-Herttuala S and Alitalo K: Lymphangiogenic growth factor
responsiveness is modulated by postnatal lymphatic vessel
maturation. Am J Pathol. 169:708–718. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gu Y, Qi X and Guo S: Lymphangiogenesis
induced by VEGF-C and VEGF-D promotes metastasis and a poor outcome
in breast carcinoma: A retrospective study of 61 cases. Clin Exp
Metastasis. 25:717–725. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cherng JM, Lin CM, Lin CL, Huang SM, Chang
HL, Lee CC, Chiang LC and Chang PY: Effects of VEGF121 and/or
VEGF165 gene transfection on collateral circulation development. J
Formos Med Assoc. 99:603–611. 2000.PubMed/NCBI
|
|
27
|
Harris NC, Davydova N, Roufail S,
Paquet-Fifield S, Paavonen K, Karnezis T, Zhang YF, Sato T,
Rothacker J, Nice EC, et al: The propeptides of VEGF-D determine
heparin binding, receptor heterodimerization, and effects on tumor
biology. J Biol Chem. 288:8176–8186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Okamoto K, Oshika Y, Fukushima Y, Ohnishi
Y, Tokunaga T, Tomii Y, Kijima H, Yamazaki H, Ueyama Y, Tamaoki N,
et al: Xenografts of human solid tumors frequently express
cellular-associated isoform of vascular endothelial growth factor
(VEGF) 189. Oncol Rep. 6:1201–1204. 1999.PubMed/NCBI
|
|
29
|
Karnezis T, Shayan R, Caesar C, Roufail S,
Harris NC, Ardipradja K, Zhang YF, Williams SP, Farnsworth RH, Chai
MG, et al: VEGF-D promotes tumor metastasis by regulating
prostaglandins produced by the collecting lymphatic endothelium.
Cancer Cell. 21:181–195. 2012. View Article : Google Scholar : PubMed/NCBI
|