Introduction
Chronic myeloid leukemia (CML), a clonal
myeloproliferative disorder, is characterized by the Philadelphia
(Ph) chromosome which originates from the t(9;22)(q34;q11)
reciprocal translocation and leads to the BCR-ABL chimeric
oncoprotein (1–3). This BCR-ABL oncoprotein bears
constitutive tyrosine kinase activity and therefore promotes
uncontrolled growth and proliferation of leukemia cells (4). Development of inhibitors targeting
BCR-ABL has been generally successful in the past 15 years, with
highly decreased mortality for CML patients. However, resistance to
these drugs has been increasingly reported. CML remains an
incurable disease (5,6). Therefore, novel drugs with targets
other than BCR-ABL are expected.
Idelalisib, also named CAL101, was approved by the
US Food and Drug Administration in July, 2014. It is a
first-in-class oral PI3K inhibitor that has shown substantial and
sustained antileukemia efficacy in patients with
relapsed/refractory chronic lymphocytic leukemia (CLL) (7,8).
Compared with other chemotherapy regimens, idelalisib showed
advantages of long-term efficacy and reduced toxicity (9).
It has been reported that PI3K signaling contributes
to BCR-ABL transformation and is essential for in vivo
leukemogenesis of CML (10).
Furthermore, the PI3K p110δ isoform is preferentially expressed in
hematopoietic cells (11),
suggesting that idelalisib which targets p110δ shows favorable
antitumor efficacy against CML.
Therefore, in the present study, we investigated the
antileukemia activities of idelalisib in CML K562 cells.
Materials and methods
Reagents
Idelalisib and imatinib were purchased from Selleck
(London, ON, Canada).
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT) reagent was purchased from Amresco (Solon, OH, USA).
Antibodies against Akt, phospho-Akt (Ser473), phospho-GSK-3β
(Ser9), caspase-3, −8 and −9, poly(ADP-ribose) polymerase (PARP),
β-actin, and anti-mouse and anti-rabbit HRP-conjugated secondary
antibodies were obtained from Cell Signaling Technology (Danvers,
MA, USA). Antibodies against phospho-pRb (pS780), cyclin D1 and p27
were obtained from BD Biosciences Pharmingen (San Jose, CA, USA).
Antibodies against lamin B, p21, Bad, Bcl-2 and Bax were purchased
from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture
The human CML K562 cell line was purchased from the
Cell Resource Center, Peking Union Medical College (Beijing,
China). The cells were routinely maintained in RPMI-1640 medium
supplemented with 10% fetal bovine serum, 1% kanamycin (100 µg/ml),
and 1% glutamine (0.44 µg/ml) at 37°C in a humidified atmosphere
containing 5% CO2.
MTT assay
The MTT assay was performed to assess cell viability
as previously described (12,13).
Briefly, cells (2×104 cells/ml) were cultured in a
96-well plate for 48 h in the presence of 0, 1, 5, 10, 20, 50, 100,
150 and 200 µM of idelalisib. After addition of MTT (5 mg/ml) to
each well, the cells were further incubated for 4 h. The produced
formazan blue was dissolved with dimethyl sulfoxide (DMSO), and the
absorbance was measured at 490 nm using the microplate reader iMark
(Bio-Rad, Hercules, CA, USA).
Soft agar assay
The soft agar assay was carried out as previously
described (14) with a small
modification. K562 cells were treated with 0, 20, 50 and 100 µM of
idelalisib for 48 h. Then, the treated cells were seeded on
solidified agarose in 60-mm dishes (1.2×104 cells/dish).
After incubation for 10 days at 37°C, the cells were fixed with 4%
paraformaldehyde and stained with 0.5% crystal violet for 30 min.
Colonies were counted under a microscope. Each assay was performed
3 times.
Flow cytometric analysis of cell cycle
distribution
Cell cycle analysis was carried out as previously
described (15). The cell
suspension (4×105 cells/2 ml/well) of K562 cells was
planted in a 6-well plate and exposed to various concentrations of
idelalisib for 48 h. The cells were harvested, washed with ice-cold
phosphate-buffered saline (PBS), and fixed with 75% ethanol. After
centrifugation, the fixed cells were resuspended in propidium
iodide (PI) solution (25 µg/ml), and incubated in the dark for 30
min at 4°C to be available for analysis by BD Accuri C6 flow
cytometer (BD Biosciences, San Jose, CA, USA). Data were analyzed
using FlowJo 7.6 software.
Flow cytometric analysis of apoptosis
with Annexin V-FITC/PI staining
Analysis of apoptosis was carried out by Annexin
V-FITC/PI double staining as previously described (16). K562 cells treated with or without
idelalisib in a 6-well plate for 48 h were collected, washed with
ice-cold PBS, and then stained with 2.5 µl of Annexin V-FITC and
2.5 µl of PI (5 µg/ml) in binding buffer for 15 min at room
temperature in the dark. Flow cytometric analysis was conducted
using BD FACSVerse flow cytometer (BD Biosciences).
Synergism assay
Synergism was determined by the isobologram and
Fa-CI plot based on Chou and Talalay method (17,18).
K562 cells seeded in a 96-well plate were exposed to DMSO (as
control), idelalisib, imatinib or their combination at a fixed
ratio of IC50 idelalisib to IC50
imatinib (390:1) for 48 h. Cell growth inhibition was
determined using the MTT assay, and the IC50 values were
calculated. The combination index (CI) was calculated using
CalcuSyn software according to the method of Chou and Talalay: CI
<1 is defined as synergism; CI =1 is defined as an additive
effect; and CI >1 is defined as antagonism. All experiments were
carried out in triplicate.
Western blot analysis
Cell lysate preparation and western blot analysis
were performed as previously described (19,20).
Briefly, total and nuclear proteins were prepared using RIPA lysis
buffer (Roche Diagnostics, Basel, Switzerland) and NE-PER Nuclear
and Cytoplasmic Extraction kit (Thermo Fisher Scientific, Waltham,
MA, USA), respectively. Cell lysates with equal amount of protein
were subjected to 10% SDS-polyacrylamide gel electrophoresis
(PAGE), and the separated proteins were transferred onto a
polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA,
USA). The membrane was blocked in 5% non-fat dried milk, exposed to
the specified primary antibodies overnight at 4°C, and then to the
respective secondary antibodies. The blots were visualized using
enhanced chemiluminescence (ECL) reagents and digitalized by
scanning.
Statistical analysis
Data are presented as mean ± standard deviation (SD)
from 3 independent experiments. The Student's t-test was carried
out for analysis of statistical significance using GraphPad Prism 5
software (GraphPad, San Diego, CA, USA). p<0.05 was regarded as
indicative of a statistically significant difference.
Results
Idelalisib inhibits the proliferation
of K562 cells
The inhibitory activity of idelalisib on the
proliferation of K562 cells was assessed by MTT and soft agar
assays, respectively. First, the MTT assay was utilized. As shown
in Fig. 1A, after exposure to
idelalisib at concentrations from 1 to 200 µM for 48 h, K562 cells
showed a dose-dependent decrease in viability, with an
IC50 value of 71.4 µM.
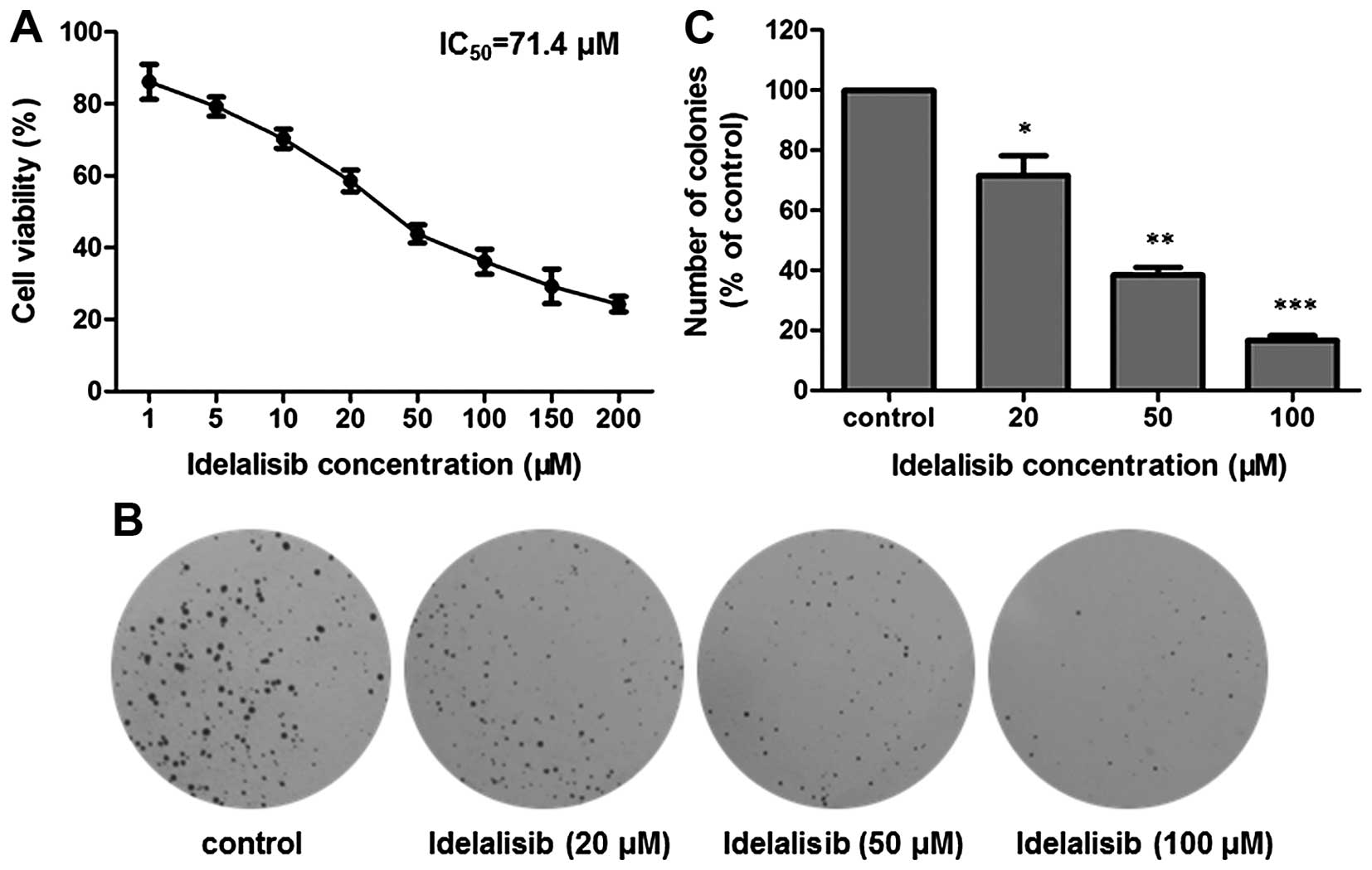 | Figure 1.Idelalisib inhibits K562 cell
proliferation. (A) MTT assay. Cells were exposed to different
concentrations (0, 1, 5, 10, 20, 50, 100, 150 and 200 µM) of
idelalisib for 48 h. Cell viability was measured by determination
of the absorbance at 490 nm after addition of MTT reagent. (B) Soft
agar assay. K562 cells treated with different concentrations (0,
20, 50 and 100 µM) of idelalisib for 48 h, were grown in soft agar
for 10 days at 37°C, fixed with 4% paraformaldehyde and stained
with 0.5% crystal violet for 30 min. The resulting colonies were
counted under a microscope. (C) Bar graph showing the number
(percentage of control) of colonies formed by K562 cells with or
without idelalisib treatment. Data are mean ± SD, representative of
3 independent experiments; *p<0.05, **p<0.01, ***p<0.001
compared with the control. |
We further examined the antiproliferative activity
of idelalisib by use of soft agar assay, which is a suitable method
for monitoring anchorage-independent cell growth (21). The cells treated with 0, 20, 50 and
100 µM of idelalisib for 48 h were grown in soft agar for 10 days.
As shown in Fig. 1B and C,
treatment with idelalisib decreased both the number and size of the
cell colonies, confirming that idelalisib dose-dependently
inhibited K562 cell proliferation.
Idelalisib induces cell cycle arrest
in the G1 phase in the K562 cells
To determine whether the suppression of K562 cell
proliferation by idelalisib is attributed to cell cycle arrest,
cell cycle distribution was examined by flow cytometry after
idelalisib treatment for 48 h. As shown in Fig. 2A and B, idelalisib induced
accumulation of the cell population in the G1 phase, with 60.8% for
50 µM idelalisib-treated cells vs. 47.4% for control cells. Then,
we investigated the effect of idelalisib on cell cycle-related
proteins. Fig. 2C indicates that
treatment with idelalisib decreased the expression of cyclin D1 and
the phosphorylation of pRb, but increased the expression of p27 and
p21. To further investigate the mechanism of idelalisib in G1 cell
cycle arrest, we examined the effect on the phosphorylation of Akt
and GSK-3β, which are known to regulate the cell cycle downstream
of the PI3K/Akt pathway (22).
Phosphorylation of Akt and GSK-3β was dose-dependently inhibited by
idelalisib, suggesting that the cell cycle arrest effect of
idelalisib may be attributed to its blockade of the PI3K/Akt
pathway.
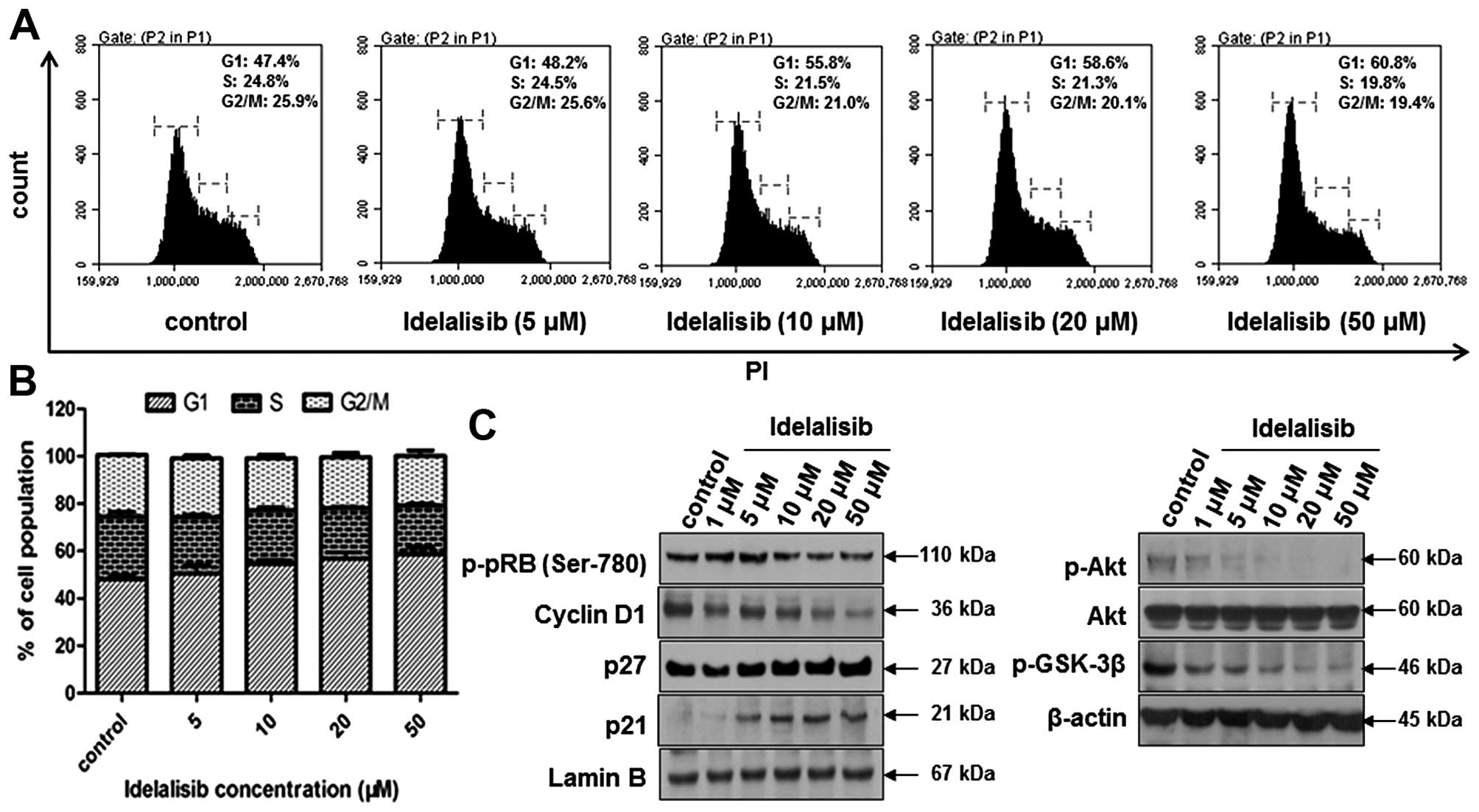 | Figure 2.Idelalisib induces K562 cell cycle
arrest in the G1 phase. (A) Cell cycle distribution analyzed by
flow cytometry. Cells were treated with idelalisib at 0, 5, 10, 20
and 50 µM for 48 h, stained with PI and subjected to flow
cytometry. (B) Bar graph showing the percentage of K562 cells in
the G1, S and G2/M phase, respectively. Data represent mean ± SD of
3 independent experiments. (C) Effect of idelalisib on cell
cycle-related proteins (left panel) and downstream effectors of the
PI3K/Akt pathway (right panel). K562 cells were treated with the
indicated concentrations of idelalisib (0, 1, 5, 10, 20 and 50 µM)
for 48 h. Cell lysates were prepared respectively to be available
for analysis of protein levels in the nucleus (p-pRb, cyclin D1,
p21 and p27) or whole cell (p-Akt, Akt and p-GSK-3β) by western
blotting. |
Idelalisib induces apoptosis in the
K562 cells
Since apoptosis may contribute to a decrease in cell
viability, we also examined whether idelalisib induces apoptosis in
the K562 cells. Flow cytometric analysis was carried out after
Annexin V-FITC/PI staining of K562 cells with or without idelalisib
treatment. As indicated in Fig. 3A and
B, the cell population in the upper- and lower-right quadrants
was increased in a dose-dependent manner after idelalisib
treatment, with 22.94% for cells treated with 100 µM idelalisib vs.
7.55% for control cells, suggesting that idelalisib induced
apoptosis in the K562 cells. Notably, the increased apoptotic cells
were mainly in the early-stage (lower-right quadrant; 19.8% for
cells treated with 100 µM idelalisib vs. 5.69% for control
cells).
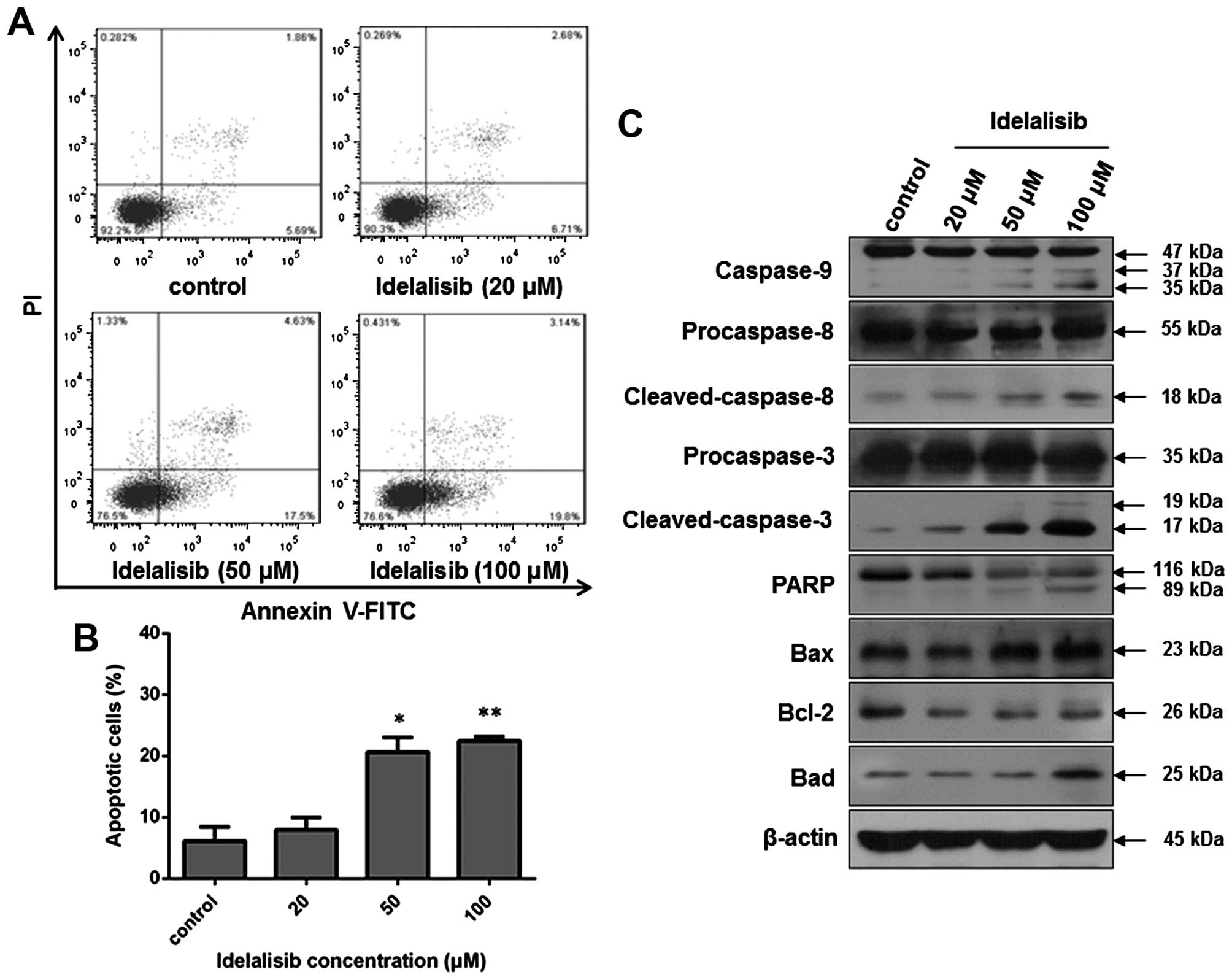 | Figure 3.Idelalisib induces apoptosis in the
K562 cells. (A) Cell apoptosis analyzed by flow cytometry. Cells
were treated with indicated concentrations of idelalisib (0, 20, 50
and 100 µM) for 48 h, double-stained with Annexin V-FITC/PI and
subjected to flow cytometric analysis. (B) Bar graph showing the
percentage of apoptotic K562 cells. Data represent mean ± SD of 3
independent experiments; *p<0.05, **p<0.01, compared with the
control. (C) Effect of idelalisib on apoptosis-related proteins.
K562 cells were treated with idelalisib (0, 20, 50 and 100 µM) for
48 h. The levels of cleaved caspase-3, −8 and −9, and PARP, as well
as the expression of Bcl-2, Bax and Bad were examined by western
blotting. |
Then, we investigated the effect on
apoptosis-related proteins by western blot analysis. As shown in
Fig. 3C, idelalisib treatment
increased the level of cleaved caspase-9, −8 and −3, and PARP, as
well as the expression of Bad and Bax. In contrast, the expression
of anti-apoptotic protein Bcl-2 was reduced. These results suggest
that the apoptosis induction by idelalisib in K562 cells may be
related to the Bad/Bcl-2/Bax family and the cleavage of caspases
and PARP.
Synergistic effect of idelalisib and
imatinib in K562 cells
A well-designed drug combination may enhance
efficacy while reducing toxicity. To investigate whether idelalisib
can enhance the antileukemia activity of the first-line drug
imatinib, we carried out a combination study using Chou and Talalay
method. K562 cells were treated with idelalisib and imatinib alone
or in combination for 48 h, respectively. MTT assay was conducted
to determine the inhibitory activities of each drug and the
combination. The IC50 of imatinib was calculated to be
0.183 µM (Fig. 4A). A synergism
study was performed using a series of drug combinations (20, 40,
60, 80 and 100% of IC50 value of each drug) with a fixed
ratio of IC50 idelalisib to IC50 imatinib
(390:1). As shown in Fig. 4B,
co-treatment with the two drugs led to an enhanced cell growth
inhibition compared to either treatment alone. Analysis of the data
by CalcuSyn software indicated a synergistic effect for the
combination, since CI values were <1 at all fraction affected
(Fa) levels (Fig. 4C). All of the 3
data points (ED50, ED75 and ED90)
were far below the additivity line (Fig. 4D), with the respective CI values as
0.62, 0.47 and 0.35 (Table I),
suggesting strong synergy of the combination in regards to the
growth inhibition of K562 cells.
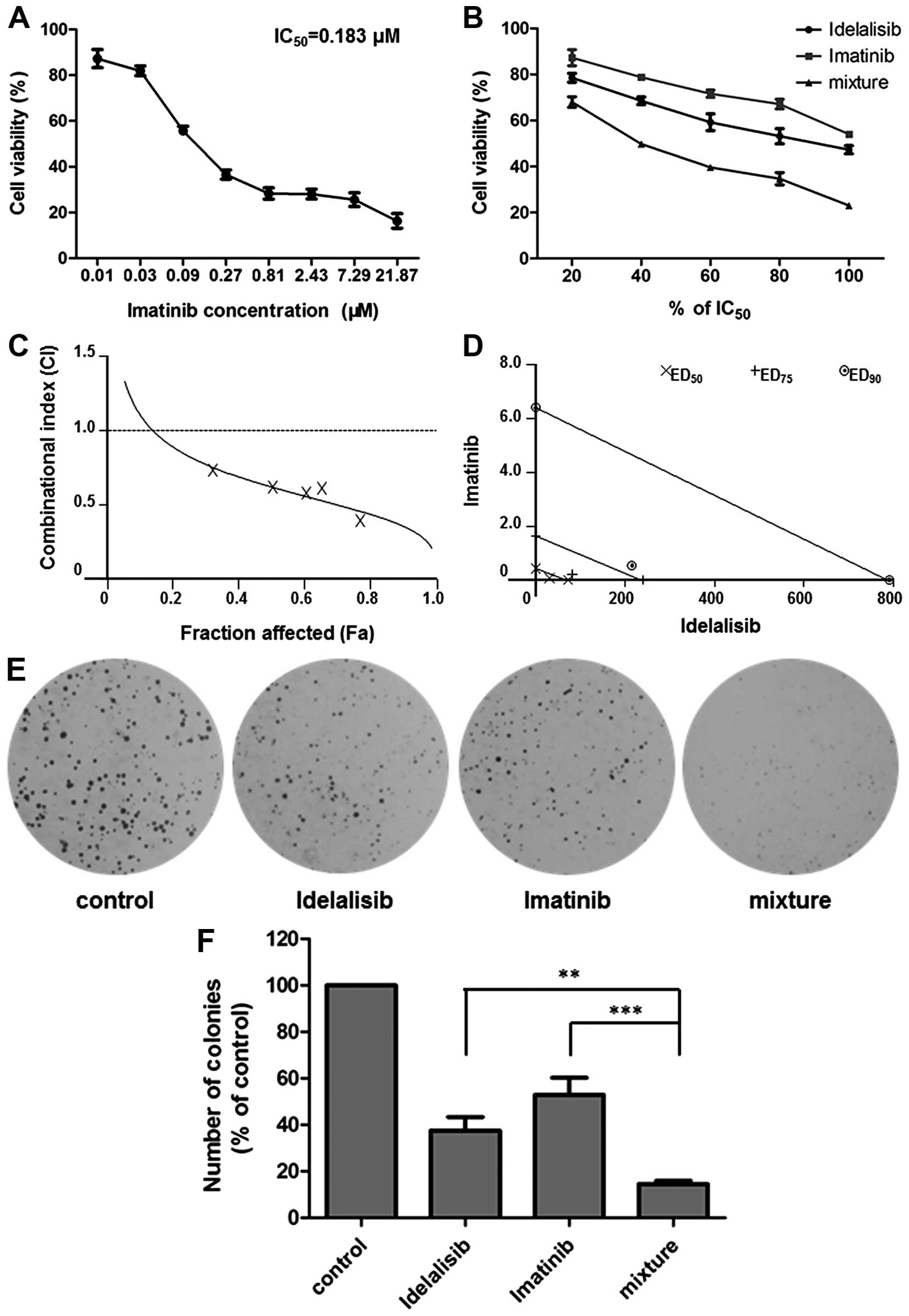 | Figure 4.Combination effect of idelalisib and
imatinib on K562 cell proliferation. (A) Imatinib inhibited the
proliferation of K562 cells. Cells were treated with imatinib (0,
0.01, 0.03, 0.09, 0.27, 0.81, 2.43, 7.29 and 21.87 µM). Cell
viability was determined by MTT assay. IC50 value of
imatinib was calculated to be 0.183 µM. Data are mean ± SD,
representative of 3 independent experiments. (B) Combination with
idelalisib enhanced the inhibitory activity of imatinib. K562 cells
were exposed to a series of concentrations of idelalisib and
imatinib (20, 40, 60, 80 and 100% IC50 of each drug),
alone or in combination. Cell viability was determined by MTT
assay. Data are mean ± SD, representative of 3 independent
experiments. (C) Analysis of the combinational effect using
CalcuSyn software. Combinational index (CI) values of drug
combinations below the horizontal line (CI=1) represent synergy.
Fa, fraction affected. (D) Isobologram of idelalisib and imatinib
combination. Data points of growth inhibition at 50%
(ED50), 75% (ED75) and 90% (ED90)
are on the left side of the respective lines, indicating a
synergistic effect. (E and F) Combination of idelalisib and
imatinib enhanced the inhibitory activity on K562 proliferation
which was determined by soft agar assay. The cells treated with
idelalisib (50 µM) and imatinib (0.128 µM) alone or in combination
for 48 h, were grown in soft agar for 10 days at 37°C. Colonies
were counted under a microscope. Data are mean ± SD, representative
of 3 independent experiments; **p<0.01, ***p<0.001, compared
with mixture (combination). |
 | Table I.Combination indices (CI) for
idelalisib and imatinib. |
Table I.
Combination indices (CI) for
idelalisib and imatinib.
|
|
|
|
| CI value (mean ±
SD) |
|---|
|
|
|
|
|
|
|---|
| Cell line | Drug(s) | IC50
(µM) | r |
ED50 |
ED75 |
ED90 |
|---|
| K562 | Idelalisib | 71.4 | 0.982 | 0.62±0.09 | 0.47±0.05 | 0.35±0.09 |
|
| Imatinib | 0.183 | 0.998 |
|
|
|
|
| Idelalisib +
imatinib | – | 0.987 |
|
|
|
Then, we further confirmed the combinational effect
of idelalisib and imatinib on K562 cells using various assay
methods. Soft agar assay showed that co-treatment with idelalisib
(50 µM) and imatinib (0.128 µM) decreased cell proliferation more
potently than either drug alone (Fig.
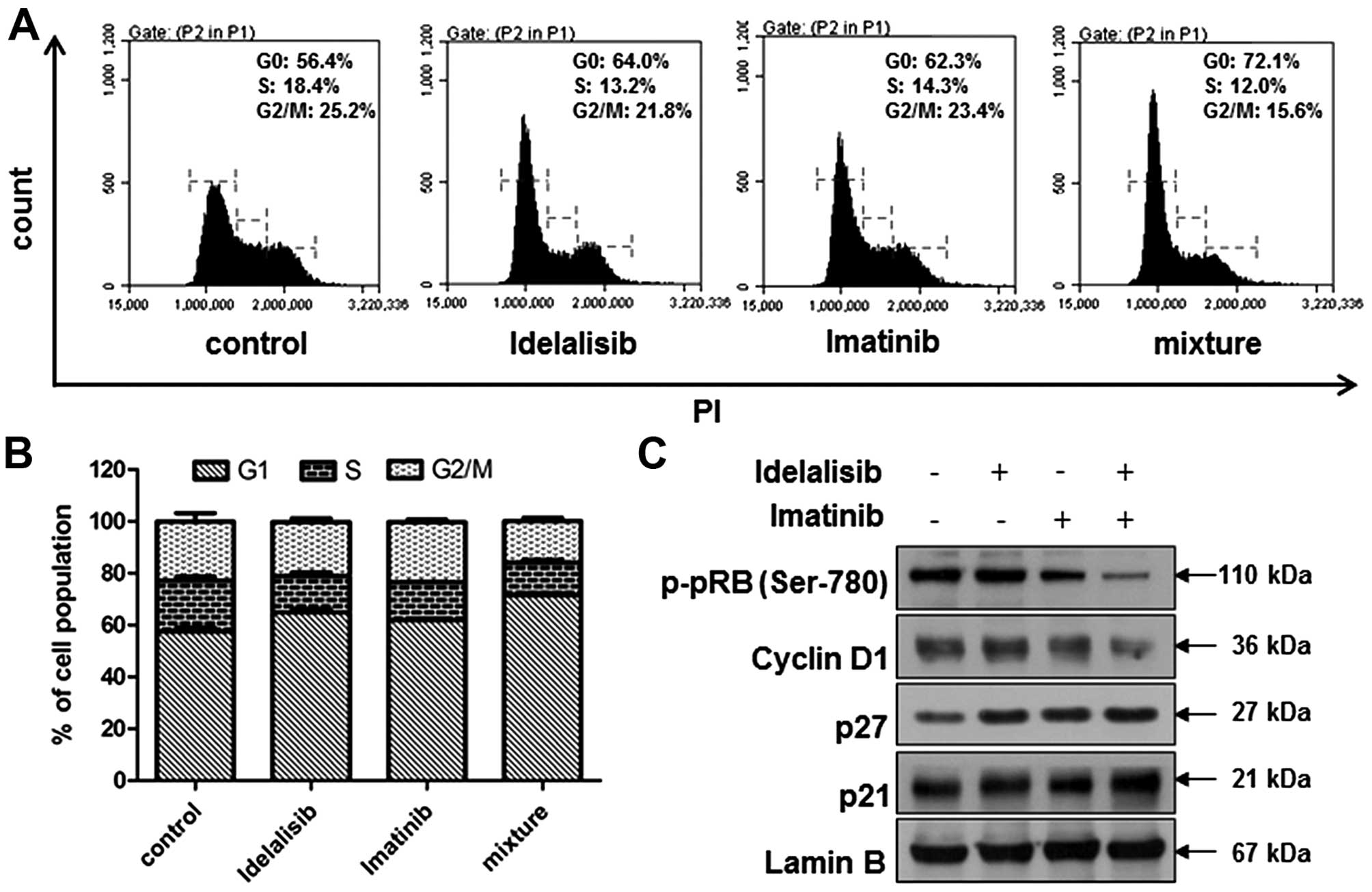
4E and F). Cell cycle distribution analysis indicated an
increased G1 arrest (Fig. 5A and
B), accompanied by further reduction in the level of p-pRb and
cyclin D1, and further enhancement in p21 and p27 expression
(Fig. 5C). In addition, combined
treatment with idelalisib (50 µM) and imatinib (0.128 µM) led to
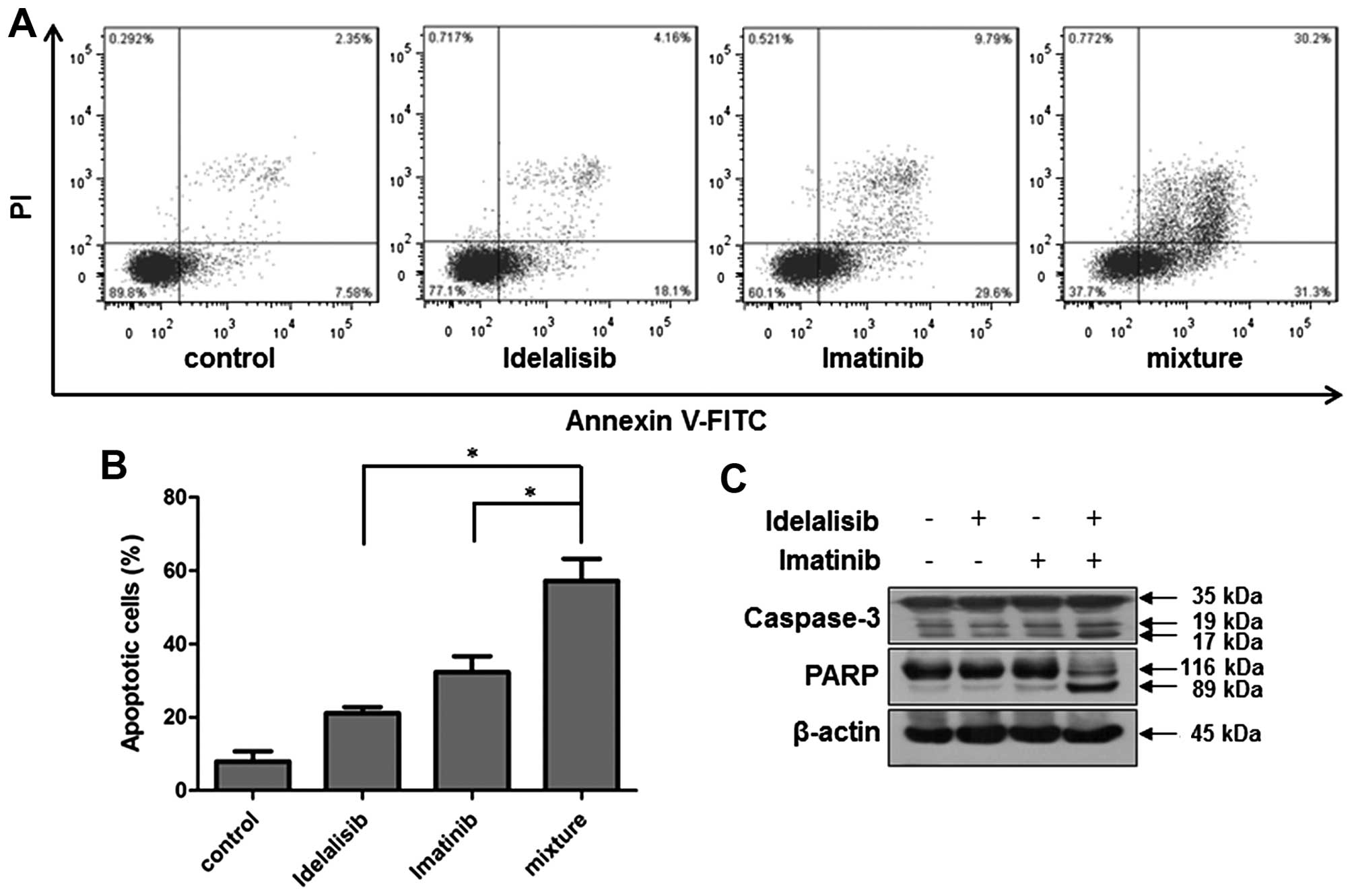
increased cell apoptosis than that induced by either drug alone
(Fig. 6A and B). Notably,
co-treatment with the two drugs induced highly increased cell
population in both the upper- and lower-right quadrants, suggesting
that the combination of idelalisib and imatinib treatment induced
apoptosis in both late and early stages. Consistently, the levels
of cleaved caspase-3 and PARP were significantly increased
following the combination treatment, as compared with each drug
alone (Fig. 6C).
Discussion
Phosphatidylinositol 3-kinases (PI3Ks), consisting
of 3 classes (class I, II and III), are closely involved in cell
growth and survival (23,24). Among these 3 classes, class I PI3K
is the most studied and is closely related to signaling in
hematopoietic cells (25). Class I
PI3K is further divided into 2 subtypes as class IA and class IB.
Class IA PI3K comprises a regulatory subunit and a catalytic
subunit (p110α, p110β or p110δ), whereas class IB PI3K comprises a
p101 regulatory subunit and a p110γ catalytic subunit. Catalytic
isoform p110δ is preferentially expressed in hematopoietic cells
(26), suggesting that targeting
p110δ may be a promising strategy for leukemia therapy.
Furthermore, it has been reported that PI3K signaling contributes
to BCR-ABL transformation and is essential for leukemogenesis of
chronic myeloid leukemia (CML) (10). In addition, sustained activation of
the PI3K/Akt signaling pathway may contribute to drug resistance
due to enhanced drug efflux by ATP-binding cassette transporters
such as P-gp (27,28). Occurrence of resistance has become a
big challenge in the chemotherapy of CML in recent years.
Therefore, targeting PI3K p110δ may be an alternative approach for
CML treatment.
In the present study, the antileukemia activity of
idelalisib, a specific inhibitor of PI3K p110δ, on CML K562 cells
was investigated. Our results demonstrated that idelalisib
dose-dependently inhibited K562 cell proliferation, which were
supported by MTT and soft agar assays. Both MTT and soft agar
assays are well known assays which are used to evaluate cell
proliferation in vitro, whereas the latter one can
additionally predict tumorigenic ability which is correlated with
anchorage-independent growth in vivo (14). G1 cell cycle arrest was induced by
idelalisib treatment, accompanied by the decreased expression of
cyclin D1 and phosphorylation of pRb in contrast to the increased
expression of cyclin-dependent kinase (CDK)-inhibitors p27 and p21.
Meanwhile, idelalisib treatment blocked the phosphorylation of Akt
and GSK-3β in a dose-dependent manner. Since Akt is known to
promote cell cycle progression by upregulating GSK-3β and cyclin D1
(29), and downregulating CDK
inhibitors p27 and p21 (30), the
cell cycle arrest effect of idelalisib may be attributed to the
blockade of the PI3K/Akt pathway.
Apoptotic cell death is triggered either by the
mitochondrial pathway or the death receptor pathway. The former is
mainly regulated by the Bcl-2 family which comprises pro-apoptotic
proteins such as Bax and Bak, anti-apoptotic proteins such as
Bcl-2, and the BH3-only proteins such as Bad, while the latter is
controlled by cell surface death receptors such as Fas (31). In the mitochondrial pathway, Bax and
Bak are known to disrupt mitochondrial outer membrane integrity
through multimerization and therefore release cytochrome c
into the cytosol, which in turn activates caspases including
caspase-9 and −3, finally leading to apoptosis (32). Bcl-2 exhibits an anti-apoptotic
effect by inhibiting formation of the Bax/Bak complex, while Bad
promotes apoptosis via neutralizing the inhibitory activity of
Bcl-2 on Bax/Bak (32). For the
death receptor pathway, Fas induces activation of caspase-8, which
cleaves downstream caspases such as caspase-3, promoting apoptosis
(31). In the present study, after
treatment with idelalisib, expression of Bad and Bax was increased
while that of Bcl-2 was reduced; the levels of cleaved caspase-9,
−8 and −3 and PARP were enhanced. Akt is known to upregulate Bcl-2
via inhibition of the antagonist Bad in the mitochondrial pathway
(33), and to mediate Fas via
regulation of FoxO in the cell death receptor pathway (34). Therefore, idelalisib promoted cell
apoptosis by activating both the mitochondrial and death receptor
pathways, in which targeting PI3K p110δ and the downstream
effectors may be involved.
Development of imatinib for CML treatment has met
with great success. However, the efficacy has been challenged by
the increasing occurrence of acquired resistance, and the original
insensitivity of a population of patients. The present study
indicates that idelalisib could highly enhance the antileukemia
activity of imatinib on K562 cells, suggesting the possibility for
the combinational use of the two drugs in the future.
In conclusion, idelalisib, a novel PI3Kδ-specific
inhibitor, alone or in combination with imatinib, exhibited
potential antileukemia activity against CML K562 cells, suggesting
the possible future application in the treatment of CML
patients.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81373441,
81202542 and 81402901), the Natural Science Foundation of Tianjin
(12JCZDJC25800 and 13JCYBJC24800), the China Postdoctoral Science
Foundation Funded Project (2014M551035 and 2014M551037), and the
Tianjin Medical University Research Fund (2013ky07).
References
|
1
|
Maru Y: Molecular biology of chronic
myeloid leukemia. Cancer Sci. 103:1601–1610. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Holyoake TL and Helgason GV: Do we need
more drugs for chronic myeloid leukemia? Immunol Rev. 263:106–123.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rangatia J and Bonnet D: Transient or
long-term silencing of BCR-ABL alone induces cell cycle and
proliferation arrest, apoptosis and differentiation. Leukemia.
20:68–76. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chereda B and Melo JV: Natural course and
biology of CML. Ann Hematol. 94:(Suppl 2). S107–S121. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Larson RA: Is there a best TKI for chronic
phase CML? Blood. 126:2370–2375. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu N, Zang S, Liu Y, Wang Y, Li W, Liu Q,
Ji M, Ma D and Ji C: FZD7 regulates BMSC-mediated protection of CML
cells. Oncotarget. 7:6175–6187. 2016.PubMed/NCBI
|
|
7
|
Brown JR: Idelalisib has CLL on the run!
Blood. 126:2656–2657. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brown JR, Byrd JC, Coutre SE, Benson DM,
Flinn IW, Wagner-Johnston ND, Spurgeon SE, Kahl BS, Bello C, Webb
HK, et al: Idelalisib, an inhibitor of phosphatidylinositol
3-kinase p110δ, for relapsed/refractory chronic lymphocytic
leukemia. Blood. 123:3390–3397. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morabito F, Gentile M, Seymour JF and
Polliack A: Ibrutinib, idelalisib and obinutuzumab for the
treatment of patients with chronic lymphocytic leukemia: Three new
arrows aiming at the target. Leuk Lymphoma. 56:3250–3256. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kharas MG, Janes MR, Scarfone VM, Lilly
MB, Knight ZA, Shokat KM and Fruman DA: Ablation of PI3K blocks
BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor
prevents expansion of human BCR-ABL+ leukemia cells. J
Clin Invest. 118:3038–3050. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Byrd JC, Woyach JA and Johnson AJ:
Translating PI3K-delta inhibitors to the clinic in chronic
lymphocytic leukemia: The story of CAL-101 (GS1101). Am Soc Clin
Oncol Educ Book. 2012.691–694. 2012.doi:
10.14694/EdBook_AM.2012.32.691. PubMed/NCBI
|
|
12
|
Chen X, Tang SA, Lee E, Qiu Y, Wang R,
Duan HQ, Dan S, Jin M and Kong D: IVSE, isolated from Inula
japonica, suppresses LPS-induced NO production via NF-κB and MAPK
inactivation in RAW264.7 cells. Life Sci. 124:8–15. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang X, Tang SA, Wang R, Qiu Y, Jin M and
Kong D: Inhibitory effects of JEUD-38, a new sesquiterpene lactone
from Inula japonica thunb, on LPS-induced iNOS expression in
RAW264.7 cells. Inflammation. 38:941–948. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Y, Zhang X, Wang J, Yang J and Tan WF:
JNK is required for maintaining the tumor-initiating cell-like
properties of acquired chemoresistant human cancer cells. Acta
Pharmacol Sin. 36:1099–1106. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang SA, Zhou Q, Guo WZ, Qiu Y, Wang R,
Jin M, Zhang W, Li K, Yamori T, Dan S, et al: In vitro antitumor
activity of stellettin B, a triterpene from marine sponge Jaspis
stellifera, on human glioblastoma cancer SF295 cells. Mar Drugs.
12:4200–4213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Y, Liu J, Qiu Y, Jin M, Chen X, Fan
G, Wang R and Kong D: ZSTK474, a specific class I
phosphatidylinositol 3-kinase inhibitor, induces G1 arrest and
autophagy in human breast cancer MCF-7 cells. Oncotarget.
7:19897–19909. 2016.PubMed/NCBI
|
|
17
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Radujkovic A, Luft T, Dreger P, Ho AD,
Zeller W Jens, Fruehauf S and Topaly J: In vitro testing of drug
combinations employing nilotinib and alkylating agents with regard
to pretransplant conditioning treatment of advanced-phase chronic
myeloid leukemia. Cancer Chemother Pharmacol. 74:427–432. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kong D, Yamori T, Yamazaki K and Dan S: In
vitro multifaceted activities of a specific group of novel
phosphatidylinositol 3-kinase inhibitors on hotspot mutant PIK3CA.
Invest New Drugs. 32:1134–1143. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao W, Guo W, Zhou Q, Ma SN, Wang R, Qiu
Y, Jin M, Duan HQ and Kong D: In vitro antimetastatic effect of
phosphatidylinositol 3-kinase inhibitor ZSTK474 on prostate cancer
PC3 cells. Int J Mol Sci. 14:13577–13591. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kusakawa S, Yasuda S, Kuroda T, Kawamata S
and Sato Y: Ultra-sensitive detection of tumorigenic cellular
impurities in human cell-processed therapeutic products by digital
analysis of soft agar colony formation. Sci Rep. 5:17892–17902.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang F, Lee JT, Navolanic PM, Steelman
LS, Shelton JG, Blalock WL, Franklin RA and McCubrey JA:
Involvement of PI3K/Akt pathway in cell cycle progression,
apoptosis, and neoplastic transformation: A target for cancer
chemotherapy. Leukemia. 17:590–603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Neri LM, Cani A, Martelli AM, Simioni C,
Junghanss C, Tabellini G, Ricci F, Tazzari PL, Pagliaro P, McCubrey
JA, et al: Targeting the PI3K/Akt/mTOR signaling pathway in
B-precursor acute lymphoblastic leukemia and its therapeutic
potential. Leukemia. 28:739–748. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fruman DA and Rommel C: PI3K and cancer:
Lessons, challenges and opportunities. Nat Rev Drug Discov.
13:140–156. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Steelman LS, Pohnert SC, Shelton JG,
Franklin RA, Bertrand FE and McCubrey JA: JAK/STAT, Raf/MEK/ERK,
PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis.
Leukemia. 18:189–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sujobert P, Bardet V, Cornillet-Lefebvre
P, Hayflick JS, Prie N, Verdier F, Vanhaesebroeck B, Muller O,
Pesce F, Ifrah N, et al: Essential role for the p110delta isoform
in phosphoinositide 3-kinase activation and cell proliferation in
acute myeloid leukemia. Blood. 106:1063–1066. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mayer IA and Arteaga CL: The PI3K/AKT
pathway as a target for cancer treatment. Annu Rev Med. 67:11–28.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sui H, Pan SF, Feng Y, Jin BH, Liu X, Zhou
LH, Hou FG, Wang WH, Fu XL, Han ZF, et al: Zuo Jin Wan reverses
P-gp-mediated drug-resistance by inhibiting activation of the
PI3K/Akt/NF-κB pathway. BMC Complement Altern Med. 14:279–288.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Faes S and Dormond O: PI3K and AKT:
Unfaithful partners in cancer. Int J Mol Sci. 16:21138–21152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Warfel NA and Kraft AS: PIM kinase (and
Akt) biology and signaling in tumors. Pharmacol Ther. 151:41–49.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Radogna F, Dicato M and Diederich M:
Cancer-type-specific crosstalk between autophagy, necroptosis and
apoptosis as a pharmacological target. Biochem Pharmacol. 94:1–11.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Green DR and Llambi F: Cell death
signaling. Cold Spring Harb Perspect Biol. 7:a0060802015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ren C, Ren T, Yang K, Wang S, Bao X, Zhang
F and Guo W: Inhibition of SOX2 induces cell apoptosis and G1/S
arrest in Ewing's sarcoma through the PI3K/Akt pathway. J Exp Clin
Cancer Res. 35:44–57. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|