Introduction
Cholangiocarcinoma is the second most common type of
primary liver cancer (1). Over 90%
of cholangiocarcinoma patients have lost the opportunity for
curative surgical resection at the time of diagnosis (1). The current standard chemotherapeutic
regimen for advanced cholangiocarcinoma is systemic administration
of gemcitabine and cisplatin, but the survival benefit is far from
satisfactory. Cholangiocarcinoma patients have a poor prognosis
with a median survival of approximately one year (2). Therefore, it is urgently required to
seek therapeutic strategies that could enhance the efficacy of the
current chemotherapy regimen for the treatment of
cholangiocarcinoma.
In seeking potential agents, metformin
(dimethybiguanide), an oral biguanide drug used to treat type 2
diabetes (3), had attracted our
attention. It has long been recognized that insulin resistance is
associated with the risk for the development of several types of
human cancers (4). Moreover, the
results of a cohort study indicate that the use of metformin is
associated with a reduced incidence of cancer in patients with type
2 diabetes (5). The epidemiologic
studies have led to investigations of metformin as an anticancer
drug and the underlying mechanisms in cell culture and animal
models. Metformin has been shown to inhibit cell proliferation,
induce cell cycle arrest and promote apoptosis by activating the
AMP-activated protein kinase (AMPK) pathways (6). AMPK is a central cellular energy
sensor and a crucial factor in the interaction between metabolism
and cancer (6). Activation of AMPK
by metformin results in the inhibition of mammalian target of
rapamycin (mTOR) signaling pathways and stimulation of the p53/p21
axis (7). Several studies have
reported that metformin inhibits the growth of gastric cancer
(7), leukemia (8), prostate (9) and esophageal cancer (10), and hepatocellular carcinoma cells
(11). A recent study has
demonstrated that metformin also exhibits an antitumor effect
against cholangiocarcinoma cells (12). In addition, metformin was found to
synergize with 5-fluorouracil, epirubicin and cyclophosphamide to
suppress breast cancer cell growth (13), to enhance the efficacy of
5-fluorouracil to inhibit esophageal adenocarcinoma cell growth
(14), and to sensitize
intrahepatic cholangiocarcinoma cells to sorafenib, 5-fluorouracil
and As2O3 (15). However, it is unknown whether
metformin could also be used to strengthen the efficacy of
gemcitabine and cisplatin, the standard chemotherapeutic regimen
for cholangiocarcinoma.
Materials and methods
Cell culture
Human cholangiocarcinoma cell lines, RBE and
HCCC-9810, were obtained from the Chinese Academy of Sciences Cell
Bank (Shanghai, China). Cells were cultured at 37°C in Dulbecco's
modified Eagle's medium (DMEM) (Gibco-BRL, Grand Island, NY, USA)
supplemented with 10% fetal bovine serum.
Reagents and antibodies
Gemcitabine and cisplatin were provided by Jinan
Trio PharmaTech Co., Ltd. (Jinan, China). Metformin was purchased
from Sigma-Aldrich (Shanghai, China). Metformin, gemcitabine and
cisplatin were dissolved in water to make stock solutions of 100, 5
and 5 mM, respectively. Antibodies (Abs) against AMPK,
phosphorylated AMPK (pAMPK and Thr172), caspase-3 and cyclin D1
were purchased from Cell Signaling Technology (Beverly, MA, USA).
Abs against p21Waf1, p27kip1 and anti-β-actin
were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). An
anti-Ki-67 Ab was purchased from Abcam Inc. (Cambridge, MA,
USA).
Cell viability assay
The Cell Counting Kit-8 (CCK-8) (Dojindo Molecular
Technologies, Inc., Beijing, China) was used to determine cell
viability. Cells were seeded at 1×103 cells/well in
96-well plates. At different time points after treatments, the
culture medium was replaced with 100 µl of fresh medium containing
10 µl of CCK-8 solution. Cells were further incubated for 2 h at
37°C, and the optical density (OD) at 450 nm was measured.
Untreated cells served as controls. The proliferation inhibition
rate (%) was calculated according to the formula: (Control OD value
- experimental OD value/control OD value) × 100.
Flow cytometry for assessing cell
cycle distribution and apoptosis
Cells were seeded at 5×105 cells/well in
6-well plates, incubated with treatment reagents, and harvested at
indicated time points. The percentages of cells at the G2/M, S and
G0/G1 phases were determined using a cell cycle detection kit (BD
Biosciences, Beijing, China) by a Beckman Coulter EPICS Altra II
cytometer (Beckman Coulter, Brea, CA, USA). In addition,
1×105 cells were suspended in 100 µl binding buffer,
incubated with 5 µl of Annexin V and 5 µl of propidium iodide (PI)
for 15 min at room temperature in the dark, according to the
manufacturer's instructions (BD Biosciences, San Jose, CA, USA).
Then the cells were subjected to flow cytometry to measure the
apoptosis rate (%).
Measurement of caspase activity
The activity of caspase-3 in cell lysates was
determined using the CaspACE™ Assay system (G7220; Promega Corp.,
Madison, WI, USA).
Western blot analysis
The western blotting methods have been previously
described (16,17). Cells were homogenized in protein
lysate buffer, and debris was removed by centrifugation. Protein
concentrations were determined. Lysates were resolved on sodium
dodecyl sulfate-polyacrylamide gels and electrophoretically
transferred to polyvinylidene difluoride membranes. The membranes
were blocked, and incubated overnight with primary Abs, and
subsequently with secondary horseradish peroxidase-conjugated Abs.
They were developed with 5-bromo-4-chloro-3-indolyl phosphate
(BCIP)/nitro blue tetrazolium (NBT) (Tiangen Biotech Co., Ltd.,
Beijing, China).
Animal experimental protocols
Animal experiments were performed according to the
guidelines of the Animal Ethics Committee of Shandong University
(Jinan, China). Six- to 8-week old male nude BALB/c mice (H-2b)
were obtained from the Animal Research Center, Shandong University
(China). Mice were maintained under specific pathogen-free
conditions using a laminar airflow rack and had continuous free
access to sterilized food and autoclaved water. RBE cells
(4×106) were subcutaneously injected into the flanks of
mice, which were monitored for appearance of palpable tumors. Tumor
volume (V) was estimated by the formula: V = π/6 ×
a2 × b, where a is the short axis,
and b the long axis. When tumors reached ~150 mm3
in volume, mice were randomly assigned to 4 treatment groups (each
group had 8 mice): control, metformin, gemcitabine/cisplatin and
metformin plus gemcitabine/cisplatin. The mice in the metformin
group received intraperitoneal injections of metformin at a dose of
2 mg/kg body weight 5 times a week for 28 days, while the mice in
the control group received intraperitoneal injections of normal
saline at the same volume and frequency as metformin solution. The
mice in the gemcitabine/cisplatin group received biweekly
intraperitoneal injections of gemcitabine at a dose of 100 mg/kg
body weight, and weekly injections of cisplatin at a dose of 4
mg/kg body weight (18). The mice
in the metformin plus gemcitabine/cisplatin group received both
injections of metformin and gemcitabine/cisplatin as described
above. Thirty-five days after commencement of treatments, the mice
were sacrificed. Samples were harvested for analysis.
In situ Ki-67 proliferation index
Formalin-fixed tumor specimens were transferred to
70% ethanol, and subsequently paraffin-embedded and sectioned.
Tumor sections were blocked with 3% BSA for 2 h, and incubated with
an anti-Ki-67 Ab at 4°C overnight. They were subsequently incubated
for 30 min with the appropriate secondary Ab using the
Ultra-Sensitive TMS-P kit (Zhongshan Co., Ltd., Beijing, China),
and immunoreactivity was developed with SIGMAFAST
3,3′-diaminobenzidine tetrahydrochloride (DAB) and CoCl2
enhancer tablets (Sigma-Aldrich, Shanghai, China). Sections were
counterstained with hematoxylin, mounted and examined by
microscopy. The Ki-67-positive cells were counted in 10 randomly
selected ×400 high-power fields under microscopy. The proliferation
index was calculated according to the following formula: Number of
Ki-67-positive cells/total cell count × 100%.
In situ detection of apoptotic
cells
Tumor sections were stained with the terminal
deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) (Roche,
Shanghai, China). The TUNEL-positive cells were counted in 20
randomly selected ×200 high-power fields under microscopy. The
apoptosis index was calculated according to the following formula:
Number of apoptotic cells/total number of nucleated cells ×
100%.
Statistical analysis
The data are expressed as mean values ± standard
deviation. Comparisons were carried out with one-way analysis of
variance (ANOVA) followed by Dunnet's test. P<0.05 was
considered to indicate a statistically significant result.
Results
Metformin inhibits the proliferation
of cholangiocarcinoma cells
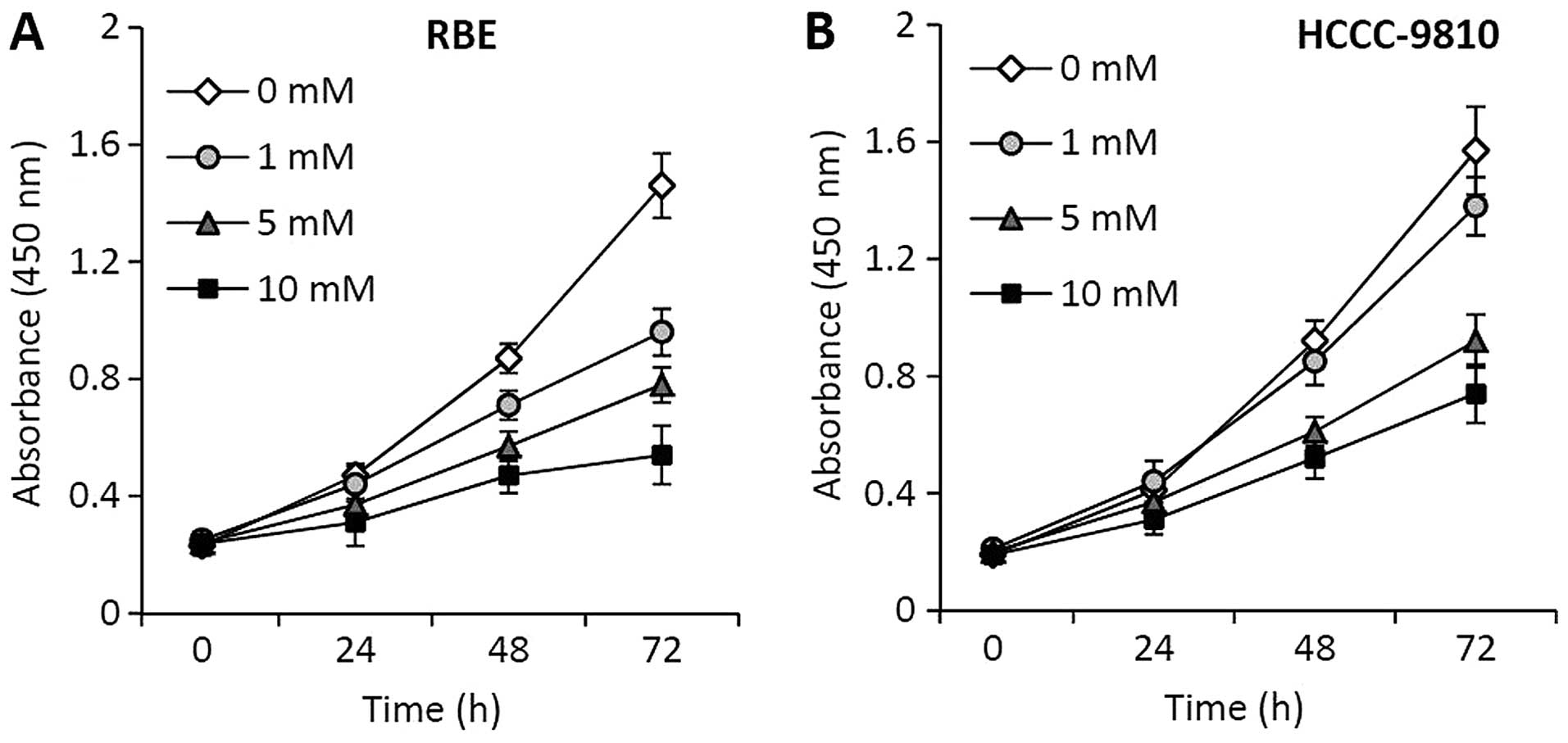
RBE and HCCC-9810 cells were incubated with
metformin at various concentrations, and their viability was
measured at 24, 48 and 72 h after incubation. Metformin showed a
strong inhibitory effect on the proliferation of RBE (Fig. 1A) and HCCC-9810 (Fig. 1B) cells in a time- and
concentration-dependent manner.
Metformin induces cell cycle
arrest
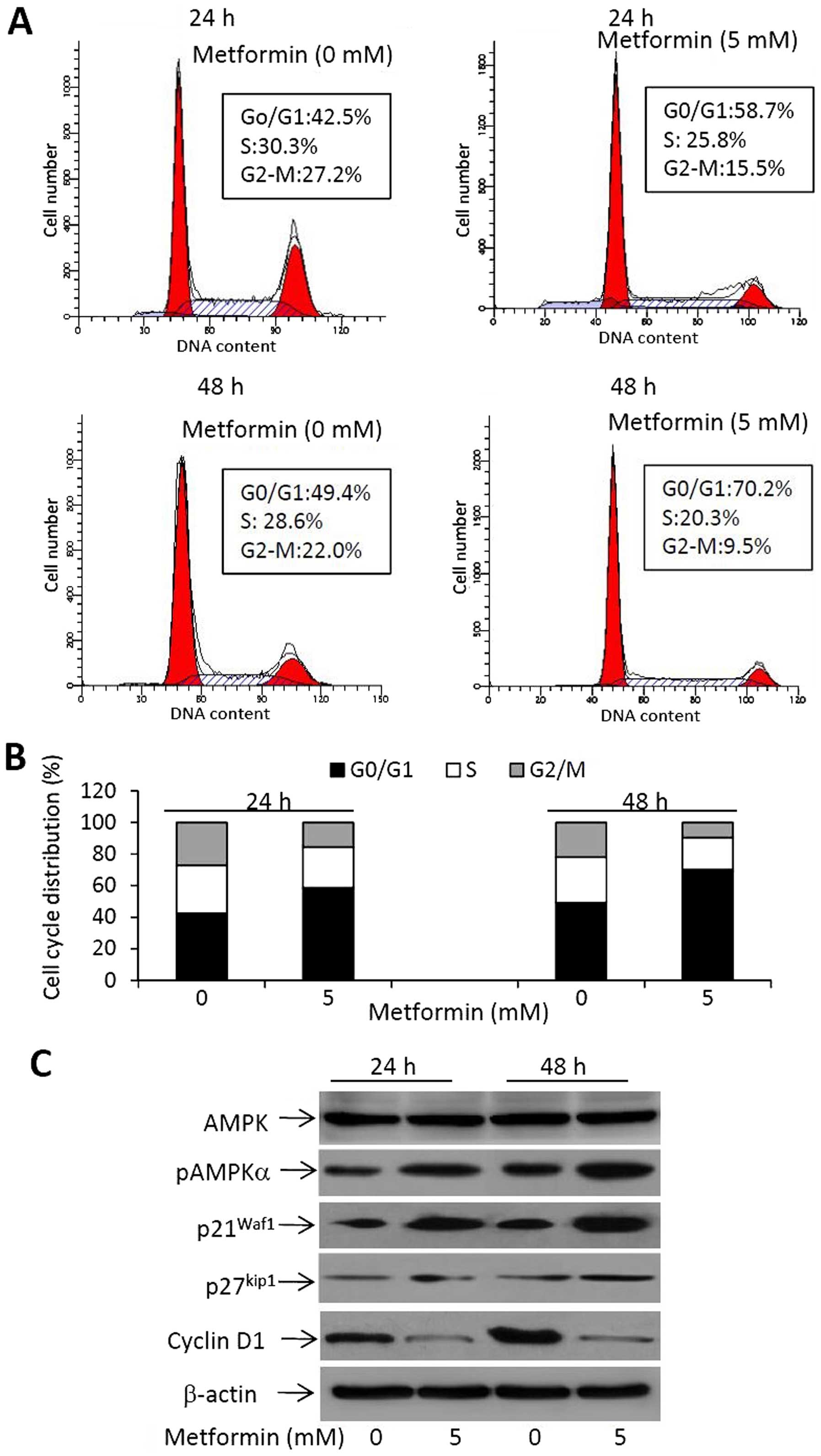
To further investigate the effects of metformin on
cell proliferation, cell cycle progression was examined by flow
cytometry. RBE cells were incubated with 0 or 5 mM of metformin for
24 or 48 h. As shown in Fig. 2A,
metformin treatment induced an increasing number of cells that
accumulated in the G0/G1 phase. Specifically, 58.7% of cells were
arrested at the G0/G1 phase 24 h after treatment, while 70.2% of
cells were arrested at the G0/G1 phase 48 h after treatment,
compared with 42.5 and 49.4% for the untreated cells, respectively
(Fig. 2A). This finding was
accompanied by reductions in the percentages of cells in the S and
G2/M phases (Fig. 2B). The results
suggest that metformin inhibits the cell cycle progression from
G0/G1 into S phases in cholangiocarcinoma cells.
We next investigated the expression of molecules
involved in the action of metformin on cell proliferation by
western blot analysis. As shown in Fig.
2C, metformin treatment induced an increased expression of
pAMPK, while the expression of AMPK remained unchanged, indicating
that metformin increased the activation of the AMPK pathway. The
downstream factors, p21Waf1 and p27kip1, were
also upregulated upon metformin treatment (Fig. 2C). Cyclin D1, a key protein required
for the progression through the G1 phase of the cell cycle, was
markedly downregulated in metformin-treated cells (Fig. 2C).
Gemcitabine and cisplatin inhibit cell
proliferation and induce apoptosis
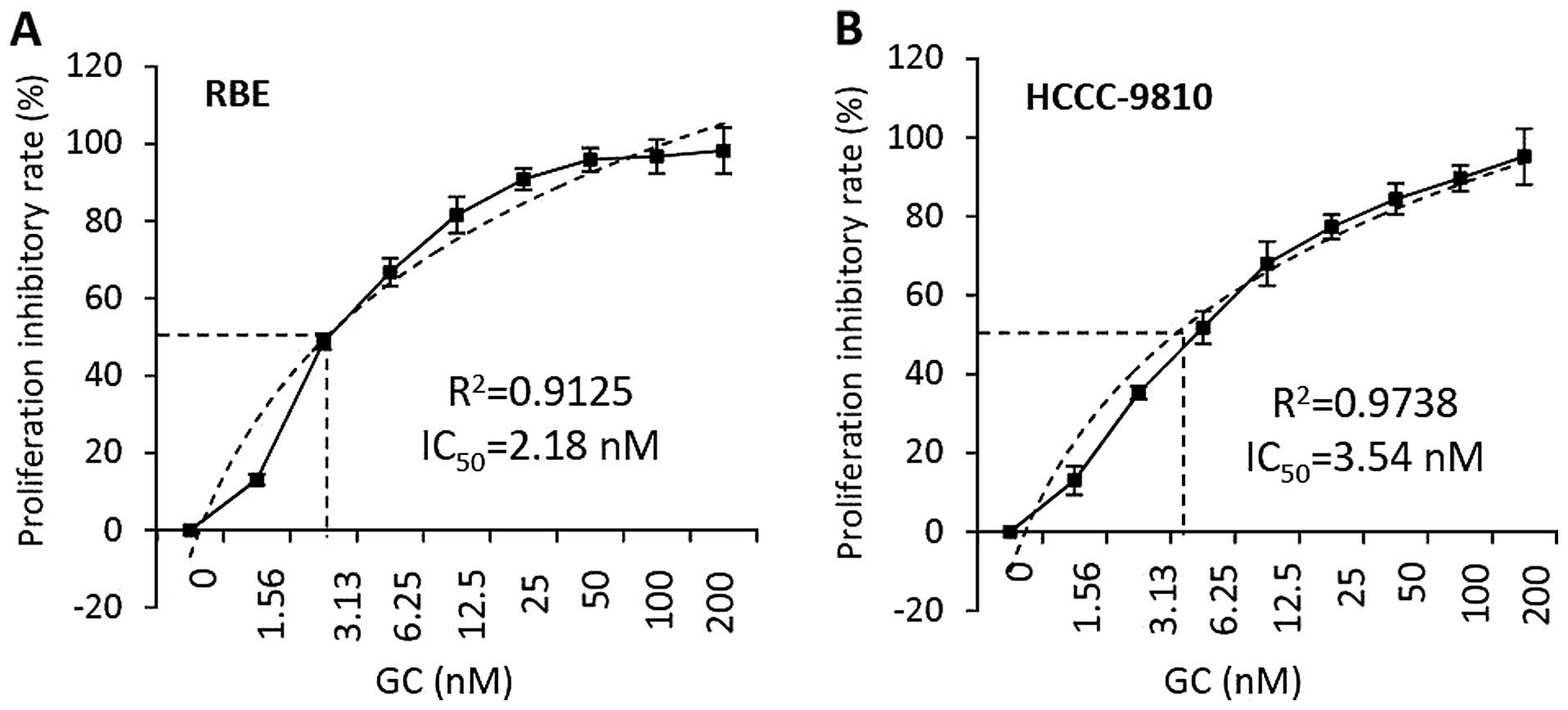
RBE and HCCC-9810 cells were incubated with the
combination of gemcitabine and cisplatin (GC) for 48 h. The
concentrations of both gemcitabine and cisplatin ranged from 1.56
to 200 nM based on independent dose finding experiments. Cell
viability was measured and the proliferation inhibitory rates were
calculated. As shown in Fig. 3, GC
showed proliferation inhibitory effects against the two cell lines
in a dose-dependent manner. With a simple linear regression
analysis, the concentration of drugs resulting in 50% maximal
proliferation inhibition (IC50) was calculated to be
2.18 nM for RBE cells, which were incubated with GC at 48 h
(Fig. 3A); while the
IC50 for HCCC-9810 cells was 3.54 nM (Fig. 3B).
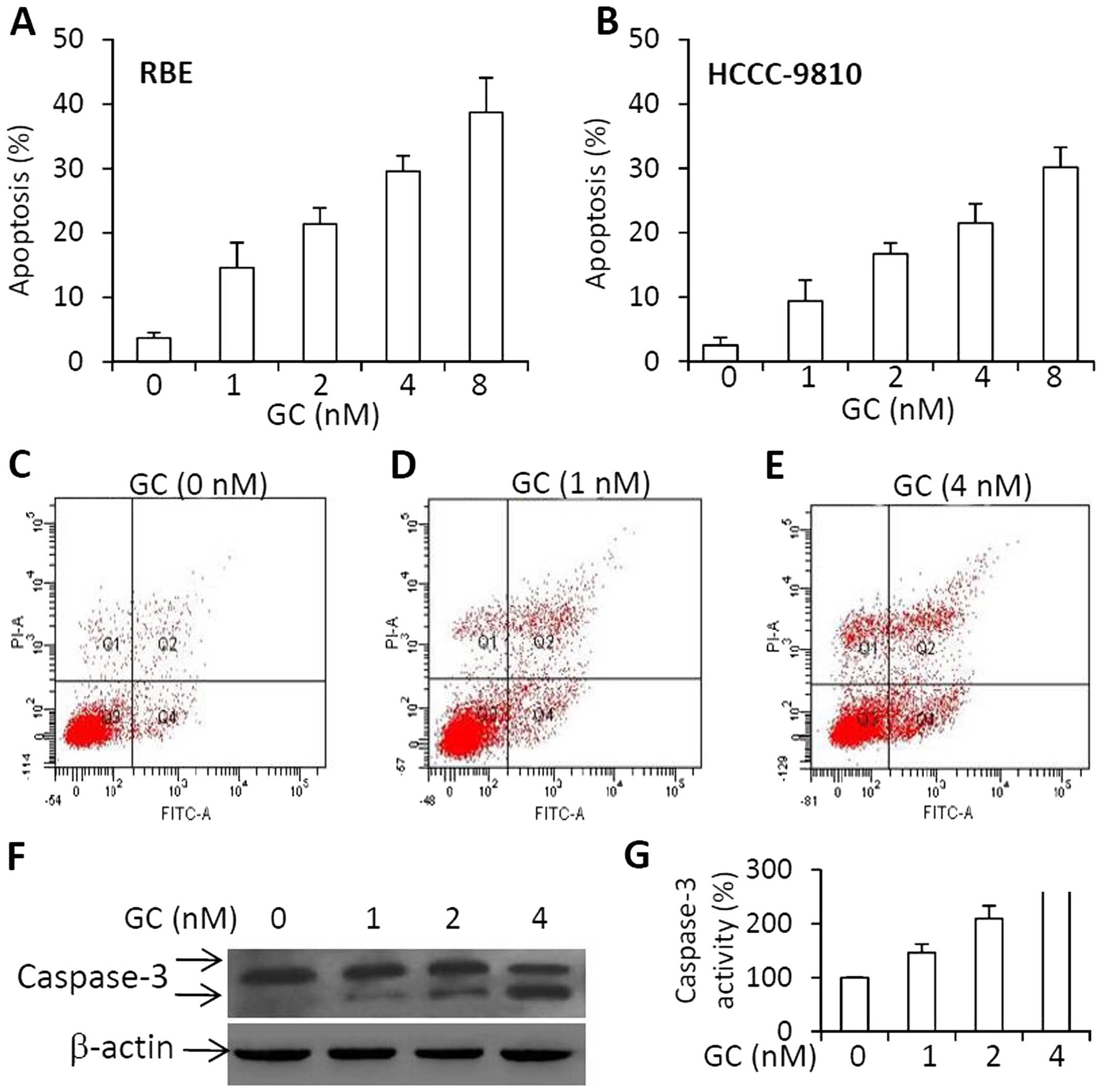
We next demonstrated whether GC could also induce
cell apoptosis. As shown in Fig. 4A and
B, GC induced apoptosis of RBE and HCCC-9810 cells in a
dose-dependent manner. The representative histograms of flow
cytometry showed that the apoptosis rates of RBE cells were 3.5,
15.1 or 25.6% when they underwent a 48-h incubation with GC at
concentrations at 0 nM (Fig. 4C), 1
nM (Fig. 4D) or 4 nM (Fig. 4E), respectively. The increased
expression of cleaved caspase-3 (Fig.
4F) and activity of caspase-3 (Fig.
4G) correlated well with the cell apoptosis rates. The results
indicate that GC induced the apoptosis of RBE cells through the
caspase-dependent pathways in accordance with previous studies
(19,20).
Metformin increases the effects of
gemcitabine and cisplatin on cholangiocarcinoma cells in vitro
We next examined whether metformin could enhance the
activities of gemcitabine and cisplatin against cholangiocarcinoma
cells. Based on the above results, a concentration of GC at 1 nM
below IC50 for both cell lines and a concentration of
metformin at 5 mM were selected for the following in vitro
experiments. Cells were incubated with GC, metformin or their
combination for 48 h. Cell viability was measured and the
proliferation inhibitory rates were calculated.
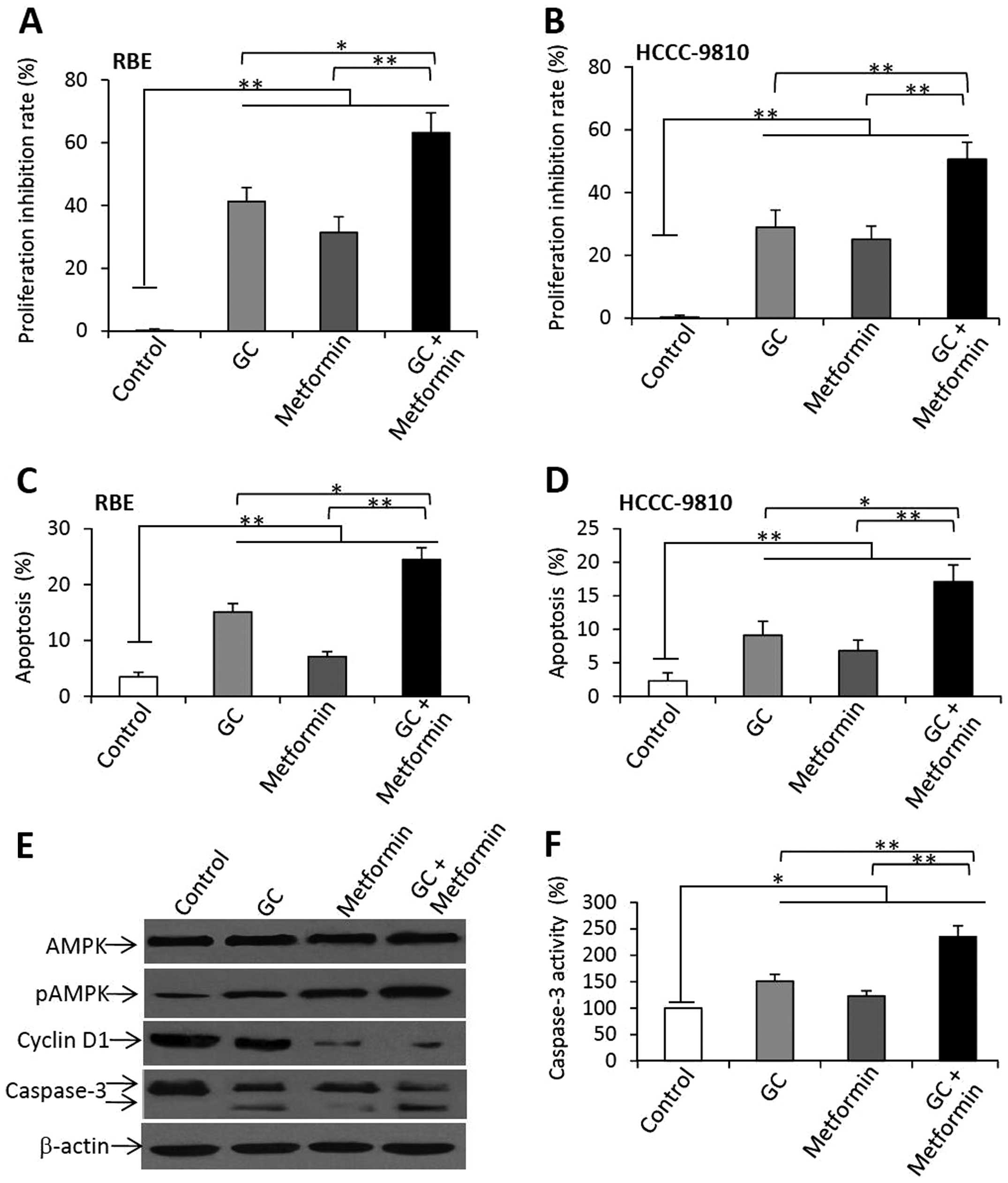
As shown in Fig. 5A
(RBE) and B (HCCC-9810), a 48-h incubation with GC caused a 41.2%
reduction in the viability of RBE cells, and a 28.9% reduction in
the viability of HCCC-9810 cells, compared to the respective
untreated cells. Metformin therapy reduced the viability of RBE
cells by 31.4%, and that of HCCC-9810 cells by 24.7%, compared to
the respective untreated controls. However, the combination of GC
and metformin further reduced the viability of RBE and HCCC-9810
cells by 63.2 and 50.6%, respectively. To investigate whether the
effects of GC and metformin are additive or synergistic, we
calculated the value of the coefficient of drug interaction (CDI)
as previously described (21). CDI
was calculated according to the formula: CDI = AB/(A × B), where AB
is the ratio of the cell viability index of the combination group
to the control group; A or B is the ratio of the viability index of
the respective GC or metformin group to the control group. A value
of CDI less than (<), equal to (=) or greater than (>) 1
indicates that the drugs are synergistic, additive or antagonistic,
respectively (21). The values of
CDI in the RBE and HCCC-9810 cells treated with GC and metformin
were all <1, indicating that the two agents had synergistic
effects in inhibiting the viability of the cells.
Similarly, GC and metformin also significantly
increased the apoptosis of the RBE (Fig. 5C) and HCCC-9810 (Fig. 5D) cells, and the combination of GC
and metformin significantly induced higher apoptosis rates for both
the RBE and HCCC-9810 cells than the rates noted in the untreated
controls as well as for GC or metformin treatments.
The above RBE cells were further analyzed by western
blot analysis to detect the alterations of key molecules involved
in cell proliferation and apoptosis. GC, metformin or their
combination had no significant effects on the expression of AMPK.
GC and metformin upregulated the expression of pAMPK, and their
combination further increased the expression of pAMPK (Fig. 5E). Both GC and metformin reduced the
expression of cyclin D1, and the combination markedly decreased the
expression of cyclin D1. Both GC and metformin significantly
increased the cleavage of caspase-3 in the RBE cells, and their
combination increased to an even greater extent the cleavage of
caspase-3 in the RBE cells (Fig.
5E). The results of caspase-3 expression were supported by that
of caspase-3 activities (Fig.
5F).
Metformin enhances the efficacy of
gemcitabine and cisplatin to suppress tumor growth in vivo
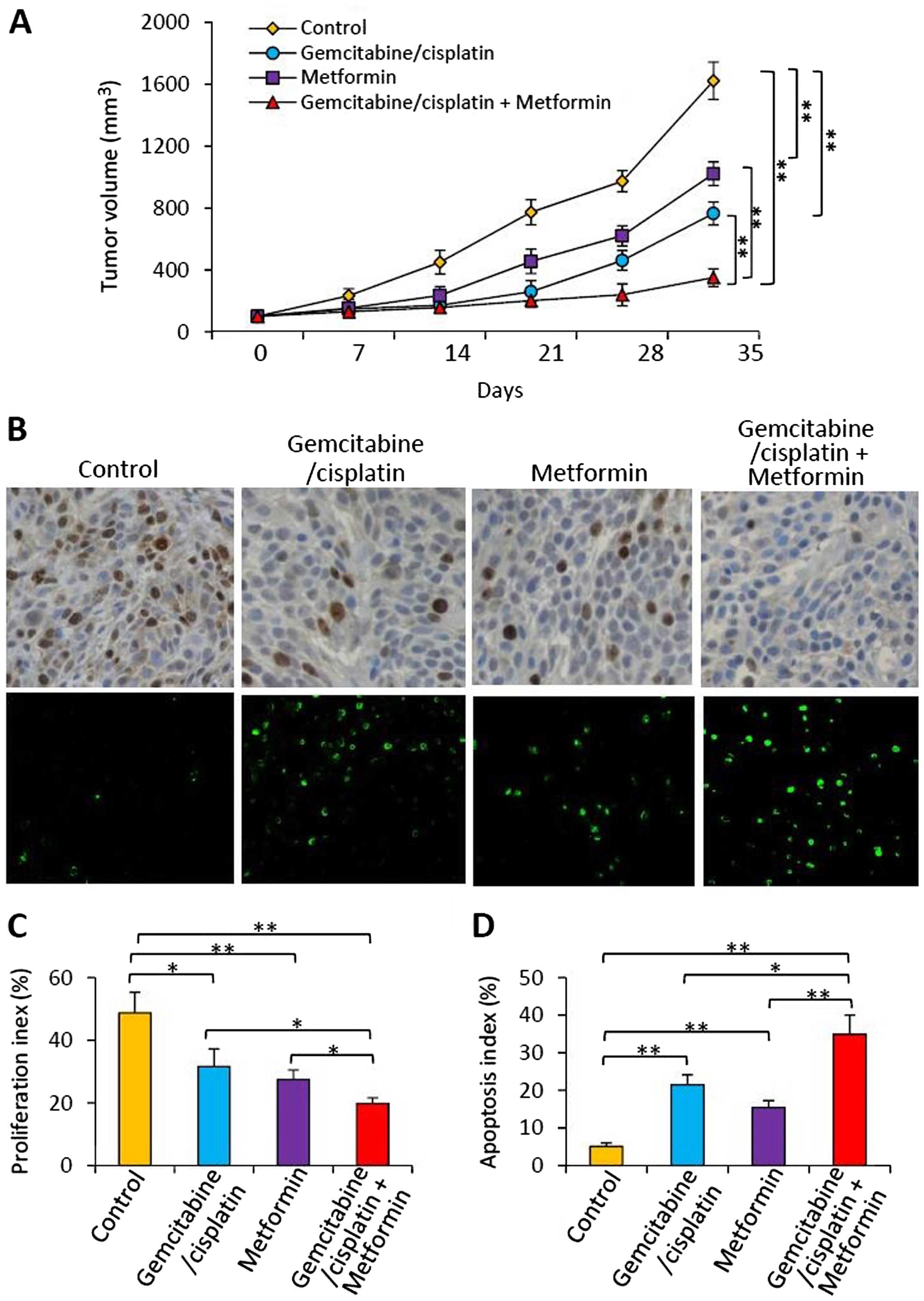
Subcutaneous RBE tumors were established in the
flanks of mice. When the tumors reached ~150 mm3, the
mice were randomly assigned to 4 treatment groups as described in
Materials and methods. As shown in Fig.
6A, the control tumors grew markedly fast reaching 1613.5±109.1
mm3 in volume 35 days after treatment. In contrast,
tumors in the gemcitabine/cisplatin-treated mice were significantly
smaller, reaching only 758.4±81.2 mm3 in volume.
Metformin therapy also significantly reduced tumor volume
(1015.3±72.6 mm3), compared with the control tumors.
Tumors treated with the combination of gemcitabine/cisplatin and
metformin reached only 349.8±57.1 mm3, and were
significantly smaller than the volumes of the control tumors, and
the tumors treated with either gemcitabine/cisplatin or metformin
(Fig. 6A). Using the method
mentioned above, we calculated the value of CDI, which was 0.76,
indicating that gemcitabine/cisplatin and metformin had a
synergistic effect in suppressing the growth of the RBE tumors.
Cell proliferation and apoptosis in
situ
Tumor sections prepared from the above tumors were
stained with an Ab which detects the cell proliferation marker
Ki-67, or the TUNEL agent to detect apoptotic cells. There were
fewer Ki-67-positive and more apoptotic cells in the tumors treated
with gemcitabine/cisplatin or metformin, when compared with the
control tumors (Fig. 6B). The
combination therapy resulted in even fewer Ki-67-positive and more
apoptotic cells (Fig. 6B). Cells
expressing Ki-67 were counted to calculate the proliferation index
(Fig. 6C) and TUNEL-positive cells
were counted to record the apoptosis index (Fig. 6D). As shown in Fig. 6C, gemcitabine/cisplatin and
metformin therapies resulted in a significant reduction in the
proliferation index by 35.1 or 43.5%, respectively, compared with
the controls. The combination therapy resulted in a highly
significant reduction in the proliferation index by 59.3%
(P<0.001) compared to the controls, and a significant reduction
compared to either gemcitabine/cisplatin or metformin treatment
(Fig. 6C). As shown in Fig. 6D, gemcitabine/cisplatin therapy
significantly increased the apoptosis index by 3-fold, and
metformin therapy by 2-fold, compared with the controls. The
apoptosis index of the tumors treated with the combinational
therapy was significantly higher by >6-fold than that of the
control tumors, and also significantly higher than either
gemcitabine/cisplatin or metformin treatment (Fig. 6D).
Discussion
Cholangiocarcinoma, derived from the epithelial
cells of biliary ducts, accounts for ~3% of all gastrointestinal
cancers. It is classified as intrahepatic, perihilar and
extrahepatic cholangiocarcinoma (22). The incidence of intrahepatic
cholangiocarcinoma has been increasing over the past 3 decades, and
most patients have locally advanced or distal metastatic diseases
at the time of presentation and lose the opportunity of curative
surgery (10). The development of
novel therapies, particularly molecular-targeted drugs, for
cholangiocarcinoma is lagging behind other cancers (23). Conventional chemotherapy continues
to play a critical role in the clinical management of
cholangiocarcinoma. However, the most commonly used chemotherapy
regimen, the combination of gemcitabine and cisplatin, only
provides a very limited beneficial effect in prolonging the
survival of patients (18).
Therefore, the development of therapeutic strategies that enhance
the efficacy of gemcitabine and cisplatin in combating
cholangiocarcinoma is urgently needed.
Metformin is one of the most commonly used drugs for
the management of type 2 diabetes worldwide. Metformin was
recommended for the treatment of diabetes following a successful
clinical trial in 1957 (24). Due
to its superior safety profile, metformin has eventually become the
first-line treatment for type 2 diabetes and it is now featured on
the World Health Organization's list of essential medicines for
both adults and children (WHO Model Lists of Essential Medicines
2015, World Health Organization). Notably, the therapeutic
potential of metformin has recently extended far beyond its
prescribed use as an anti-diabetic drug. There is a rapidly growing
body of literature demonstrating an effective role for metformin in
treating cancer and cardiovascular disease, delaying the aging
process and modulating microbiota to promote health (25).
The AMPK signaling pathway has been widely studied
in metabolic disorders and an increasing number of studies also
suggest that it plays a potential role in cancer cell biology
(26,27). AMPK activation causes cell cycle
arrest associated with stabilization of the cyclin-dependent kinase
inhibitor p21Waf1 and p27kip1 (28). The AMPK system is the key target for
metformin, and activation of AMPK contributes largely to its
anti-diabetic action (29).
Metformin is able to induce the activation of AMPK in several types
of cancer cells (12–14,30,31).
Upregulation of p21Waf1 and p27kip1 plays an
important role in the inhibitory effects of metformin (10,31).
In agreement, the present study demonstrated that metformin
treatment led to increased expression levels of p-AMPKα,
p21Waf1 and p27kip1, indicating activation of
the AMPK pathway, in cholangiocarcinoma cells.
Metformin has been reported to induce cell cycle
arrest at the G0/G1 phase by downregulating cyclin D1 in cells from
gastric cancer (32), esophageal
adenocarcinoma (10),
hepatocellular carcinoma (33), as
well as cholangiocarcinoma (12).
The present results have shown that a large proportion of
metformin-treated cells were arrested at the G0/G1 phase.
Consistently, cyclin D1, a key protein required for progression
through the G1 phase of the cell cycle (34), was markedly downregulated following
metformin treatment.
Given the fact that metformin exhibits anticancer
activities with a superior safety profile, the combination of
metformin with cytotoxic chemotherapeutic agents is expected to be
a promising strategy for cancer treatment. Diabetic patients with
breast cancer taking metformin and undergoing neoadjuvant
chemotherapy had a 3-fold higher pathologic complete response rate
than those not taking metformin (35). An epidemiological study also showed
that treatment with metformin was significantly associated with a
60% reduction in the risk of intrahepatic cholangiocarcinoma in
diabetic patients (36). However,
metformin did not show survival benefits for cholangiocarcinoma
patients with diabetes in a recent investigation, but a very small
number of patients were enrolled in this preliminary study
(37). Metformin has been shown to
increase cisplatin-induced cytotoxicity and inhibit ovarian and
gastric tumor growth (7,38). Cisplatin triggered activation of the
AMPK pathway in glioma cells (38),
and metformin enhanced the effect of cisplatin by inducing AMPK
phosphorylation (7). In accordance,
we showed here that metformin synergized with gemcitabine and
cisplatin to induce apoptosis and inhibit the proliferation of
cholangiocarcinoma cells by inducing the phosphorylation of AMPK,
and sequential downregulation of cyclin D1. Gemcitabine and
cisplatin are well known cytotoxic drugs used to treat a wide
variety of cancers, and their combination chemotherapy was
associated with a significant survival advantage for the treatment
of patients with advanced biliary cancer (2). Gemcitabine inhibits the processes
required for DNA synthesis (39),
while cisplatin interacts with DNA to form DNA adducts, leading to
the activation of apoptosis (40).
Their combination has been shown to activate caspase-3 (19,20),
in accordance with our results that gemcitabine and cisplatin
therapy markedly increased the apoptosis of cholangiocarcinoma
cells by activating caspase-3.
In summary, conventional chemotherapeutic agents
often lead to severe side-effects, such as damage to the intestine
and hematologic suppression. However, metformin has exhibited only
mild side-effects, such as abdominal discomfort, a metallic taste
and mild anorexia, and these symptoms are reversible after dose
reduction or discontinuation of the drug (41). Metformin has been demonstrated to be
a very safe drug for over half a century since it was approved for
diabetes treatment in 1957 (24).
Therefore, combining metformin with gemcitabine and cisplatin may
be advantageous, as metformin could be employed to enhance the
anticancer activities and reduce the dose of chemotherapeutic
agents to spare patients the side-effects without impairing the
antitumor efficacy. Further investigation of this possibility is
warranted.
Acknowledgements
The present study was supported by the Shandong
Provincial Natural Scientific Research Foundation (BS2015YY024 and
ZR2014HM099), the Shandong Provincial Scientific and Technology
Development Program (2014GGB14041 and 2015GGB14242), and the
Shandong Province Medical and Health Scientific and Technology
Development Program (2014WS0093, 2014WS0095 and 2014WS0096).
References
|
1
|
Bridgewater J, Galle PR, Khan SA, Llovet
JM, Park JW, Patel T, Pawlik TM and Gores GJ: Guidelines for the
diagnosis and management of intrahepatic cholangiocarcinoma. J
Hepatol. 60:1268–1289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Valle J, Wasan H, Palmer DH, Cunningham D,
Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira
SP, et al: ABC-02 Trial Investigators: Cisplatin plus gemcitabine
versus gemcitabine for biliary tract cancer. N Engl J Med.
362:1273–1281. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ben Sahra I, Laurent K, Loubat A,
Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le
Marchand-Brustel Y and Bost F: The antidiabetic drug metformin
exerts an antitumoral effect in vitro and in vivo through a
decrease of cyclin D1 level. Oncogene. 27:3576–3586. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Simon D and Balkau B: Diabetes mellitus,
hyperglycaemia and cancer. Diabetes Metab. 36:182–191. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Libby G, Donnelly LA, Donnan PT, Alessi
DR, Morris AD and Evans JM: New users of metformin are at low risk
of incident cancer: A cohort study among people with type 2
diabetes. Diabetes Care. 32:1620–1625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He H, Ke R, Lin H, Ying Y, Liu D and Luo
Z: Metformin, an old drug, brings a new era to cancer therapy.
Cancer J. 21:70–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu G, Fang W, Xia T, Chen Y, Gao Y, Jiao
X, Huang S, Wang J, Li Z and Xie K: Metformin potentiates rapamycin
and cisplatin in gastric cancer in mice. Oncotarget. 6:12748–12762.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scotland S, Saland E, Skuli N, de Toni F,
Boutzen H, Micklow E, Sénégas I, Peyraud R, Peyriga L, Théodoro F,
et al: Mitochondrial energetic and AKT status mediate metabolic
effects and apoptosis of metformin in human leukemic cells.
Leukemia. 27:2129–2138. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Giannoni E, Taddei ML, Morandi A, Comito
G, Calvani M, Bianchini F, Richichi B, Raugei G, Wong N, Tang D, et
al: Targeting stromal-induced pyruvate kinase M2 nuclear
translocation impairs oxphos and prostate cancer metastatic spread.
Oncotarget. 6:24061–24074. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fujihara S, Kato K, Morishita A, Iwama H,
Nishioka T, Chiyo T, Nishiyama N, Miyoshi H, Kobayashi M, Kobara H,
et al: Antidiabetic drug metformin inhibits esophageal
adenocarcinoma cell proliferation in vitro and in vivo. Int J
Oncol. 46:2172–2180. 2015.PubMed/NCBI
|
|
11
|
Hsieh SC, Tsai JP, Yang SF, Tang MJ and
Hsieh YH: Metformin inhibits the invasion of human hepatocellular
carcinoma cells and enhances the chemosensitivity to sorafenib
through a downregulation of the ERK/JNK-mediated NF-κB-dependent
pathway that reduces uPA and MMP-9 expression. Amino Acids.
46:2809–2822. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fujimori T, Kato K, Fujihara S, Iwama H,
Yamashita T, Kobayashi K, Kamada H, Morishita A, Kobara H, Mori H,
et al: Antitumor effect of metformin on cholangiocarcinoma: In
vitro and in vivo studies. Oncol Rep. 34:2987–2996. 2015.PubMed/NCBI
|
|
13
|
Soo JS, Ng CH, Tan SH, Malik RA, Teh YC,
Tan BS, Ho GF, See MH, Taib NA, Yip CH, et al: Metformin synergizes
5-fluorouracil, epirubicin, and cyclophosphamide (FEC) combination
therapy through impairing intracellular ATP production and DNA
repair in breast cancer stem cells. Apoptosis. 20:1373–1387. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Honjo S, Ajani JA, Scott AW, Chen Q,
Skinner HD, Stroehlein J, Johnson RL and Song S: Metformin
sensitizes chemotherapy by targeting cancer stem cells and the mTOR
pathway in esophageal cancer. Int J Oncol. 45:567–574.
2014.PubMed/NCBI
|
|
15
|
Ling S, Feng T, Ke Q, Fan N, Li L, Li Z,
Dong C, Wang C, Xu F, Li Y, et al: Metformin inhibits proliferation
and enhances chemosensitivity of intrahepatic cholangiocarcinoma
cell lines. Oncol Rep. 31:2611–2618. 2014.PubMed/NCBI
|
|
16
|
He C, Dong X, Zhai B, Jiang X, Dong D, Li
B, Jiang H, Xu S and Sun X: MiR-21 mediates sorafenib resistance of
hepatocellular carcinoma cells by inhibiting autophagy via the
PTEN/Akt pathway. Oncotarget. 6:28867–28881. 2015.PubMed/NCBI
|
|
17
|
Zhu H, Mi Y, Jiang X, Zhou X, Li R, Wei Z,
Jiang H, Lu J and Sun X: Hepatocyte nuclear factor 6 inhibits the
growth and metastasis of cholangiocarcinoma cells by regulating
miR-122. J Cancer Res Clin Oncol. 142:969–980. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rizvi S and Gores GJ: Pathogenesis,
diagnosis, and management of cholangiocarcinoma. Gastroenterology.
145:1215–1229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee EK, Jinesh GG, Laing NM, Choi W,
McConkey DJ and Kamat AM: A Smac mimetic augments the response of
urothelial cancer cells to gemcitabine and cisplatin. Cancer Biol
Ther. 14:812–822. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma Y, Yu WD, Trump DL and Johnson CS:
1,25D3 enhances antitumor activity of gemcitabine and
cisplatin in human bladder cancer models. Cancer. 116:3294–3303.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Ma Y, Jiang H, Zhu H, Liu L, Sun
B, Pan S, Krissansen GW and Sun X: Overexpression of von
Hippel-Lindau protein synergizes with doxorubicin to suppress
hepatocellular carcinoma in mice. J Hepatol. 55:359–368. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Blechacz B, Komuta M, Roskams T and Gores
GJ: Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev
Gastroenterol Hepatol. 8:512–522. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Erice O, Merino-Azpitarte M, Arbelaiz A,
Gutierrez-Larranaga M, Jiménez-Agüero R, Perugorria MJ, Bujanda L
and Banales JM: Molecular mechanisms of cholangiocarcinogenesis:
New potential targets for therapy. Curr Drug Targets. 16:12015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nattrass M and Alberti KG: Biguanides.
Diabetologia. 14:71–74. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pryor R and Cabreiro F: Repurposing
metformin: An old drug with new tricks in its binding pockets.
Biochem J. 471:307–322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vakana E, Altman JK and Platanias LC:
Targeting AMPK in the treatment of malignancies. J Cell Biochem.
113:404–409. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hardie DG, Ross FA and Hawley SA:
AMP-activated protein kinase: A target for drugs both ancient and
modern. Chem Biol. 19:1222–1236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liang J, Shao SH, Xu ZX, Hennessy B, Ding
Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al:
The energy sensing LKB1-AMPK pathway regulates p27kip1
phosphorylation mediating the decision to enter autophagy or
apoptosis. Nat Cell Biol. 9:218–224. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Del Barco S, Vazquez-Martin A, Cufí S,
Oliveras-Ferraros C, Bosch-Barrera J, Joven J, Martin-Castillo B
and Menendez JA: Metformin: Multi-faceted protection against
cancer. Oncotarget. 2:896–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ding L, Liang G, Yao Z, Zhang J, Liu R,
Chen H, Zhou Y, Wu H, Yang B and He Q: Metformin prevents cancer
metastasis by inhibiting M2-like polarization of tumor associated
macrophages. Oncotarget. 6:36441–36455. 2015.PubMed/NCBI
|
|
31
|
Rocha GZ, Dias MM, Ropelle ER,
Osório-Costa F, Rossato FA, Vercesi AE, Saad MJ and Carvalheira JB:
Metformin amplifies chemotherapy-induced AMPK activation and
antitumoral growth. Clin Cancer Res. 17:3993–4005. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kato K, Gong J, Iwama H, Kitanaka A, Tani
J, Miyoshi H, Nomura K, Mimura S, Kobayashi M, Aritomo Y, et al:
The antidiabetic drug metformin inhibits gastric cancer cell
proliferation in vitro and in vivo. Mol Cancer Ther. 11:549–560.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Miyoshi H, Kato K, Iwama H, Maeda E,
Sakamoto T, Fujita K, Toyota Y, Tani J, Nomura T, Mimura S, et al:
Effect of the anti-diabetic drug metformin in hepatocellular
carcinoma in vitro and in vivo. Int J Oncol. 45:322–332.
2014.PubMed/NCBI
|
|
34
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiralerspong S, Palla SL, Giordano SH,
Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi
GN and Gonzalez-Angulo AM: Metformin and pathologic complete
responses to neoadjuvant chemotherapy in diabetic patients with
breast cancer. J Clin Oncol. 27:3297–3302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chaiteerakij R, Yang JD, Harmsen WS,
Slettedahl SW, Mettler TA, Fredericksen ZS, Kim WR, Gores GJ,
Roberts RO, Olson JE, et al: Risk factors for intrahepatic
cholangiocarcinoma: Association between metformin use and reduced
cancer risk. Hepatology. 57:648–655. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang Z, Zhang X, Roberts RO, Roberts LR
and Chaiteerakij R: Metformin does not improve survival of
cholangiocarcinoma patients with diabetes. Hepatology. 63:667–668.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rattan R, Graham RP, Maguire JL, Giri S
and Shridhar V: Metformin suppresses ovarian cancer growth and
metastasis with enhancement of cisplatin cytotoxicity in vivo.
Neoplasia. 13:483–491. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
de Sousa Cavalcante L and Monteiro G:
Gemcitabine: Metabolism and molecular mechanisms of action,
sensitivity and chemoresistance in pancreatic cancer. Eur J
Pharmacol. 741:8–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bailey CJ and Turner RC: Metformin. N Engl
J Med. 334:574–579. 1996. View Article : Google Scholar : PubMed/NCBI
|