Introduction
Colorectal cancer (CRC) is one of the most common
gastrointestinal tract malignant carcinomas. It ranks as the
third-leading cause of cancer-related deaths around the world
(1). The increasing incidence and
mortality rate of CRC has become a primary health concern in China
(2). Currently, most patients with
CRC are cured with surgical removal of the tumor, but this is not
indicated for metastatic CRC patients. Hence, further research to
elucidate the molecular mechanisms underlying CRC cell metastasis
and a search for new targets are urgently needed for the effective
treatment of CRC.
Angiomotin (AMOT) was initially identified as an
angiostatin-binding protein and belongs to the motin family, which
includes AMOT (p80 and p130 isoforms), AMOT-like protein 1 (AMOTL1)
and AMOTL2 (3). AMOT is
characterized by a C-terminal PDZ-binding motif and conserved
coiled-coil domain and often exists in two different splicing
isoforms, p80-AMOT and p130-AMOT. In contrast to p80-AMOT, there is
an extended N-terminal domain in p130-AMOT (4). Previous studies have suggested that
AMOT can enhance endothelial cell motility and tube formation,
implying a critical role in angiogenesis (3,5).
Recently, increasing attention has shed light on the role of AMOT
in the pathogenesis of cancer (6,7).
Emerging evidence has corroborated an abnormal expression of AMOT
in several types of cancer, such as breast cancer, lung cancer and
renal carcinoma (7–10). AMOT has been proven to be highly
expressed in breast cancer (8,11).
Notably, its downregulation obviously suppressed breast cancer cell
proliferation and invasiveness by regulating the
Hippo/Yes-associated protein (YAP) pathway (8). Additionally, AMOT exhibits a
pro-proliferation role in renal cell carcinoma by retaining nuclear
YAP, a common oncogenic gene (7).
In contrast to the oncogenic role of AMOT, the antineoplastic
function of AMOT has been observed in the progression of lung
carcinoma by inhibiting cell growth and metastasis (9). To date, the functional role of AMOT in
the development of CRC remains poorly elucidated.
In this study, we aimed to investigate the
expression of AMOT and its function in cell growth, invasion and
migration. Moreover, the potential molecular mechanism involved was
also explored.
Materials and methods
Antibodies
Primary antibodies against human YAP, phospho-AKT
(p-AKT), AKT, ERK, phospho-ERK (p-ERK), N-cadherin and E-cadherin
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA). Antibodies against cyclin D1, MMP-2, MMP-9, and GAPDH were
obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The
anti-tubulin and anti-AMOT antibodies were purchased from Abcam
(Cambridge, UK). Rabbit polyclonal antibodies to human Bax and
Bcl-2 were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
Human CRC cell lines LoVo, SW620, HT29 and HCT116
were obtained from the American Type Culture Collection (ATCC;
Manassas, VA, USA). Normal human colon mucosal epithelium cell line
NCM460 was ordered from the Incell Corporation, LLC (San Antonio,
TX, USA). The CRC cells were all maintained in Dulbecco's Modified
Eagle's medium (DMEM) supplemented with 10% fetal bovine serum
(FBS), 100 U/ml penicillin and 100 µg/ml streptomycin. The NCM460
cells were cultured in an M3F base medium containing 10% FBS. All
cells were incubated at 37°C in a 5% CO2 incubator (Life
Technologies, Baltimore, MD, USA).
AMOT knockdown and overexpression
The knockdown and overexpression of AMOT in LoVo and
HCT116 cells were performed using the lentiviral expression system
provided by GeneChem Co., Ltd. (Shanghai, China). For AMOT
overexpression and inhibition, the lentiviral vectors for human
AMOT small hairpin RNA (shRNA) and vectors expressing AMOT
(LV-AMOT) were constructed, packed and purified by GeneChem Co.,
Ltd. RNA interference sequences were used as follows: sh-AMOT-1,
5′-TGCAGAGATGGTGGAATAT-3′; sh-AMOT-2, 5′-ACACATCGAAATCCGAGAT-3′;
the scrambled shRNA (sh-NC), 5′-TTCTCCGAATGTGTCACGT-3′. For
transfection, cells were plated into 24-well plates. To increase
the expression of AMOT, LoVo cells were transfected with LV-AMOT or
control lentivirus (Lv-NC) in accordance with the manufacturer's
instructions. The lentivirus encoding AMOT shRNA or sh-NC was
introduced into HCT116 cells. Approximately 8 h later, cells were
incubated with fresh DMEM. After a 48-h incubation, the stable
clones were selected for treatment with puromycin for 3 weeks. The
efficacy of the lentivirus transfection was evaluated by western
blotting.
RNAi-mediated silencing of YAP
To silence the expression of YAP in LoVo cells,
small interfering RNA (siRNA) targeting human YAP and scramble
siRNA (siR-con) were designed and synthesized by Shanghai
GenePharma Co., Ltd. (Shanghai, China). The siRNA sequences were
siR-YAP, 5′-GGUGAUACUAUCAACCAAATT-3′; siR-con,
5′-CCUACGCCACCAAUUUCGU-3′. After seeding into 6-well plates for 12
h, the cells were transfected with the siRNA duplexes (100 nM)
aforementioned, using Lipofectamine 2000 (Invitrogen Life
Technologies Carlsbad, CA, USA) according to the manufacturer's
instructions. At 48 h post-transfection, the transfection
efficiency was assessed by western blotting.
Real-time quantitative RT-PCR
(qRT-PCR)
To quantify the mRNA levels of AMOT, total RNA from
the indicated cells was extracted using RNAiso Plus (Takara Bio
Inc., Otsu, Japan) according to the manufacturer's instructions.
Then, the obtained RNA was reversely transcribed into the first
strand cDNA using the High-Capacity cDNA Reverse Transcription kits
(Applied Biosystems, Foster City, CA, USA). The specimens were then
subjected to RT-PCR with the specific primers for AMOT (sense,
5′-CCAGAATATCCCTTCAAG-3′; antisense, 5′-GAGTTCCTGGCTGACAAT-3′).
qRT-PCR was performed with a final volume of 20 µl according to the
guidelines of the SYBR Premix Ex Taq™ II kit (Takara Bio
Inc.). Human GAPDH was applied as the endogenous control. The
relative expression of the target genes was calculated using the
2−ΔΔCt method.
Western blotting
Cells from the aforementioned samples were lysed
with RIPA buffer, and the extracted protein concentrations were
quantified using the BCA protein assay kit (Beyotime Institute of
Biotechnology, Haimen, China). Then, 30 µg of lysates was separated
on SDS-PAGE and electroblotted onto PVDF membranes (Millipore,
Bedford, MA, USA). After blocking the non-specific binding with 5%
nonfat milk in TBST buffer, the membranes were incubated with the
primary antibodies against human AMOT, PCNA, ERK, p-ERK, p-AKT,
AKT, cyclin D1, Bcl-2, Bax, MMP-2, MMP-9, N-cadherin, E-cadherin
and YAP at 37°C for 1 h. After 3 washes with TBST, a secondary
antibody conjugated to horseradish peroxidase (HRP) was added for a
further 1-h incubation. The immunoreactive bands were visualized
using an ECL detection reagent (Beyotime Institute of
Biotechnology). The band intensities were quantified by Quantity
One software (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Immunoprecipitation assay
LoVo cells from the different treatments were
harvested and lysed on ice for 20 min with lysis buffer, followed
by centrifugation. For immunoprecipitation, 10 µg of anti-YAP
antibodies were pre-treated with 50 µl protein A-Sepharose beads
(Invitrogen Life Technologies) in 500 µl of lysis buffer at 4°C for
15 min. Then, the aforementioned mixture was added into the lysates
at a volume of 500 µl for a 1-h incubation. After washing with
ice-cold lysis buffer, the formed immunocomplex was eluted by
heating for 5 min at 95°C. The immunoprecipitated proteins were
separated by SDS-PAGE, then subjected to immunoblotting with the
specific antibodies against AMOT.
Cell viability as detected by the
3-(4,5-dimethylthiazolyl-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) assay. Cell proliferation was detected using the MTT
assay
Briefly, cells were seeded into a 96-well plate and
incubated for 1–4 days after transfection. Then, the culture medium
was removed and replaced with fresh medium including 5 mg/ml MTT
(Sigma-Aldrich). After further incubation for 5 h at 37°C, the
remaining supernatant was discarded and 200 µl of DMSO was
introduced to dissolve the formed crystal formazan. The color
reaction was detected by measuring the absorbance at 570 nm with an
enzyme immunoassay analyzer (Bio-Rad Laboratories, Inc.).
Measuring cell apoptosis by flow
cytometric analysis
The cell apoptosis ratio was evaluated using an
Annexin V-FITC/PI Apoptosis Detection kit (Beyotime Institute of
Biotechnology). At 48 h after transfection, cells were collected by
trypsinization and suspended in 500 µl of binding buffer. Then, 10
µl Annexin V-FITC and 5 µl PI (Sigma-Aldrich) were added for a
15-min incubation at room temperature in the dark. The samples were
subjected to flow cytometry (FACScan; BD Biosciences, San Jose, CA,
USA) in a device equipped with CellQuest software to assess the
apoptotic cells.
Migration and invasion assays
Cell invasion and migration assays were performed
with or without Matrigel-coated inserts in the Transwell chambers
(Millipore) according to the manufacturer's instructions. After
treatment with the indicated conditions, cells in 200 µl serum-free
medium were seeded into the upper chamber. The medium including 10%
FBS was used as a chemoattractant to add into the lower chamber.
Approximately 24 h later, the non-invading or non-migrated cells
were removed by scrapping with a cotton swab. After fixation with
100% methanol, the invasive or migratory cells were stained with
0.5% crystal violet (Sigma-Aldrich) for 15 min at room temperature.
The number of cells that had migrated or invaded through the
membrane was quantified by counting the cells from at least three
random fields with an inverted microscope.
Statistical analysis
Data were obtained from at least three independent
experiments and are presented as the mean ± SD. All data analysis
was carried out using SPSS 16.0 software (SPSS, Inc., Chicago, IL,
USA). The statistical comparisons among the groups were evaluated
using a Student's t-test and ANOVA. P<0.05 was defined as
statistically significant.
Results
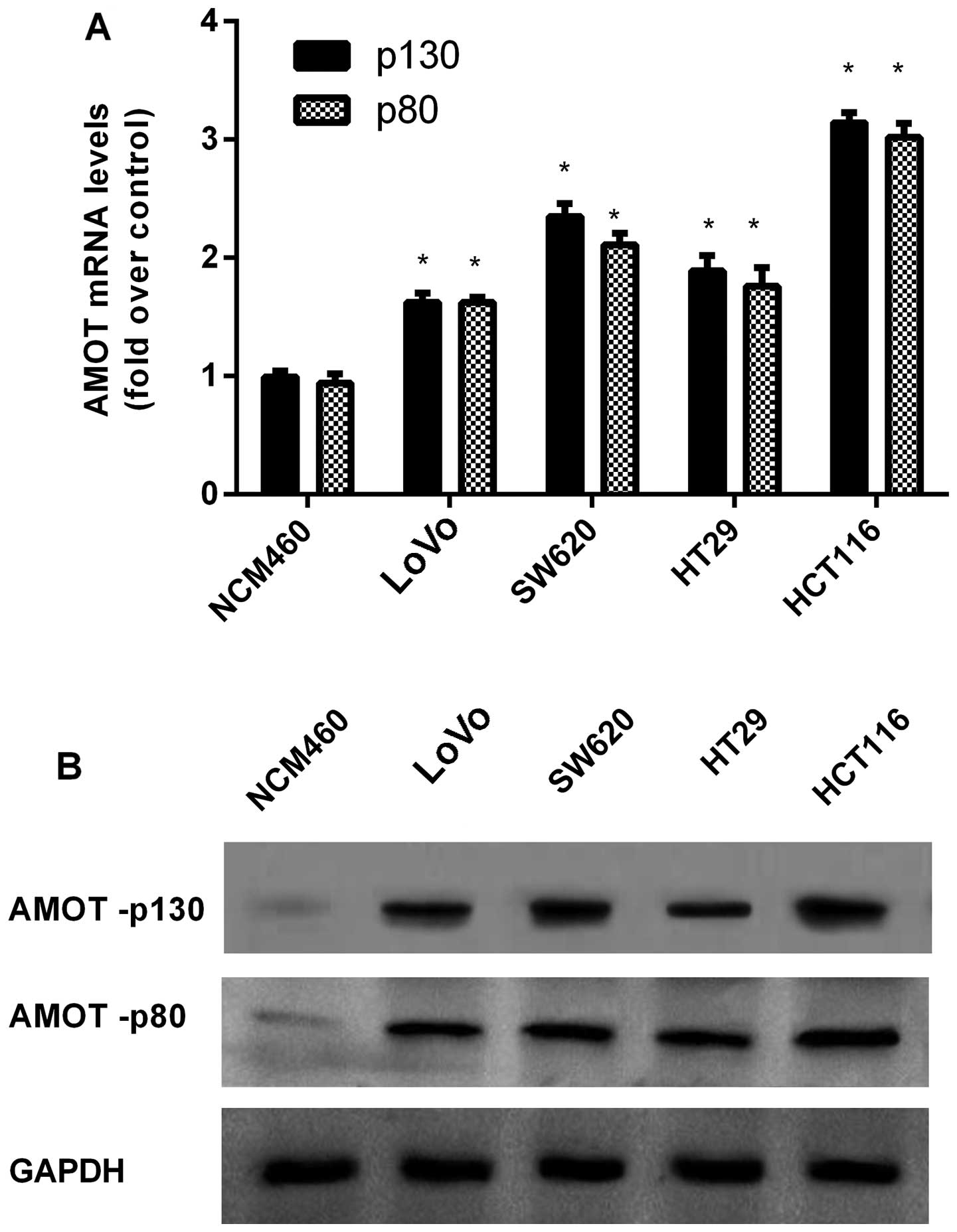
Upregulation of AMOT in CRC cell
lines
In order to explore the function of AMOT in the
progression of CRC, a preliminary cohort study was performed in a
series of CRC cell lines. In contrast to the NCM460 normal colon
epithelium cell line, the mRNA levels of AMOT were markedly higher
in the four examined CRC cell lines (Fig. 1A). Concomitantly, western blotting
showed the notable increase of AMOT protein levels in CRC cells
when compared to NCM460 cells (Fig.
1B).
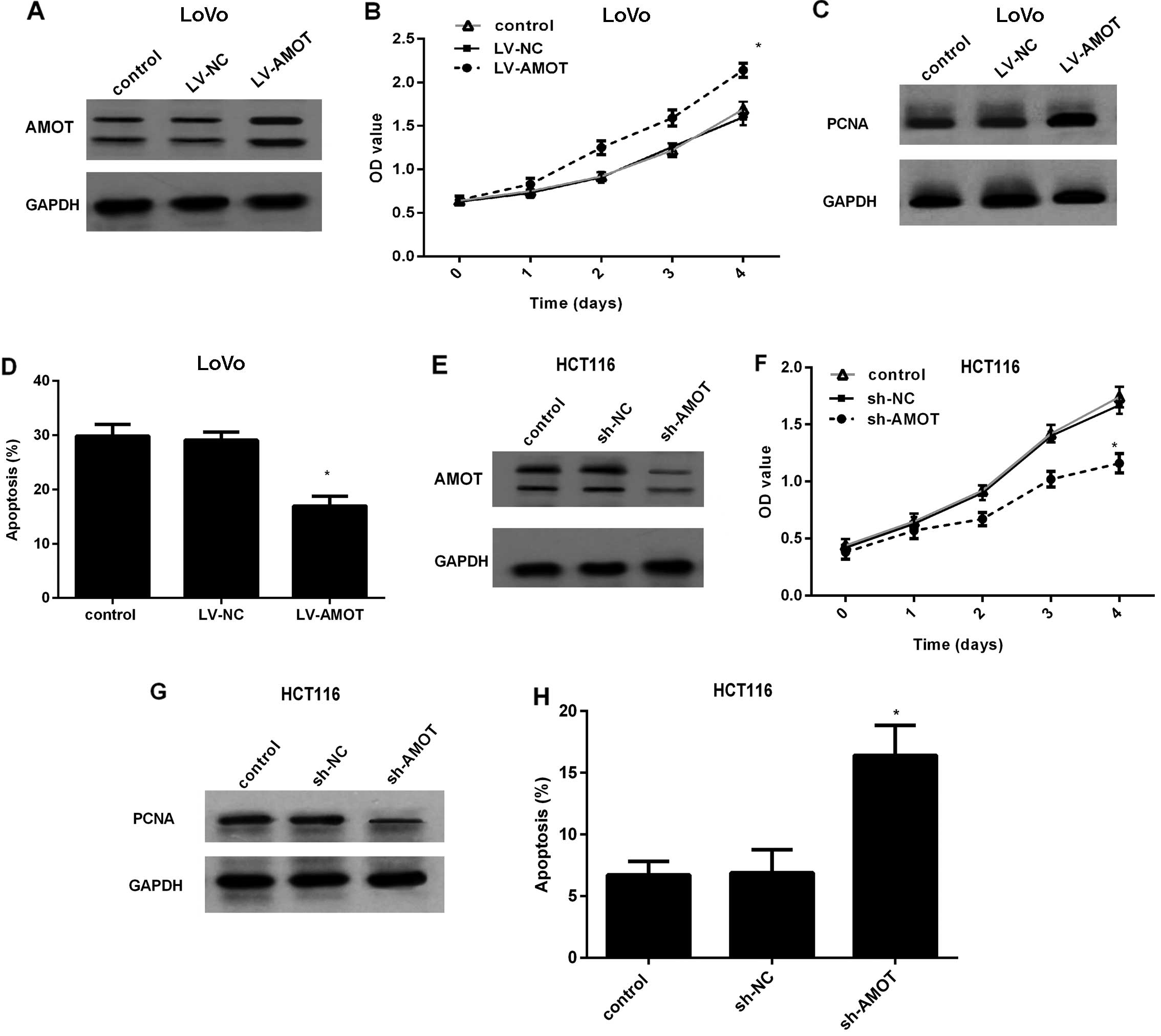
Effect of AMOT expression on CRC cell
growth
To dissect the effect of AMOT in CRC cells, AMOT
protein levels were effectively increased in LoVo cells by LV-AMOT
transfection (Fig. 2A). An MTT
assay corroborated that overexpression of AMOT markedly increased
the proliferation rate of the LoVo cells (Fig. 2B), concomitant with a similar
increase in the expression of the PCNA protein, a common marker for
cell proliferation (Fig. 2C). To
further evaluate the role of AMOT overexpression on cell apoptosis,
5-fluorouracil (5-FU) was added to induce low basal apoptosis
levels. As shown in Fig. 2D, AMOT
upregulation obviously suppressed 5-FU-induced cell apoptosis from
29.87 to 16.94%. To better investigate the function of AMOT in CRC
cell growth, we constructed the stably AMOT-silenced cell line
HCT116 by LV-AMOT shRNA transfection (Fig. 2E). In contrast, knockdown of AMOT
expression in HCT116 cells markedly dampened the cell proliferation
ability (Fig. 2F), accompanied by a
corresponding decrease in PCNA expression (Fig. 2G). Compared to the control or sh-NC
groups, AMOT silencing markedly increased the apoptotic ratio to
15.87% (Fig. 2H). Collectively,
these results indicate that AMOT promotes CRC cell growth and
protects CRC cells against apoptosis.
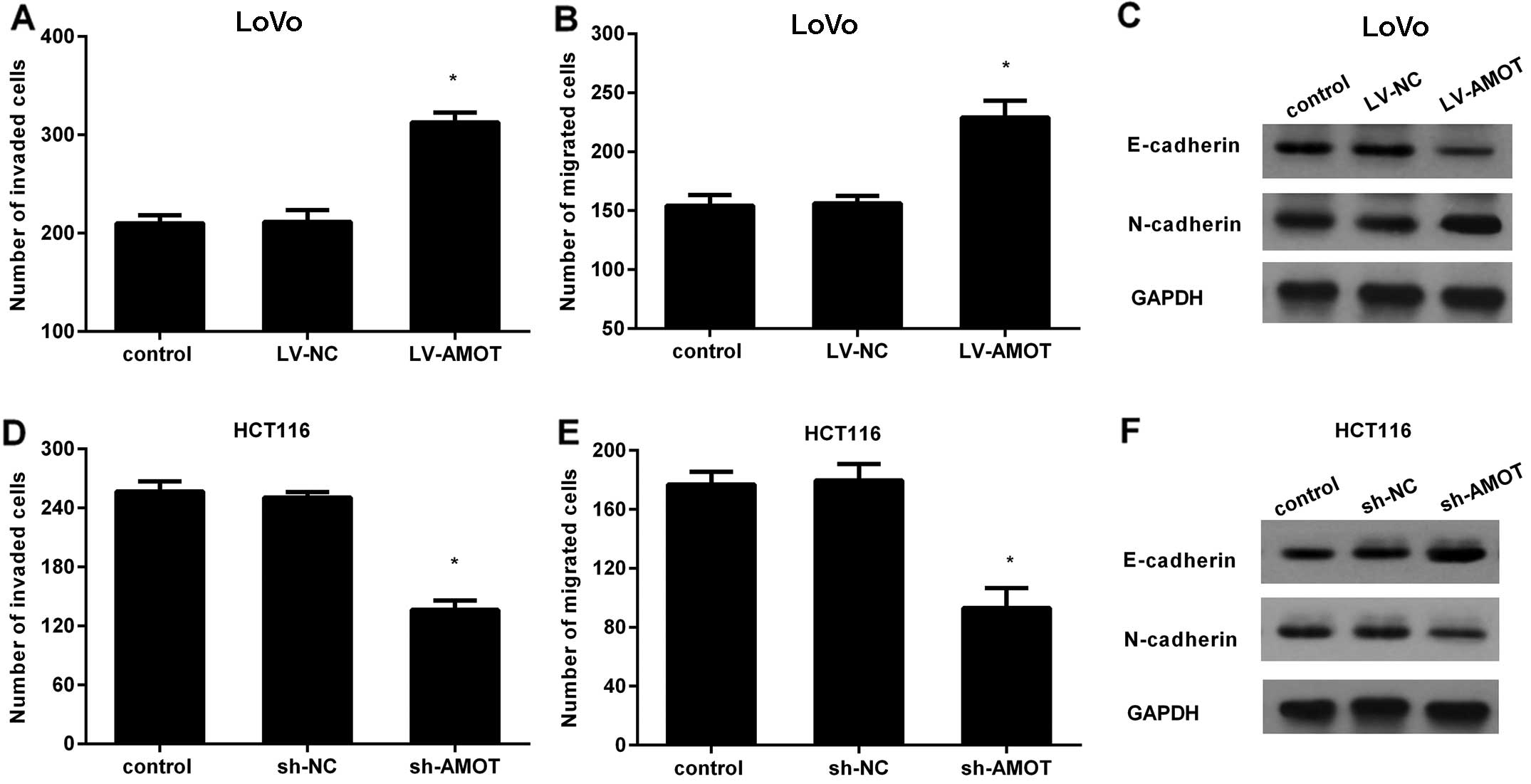
AMOT promotes CRC cell invasion,
migration and epithelial-mesenchymal transition (EMT)
Based on the pro-growth role of AMOT in CRC cell
growth, we further defined its effects on CRC cell metastatic
potential by detecting cell invasion, migration and EMT. As shown
in Fig. 3A, LV-AMOT-treated LoVo
cells exhibited a statistically significant increase in cell
invasion, compared with the control groups. Furthermore, an
increase in AMOT markedly enhanced the number of LoVo cells
migrating through the membrane (Fig.
3B). EMT is widely accepted as a major contributor to cancer
cell metastasis. As expected, AMOT overexpression also reduced the
expression of epithelial marker E-cadherin and increased the
expression of mesenchymal marker N-cadherin (Fig. 3C). To further corroborate the
aforementioned effects, the expression of AMOT was knocked down by
LV-AMOT shRNA treatment. Blocking AMOT expression significantly
antagonized the invasion ability of HCT116 cells (Fig. 3D). Simultaneously, the number of
migrating HCT116 cells was also notably attenuated when cells were
transfected with LV-AMOT shRNA (Fig.
3E). Notably, downregulation of AMOT inhibited EMT by
increasing the expression levels of E-cadherin and decreasing the
levels of N-cadherin (Fig. 3F).
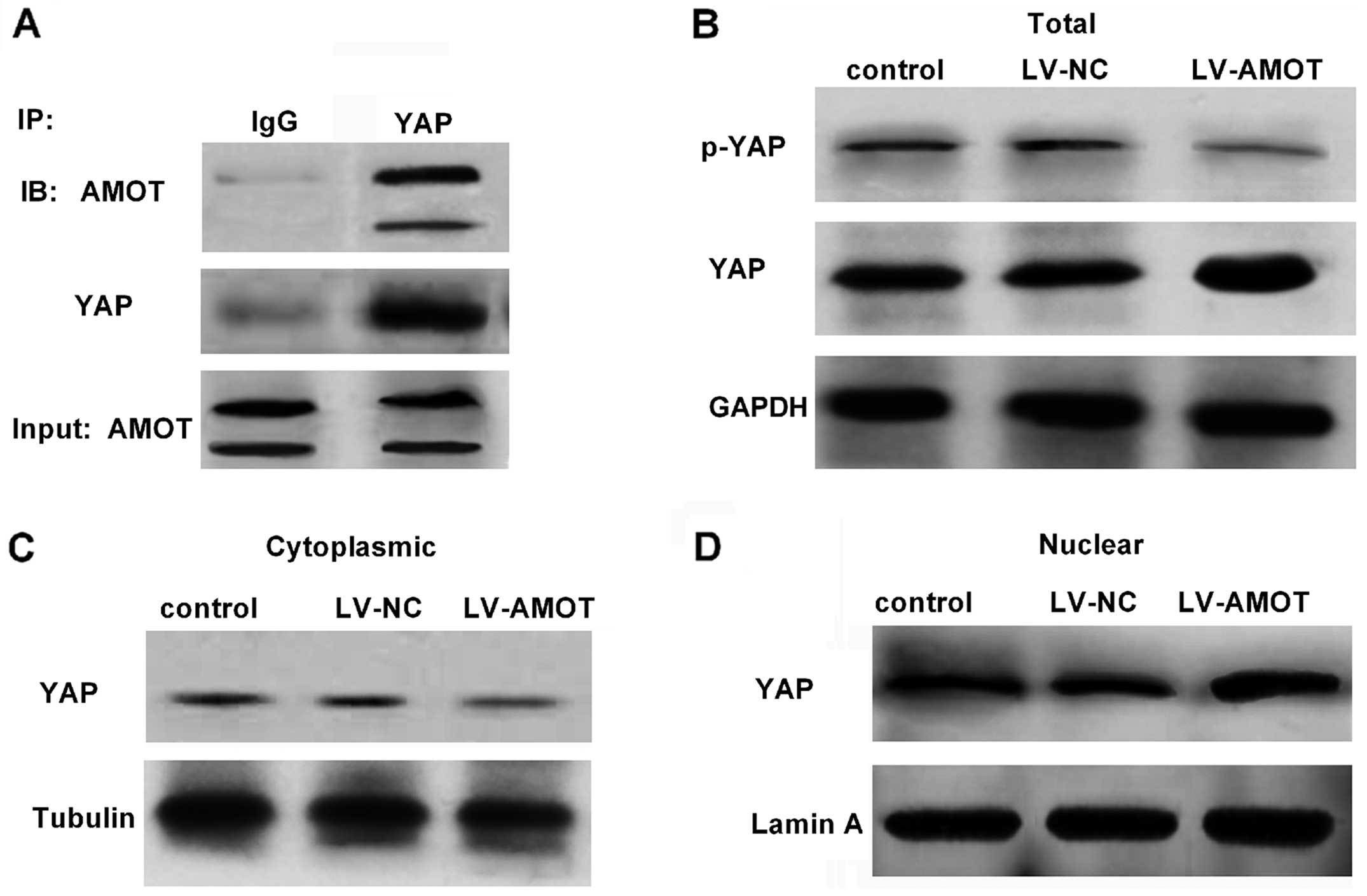
Upregulation of AMOT induces the
activation of YAP by increasing the nuclear localization of
YAP
AMOT has been reported to interact with YAP to
stimulate its activity (7). Upon
dephosphorylation, YAP is activated and then translocated to the
nucleus to exert its oncogenic potential by activating downstream
signaling (12). We further
clarified the relationship between AMOT and YAP in CRC. In HCT116
cells, an antibody against YAP efficiently knocked down AMOT
(Fig. 4A). To explore the effect of
AMOT on YAP activity, the expression levels of phosphorylated YAP
(p-YAP) were analyzed. As shown in Fig.
4B, p-YAP levels were mitigated in AMOT-overexpressing HCT116
cells (Fig. 4B). Moreover,
upregulation of AMOT reduced the expression of cytoplasmic YAP
(Fig. 4C). Notably, treatment with
LV-AMOT significantly increased the relative expression of nuclear
YAP (Fig. 4D). These data indicated
that AMOT induces the activity of YAP in CRC cells.
YAP is required for the oncogenic
effect of AMOT overexpression in CRC cells
To elucidate the underlying molecular mechanism
involved in the AMOT-induced oncogenic effect in HCT116 cells, we
explored the role of YAP during this process. Western blotting
illustrated the knockdown of YAP in HCT116 cells transfected with
YAP siRNA (Fig. 5A). A function
assay validated that silencing of YAP markedly antagonized the
pro-proliferation effect of AMOT overexpression in HCT116 cells
(Fig. 5B). Furthermore, AMOT
upregulation notably enhanced cell resistance to 5-FU-induced
apoptosis, which was obviously attenuated following YAP
downregulation (Fig. 5C).
Concomitantly, knockdown of YAP significantly attenuated the
invasion (Fig. 5D) and migration
(Fig. 5E) of HCT116 cells triggered
by AMOT upregulation. Collectively, these results revealed that
AMOT may promote CRC cell growth, invasion and migration mainly
through YAP.
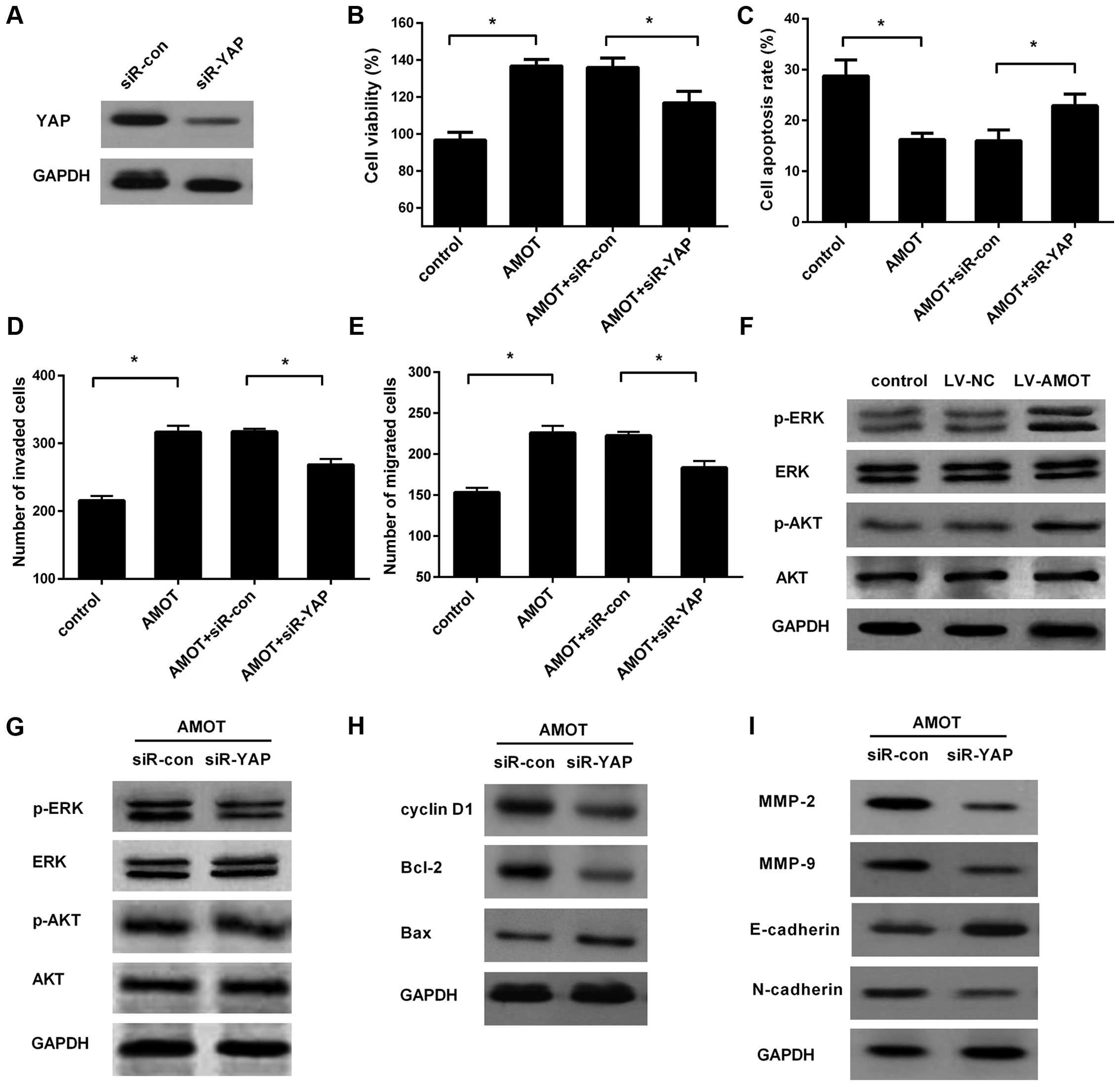 | Figure 5.AMOT promotes CRC cell growth,
invasion and migration through the YAP-ERK/PI3K-AKT signaling
pathway. HCT116 cells were transfected with YAP siRNA, prior to
infection with LV-AMOT. (A) The efficacy of YAP silencing was
analyzed. (B-E) Evaluation the effects of YAP knockdown on cell
viability, apoptosis, invasion and migration triggered by AMOT
overexpression. (F) After infection with LV-AMOT, the activation of
the ERK and PI3K/AKT pathways was assessed by western blotting. (G)
Effects of YAP silencing on AMOT-induced activation of the ERK and
PI3K/AKT signaling pathways. (H) The corresponding expression of
downstream effectors related to cell proliferation (cyclin D1,
Bcl-2 and Bax). (I) After preconditioning with YAP siRNA, the
expression of ERK and PI3K/AKT signaling proteins involved in cell
metastatic potential (MMP-2, MMP-9, E-cadherin and N-cadherin) was
monitored in AMOT-overexpressed HCT116 cells. *P<0.05. AMOT,
angiomotin; CRC, colorectal cancer; YAP, Yes-associated protein;
siRNA, small interfering RNA. |
Increase in AMOT activates the
ERK/PI3K-AKT signaling pathway in a manner mediated by YAP
Abnormal activation of the ERK and PI3K/AKT pathways
has been reported in various carcinomas and they play critical
roles in carcinogenesis by regulating cell proliferation, invasion
and migration (13–16). To further elucidate the mechanism
involved in AMOT-mediated oncogenic potential, both of these two
pathways were explored further. In line with our hypothesis,
overexpression of AMOT markedly induced the expression of p-ERK and
p-AKT, but not the total ERK and AKT (Fig. 5F). A previous study confirmed that
YAP can transcriptionally activate the PI3K/AKT and ERK signaling
pathways in human cutaneous squamous cell cancer (12). Further analysis of the mechanism
demonstrated that silencing of YAP expression markedly attenuated
AMOT-induced expression of p-ERK and p-AKT (Fig. 5G), accompanied by the decrease in
the subsequent expression of cyclin D1, Bcl-2 and the increase in
the expression of Bax (Fig. 5H).
Additionally, the downstream increases in MMP-2 and MMP-9 levels in
AMOT-elevated cells were obviously attenuated after YAP siRNA
transfection (Fig. 5I).
Consistently, YAP knockdown also inhibited AMOT-induced EMT by
increasing E-cadherin expression and suppressing N-cadherin levels.
All of these results implied that AMOT could induce the activation
of the YAP-ERK/PI3K-AKT signaling pathway.
Discussion
Recent studies have demonstrated the aberrant
alteration of AMOT in the progression of cancer (7,9,10).
However, its role in carcinogenesis is controversial in various
types of cancer. For example, AMOT exerts oncogenic effects in
breast cancer by promoting cell proliferation and invasion through
the Hippo/YAP pathway (8). Notably,
knockout of AMOT in the livers of mice led to reduced hepatic ‘oval
cell’ proliferation and tumorigenesis (10). Other evidence substantiates that
knockdown of AMOT initiates lung cancer cell proliferation,
migration, invasion and ultimately acts as a tumor suppressor in
lung carcinoma progression by sequestering oncogenic YAP/TAZ
(9). In the present study, the
higher expression of AMOT was detected in CRC cells. Importantly,
overexpression of AMOT promoted LoVo cell proliferation and
resistance to 5-FU-induced apoptosis. Moreover, its upregulation
also enhanced cell invasion, migration and EMT. Consistently,
blocking AMOT expression consistently inhibited HCT116 cell growth
and metastatic potential. Therefore, these findings suggest that
AMOT may elicit the oncogenic function in the carcinogenesis of
CRC.
Accumulating evidence has revealed that aberrant
alteration of key components of the Hippo signaling pathway can
result in uncontrolled cell growth and cancer progression (17,18).
As a major downstream transcription activator of the Hippo pathway,
YAP can bind to several transcription factors to regulate the
development of cancer. Indeed, high expression of YAP has been
noted in a wide variety of cancers, including CRC (12,19).
However, YAP is inactivated and sequestered in the cytoplasm when
phosphorylated by the kinase of the Hippo pathway (20). Once dephosphorylated, it becomes
activated and translocates to the nucleus, where it can act as an
oncogenic regulator for tumor cell growth, invasion and migration
(12,21). Recent studies confirmed the
correlation between AMOT and YAP, however, this is controversial
(7,22). In the liver, AMOT can function as a
YAP cofactor to prevent YAP phosphorylation and increase its
activity toward a specific set of genes that facilitate
tumorigenesis (10). In contrast to
the aforementioned finding, AMOT can interact with YAP to sequester
YAP in the cytoplasm, which finally mitigates the development of
lung cancer (9). Whether AMOT
stimulates or suppresses YAP activity in CRC remains undefined. In
our study, AMOT interacted with YAP in HCT116 cells. More
importantly, AMOT decreased the levels of p-YAP. Furthermore,
overexpression of AMOT attenuated the expression of YAP in the
cytoplasm and increased its levels in the nucleus. All of these
data indicated that AMOT could promote YAP activity in CRC cells,
implying a potential oncogenic function of AMOT in the progression
of CRC.
It is believed that the progression and metastasis
of malignant tumors are complicated processes involved in multiple
cellular events, including cell growth, invasion and migration.
Here, we demonstrated that AMOT upregulation enhanced CRC cell
proliferation, apoptosis resistance, invasion, migration and EMT.
Simultaneously, blocking AMOT expression also inhibited CRC cell
growth and motility. As an oncogene, YAP is highly expressed in CRC
and acts as a predictor of poor prognosis in CRC patients (23). Moreover, YAP mediated the high
proliferation and metastasis in CRC cells. Notably, our study
confirmed the increased activity and expression of YAP in
AMOT-overexpressing CRC cells. All of these findings led us to
speculate as to whether YAP is involved in the AMOT-mediated
oncogenic function in CRC. To address this hypothesis, we silenced
YAP expression. As expected, knockdown of YAP markedly attenuated
the malignancy of cell growth, invasion and migration upon AMOT
overexpression, indicating a critical role of YAP in the
AMOT-triggered metastasis-promoting process of CRC.
The ERK and PI3K/AKT pathways are often
simultaneously activated in various carcinomas and have therefore
been the focus of numerous investigations in CRC (13,14).
It is widely accepted that both the ERK and the PI3K/AKT pathways
stimulate a cascade of responses from cell survival to metastasis
by their downstream effectors, which ultimately facilitates the
development and progression of CRC (13–16).
Results from previous studies demonstrated that AMOT upregulation
increased the phosphorylation of ERK and AKT (17,24).
Notably, a recent study revealed that YAP contributed to the
development of human cutaneous squamous cell cancer by activating
the RAS-mediated AKT and ERK signaling pathways (12). Moreover, YAP overexpression was
found to increase EMT in pancreatic cancer cells by activating the
AKT cascade (25). YAP also
enhanced the ERK/AKT signaling pathway in renal cell carcinoma
cells (7). Does AMOT regulate the
activation of the ERK/AKT pathway through YAP? To answer this
question, we explored the effects of YAP silencing during this
process. As we hypothesized, YAP knockdown significantly attenuated
AMOT-induced activation of the ERK/AKT pathway, as well as the
activation of their downstream targets associated with tumor cell
growth and metastasis. These findings support the hypothesis that
AMOT induces the activation of the AKT/ERK pathway through YAP,
which facilitates the tumorigenesis of CRC.
In conclusion, this study confirmed the upregulation
of AMOT in CRC cells. Notably, overexpression of AMOT promoted the
cell growth and metastatic potential in CRC cells mainly by
activating the YAP-ERK/AKT signaling pathway. Accordingly, our
study may provide new insight concerning how AMOT acts as an
oncogene in the progression of CRC, presenting a promising
therapeutic agent against CRC.
Acknowledgements
Financial support was provided by a project
supported by the Natural Science Basic Research Plan in Shaanxi
Province of China (no. 2014JM2-8201) and the National High
Technology Research and Development Program (‘863’ Program) (no.
2014AA022304).
References
|
1
|
Weitz J, Koch M, Debus J, Höhler T, Galle
PR and Büchler MW: Colorectal cancer. Lancet. 365:153–165. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hu F, Li D, Wang Y, Yao X, Zhang W, Liang
J, Lin C, Ren J, Zhu L, Wu Z, et al: Novel DNA variants and
mutation frequencies of hMLH1 and hMSH2 genes in colorectal cancer
in the Northeast China population. PLoS One. 8:e602332013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Troyanovsky B, Levchenko T, Månsson G,
Matvijenko O and Holmgren L: Angiomotin: An angiostatin binding
protein that regulates endothelial cell migration and tube
formation. J Cell Biol. 152:1247–1254. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ernkvist M, Aase K, Ukomadu C,
Wohlschlegel J, Blackman R, Veitonmäki N, Bratt A, Dutta A and
Holmgren L: p130-angiomotin associates to actin and controls
endothelial cell shape. FEBS J. 273:2000–2011. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bratt A, Birot O, Sinha I, Veitonmäki N,
Aase K, Ernkvist M and Holmgren L: Angiomotin regulates endothelial
cell-cell junctions and cell motility. J Biol Chem.
280:34859–34869. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moleirinho S, Guerrant W and Kissil JL:
The angiomotins - from discovery to function. FEBS Lett.
588:2693–2703. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lv M, Li S, Luo C, Zhang X, Shen Y, Sui
YX, Wang F, Wang X, Yang J, Liu P, et al: Angiomotin promotes renal
epithelial and carcinoma cell proliferation by retaining the
nuclear YAP. Oncotarget. 7:12393–12403. 2016.PubMed/NCBI
|
|
8
|
Lv M, Lv M, Chen L, Qin T, Zhang X, Liu P
and Yang J: Angiomotin promotes breast cancer cell proliferation
and invasion. Oncol Rep. 33:1938–1946. 2015.PubMed/NCBI
|
|
9
|
Hsu YL, Hung JY, Chou SH, Huang MS, Tsai
MJ, Lin YS, Chiang SY, Ho YW, Wu CY and Kuo PL: Angiomotin
decreases lung cancer progression by sequestering oncogenic YAP/TAZ
and decreasing Cyr61 expression. Oncogene. 34:4056–4068. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yi C, Shen Z, Stemmer-Rachamimov A, Dawany
N, Troutman S, Showe LC, Liu Q, Shimono A, Sudol M, Holmgren L, et
al: The p130 isoform of angiomotin is required for Yap-mediated
hepatic epithelial cell proliferation and tumorigenesis. Sci
Signal. 6:ra772013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang H and Fan Q: MicroRNA-205 inhibits
the proliferation and invasion of breast cancer by regulating AMOT
expression. Oncol Rep. 34:2163–2170. 2015.PubMed/NCBI
|
|
12
|
Jia J, Li C, Luo S, Liu-Smith F, Yang J,
Wang X, Wang N, Lai B, Lei T, Wang Q, et al: Yes-associated protein
contributes to the development of human cutaneous squamous cell
carcinoma via activation of RAS. J Invest Dermatol. 136:1267–1277.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ye Q, Cai W, Zheng Y, Evers BM and She QB:
ERK and AKT signaling cooperate to translationally regulate
survivin expression for metastatic progression of colorectal
cancer. Oncogene. 33:1828–1839. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huynh N, Liu KH, Baldwin GS and He H:
P21-activated kinase 1 stimulates colon cancer cell growth and
migration/invasion via ERK- and AKT-dependent pathways. Biochim
Biophys Acta. 1803:1106–1113. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao D, Sui Y and Zheng X: MiR-331-3p
inhibits proliferation and promotes apoptosis by targeting HER2
through the PI3K/Akt and ERK1/2 pathways in colorectal cancer.
Oncol Rep. 35:1075–1082. 2016.PubMed/NCBI
|
|
16
|
Huang J, Che MI, Lin NY, Hung JS, Huang
YT, Lin WC, Huang HC, Lee PH, Liang JT and Huang MC: The molecular
chaperone cosmc enhances malignant behaviors of colon cancer cells
via activation of Akt and ERK. Mol Carcinog. 53:(Suppl 1). E62–E71.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Harvey KF, Zhang X and Thomas DM: The
Hippo pathway and human cancer. Nat Rev Cancer. 13:246–257. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin L, Sabnis AJ, Chan E, Olivas V, Cade
L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, et al: The Hippo
effector YAP promotes resistance to RAF- and MEK-targeted cancer
therapies. Nat Genet. 47:250–256. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li H, Wang S, Wang G, Zhang Z, Wu X, Zhang
T, Fu B and Chen G: Yes-associated protein expression is a
predictive marker for recurrence of hepatocellular carcinoma after
liver transplantation. Dig Surg. 31:468–478. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao B, Lei QY and Guan KL: The Hippo-YAP
pathway: New connections between regulation of organ size and
cancer. Curr Opin Cell Biol. 20:638–646. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dai XY, Zhuang LH, Wang DD, Zhou TY, Chang
LL, Gai RH, Zhu DF, Yang B, Zhu H and He QJ: Nuclear translocation
and activation of YAP by hypoxia contributes to the chemoresistance
of SN38 in hepatocellular carcinoma cells. Oncotarget. 7:6933–6947.
2016.PubMed/NCBI
|
|
22
|
Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q
and Guan KL: Angiomotin is a novel Hippo pathway component that
inhibits YAP oncoprotein. Genes Dev. 25:51–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang L, Shi S, Guo Z, Zhang X, Han S, Yang
A, Wen W and Zhu Q: Overexpression of YAP and TAZ is an independent
predictor of prognosis in colorectal cancer and related to the
proliferation and metastasis of colon cancer cells. PLoS One.
8:e655392013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ranahan WP, Han Z, Smith-Kinnaman W,
Nabinger SC, Heller B, Herbert BS, Chan R and Wells CD: The adaptor
protein AMOT promotes the proliferation of mammary epithelial cells
via the prolonged activation of the extracellular signal-regulated
kinases. Cancer Res. 71:2203–2211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yuan Y, Li D, Li H, Wang L, Tian G and
Dong Y: YAP overexpression promotes the epithelial-mesenchymal
transition and chemoresistance in pancreatic cancer cells. Mol Med
Rep. 13:237–242. 2016.PubMed/NCBI
|