Introduction
With the development of medical imaging technology
and molecular diagnostics, metastatic brain tumors are being
detected and treated at earlier stages. The most common treatments
for metastatic brain tumors include surgery, chemotherapy and
radiotherapy. Radiosurgery is frequently used in patients with
metastatic brain tumors. However, when a tumor is relatively large,
developed in the brain stem, or close to important nerves such as
the optic nerve, it cannot be treated with a high dose of
radiation. In these situations, it is imperative to increase the
radiosensitivity of cancer cells in order to produce the desired
outcome with fewer doses of radiation. Therefore, it is essential
to develop radiosensitizers which are non-toxic, effective and
molecularly targeted.
The JNK pathway is one subtype of MAP kinase
signaling that is activated primarily by cytokines and exposure to
environmental stress (1). JNK
participates in every type of cellular response, including
apoptosis (2,3). JNK also participates in the
phosphorylation of H2AX after radiation (4). Based on this finding, Yue et al
reported that inhibition of JNK activity enhanced radiosensitivity
and apoptosis in vestibular schwannoma (5). Accumulating evidence has demonstrated
the important role of JNK in the process of DNA repair after
radiation. However, to the best of our knowledge, no study has
reported the effects of JNK small-molecule inhibitor SP600125 on
lung and breast cancer cells treated with radiotherapy. On the
basis of its efficacy in vestibular schwannoma, it is plausible
that inhibition of JNK activity could also increase the
radiosensitivity of lung and breast cancer cells.
Currently, the JNK inhibitor, PGL5001, is used to
treat inflammatory endometriosis, and has undergone a phase II
clinical trial (clinicaltrials.gov ID NCT01630252). The results of
that study indicated that the JNK inhibitor has the potential to be
useful in the treatment of various clinical conditions. The JNK
small-molecule inhibitor SP600125
(anthra[1,9-cd]pyrazol-6(2H)-one) specifically inhibited
phosphorylation of c-Jun (6,7). In
the present study, we investigated the effects of the JNK inhibitor
SP600125 in combination with radiation in vitro and in
vivo. Our results demonstrated that JNK inhibition increased
the radiosensitivity in lung and breast cancer cell lines. Overall,
the present study showed that the combination of SP600125 with
radiotherapy appears to have promising antitumor activity, and
provides therapeutic benefits in the clinical setting for
metastatic brain tumor.
Materials and methods
Cell cultures and treatment
conditions
The murine Lewis lung cancer (LLC) and mammary
carcinoma (4T1) cell lines were obtained from the American Type
Culture Collection (ATCC; Manassas, VA, USA). The cell lines were
cultured in Dulbeccos modified Eagles medium (DMEM) supplemented
with 10% fetal bovine serum (FBS) (both from Gibco-BLR,
Gaithersburg, MD, USA) at 37°C in a 5% CO2/95% air
incubator. SP600125 was kindly provided by Sigma (St. Louis, MO,
USA), constituted in dimethyl sulfoxide (DMSO) (10 mM), and stored
at −80°C. Cellular exposure to SP600125 was performed 2 h before
irradiation with Gamma Knife Perfexion.
MTT assay
The toxicity of SP600125 was monitored using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltertazolium bromide (MTT)
assay. The cells (103 cells/well) were seeded into
96-well plates and cultured under standard conditions (10% FBS at
37°C and 5% CO2/95% air). At every 24 h, an aliquot of
10 µl MTT [5 mg/ml dissolved in phosphate-buffered saline (PBS)]
was added to each well. The medium was removed from each well after
2 h. MTT formazan was solubilized in 200 µl DMSO. Optical density
was read at 570 nm.
Clonogenic assay
For the colony formation assay, LLC and 4T1 cell
lines were plated in 6-well culture dishes
(102-1.4×104 cells/well) and exposed to
compound and γ-irradiation followed by incubation at 37°C for 8–10
days. Cells were fixed in methanol for 5 min and stained with
toluidine blue (0.1%; Sigma) for 15 min. The number of colonies
containing at least 50 cells were counted. The surviving fraction
was calculated as the number of colonies divided by the number of
cells that were initially plated. Dose enhancement factor (DEF) was
calculated at the 10% survival level. D0.1 was defined as the
quotient of D0.1 RT/D0.1 RT + drug. A DEF >1 indicates that the
drug is functioning as a radiosensitizer.
Western blotting
Western blot analysis was carried out as previously
described (8,9). Whole-cell lysates were isolated by
RIPA buffer. Lysates were separated by 8–12% SDS-PAGE and
transferred onto polyvinylidene difluoride (PVDF) membranes (Pall
Corporation, Port Washington, NY, USA). The membranes were probed
with antibodies against GAPDH, P-JNK, BAX, P-Bcl-2, cleaved
caspase-3, cleaved PARP and phospho-histone H2AX (Cell Signaling
Technology, Danvers, MA, USA) overnight at 4°C. Bound secondary
antibody was visualized by goat anti-rabbit antibody (Jackson
Immunoresearch Laboratory, West Grove, PA, USA). The imaging was
obtained using an LAS-4000 instrument (Fuji, Tokyo, Japan). GAPDH
was used as an internal control.
Immunocytochemistry
The cells were plated at a concentration of
1.2×105 cells/well into a 8-well chamber slide (Labtek
Pty Ltd., Brendale, Australia). After 6 h, the slide was fixed for
20 min with 4% paraformaldehyde, washed with PBS, and the cells
were permeabilized for 15 min with 0.8% Triton-X 100. The cells
were blocked with 5% goat serum and 0.8% Triton-X 100 for 30 min
followed by three 5-min PBS washes. The rabbit monoclonal
anti-phosphorylated Ser139 histone H2AX (1:300; Cell Signaling
Technology) antibody was added and incubated overnight at 4°C. The
cells were exposed to goat anti-rabbit secondary antibody (1:400 in
PBS) conjugated with Alexa Fluor 488 or 568 (1:400; Molecular
Probes, Sunnyvale, CA, USA). Nuclei were stained with
4′,6-diamidino-2-phenylindole (DAPI). Images were captured with an
Olympus FV10-ASW confocal laser scanning biological microscope
system. The specific immunofluorescence signal in the nucleus was
observed using Alexa Fluor 488 or 568 and DAPI fluorescence filters
(10). Quantification of the γH2AX
foci was conducted, such that cells were counted when they
contained >5 foci. At least 50 cells were counted.
Animal studies
Female 7- to 8-week-old C57BL/6 mice were obtained
from Orient Co. (Seongnam, Korea). LLC cells (5×105) in
50 µl of PBS were subcutaneously inoculated into the backside and
both hind limbs of mice (n=5) and allowed to grow for 10–11 days.
To assess the toxicity of SP600125, right hind limb tumors were
directly injected with DMSO (dissolved in PBS), and left hind limb
tumors were directly injected with 5 µM SP600125 (dissolved in DMSO
and PBS) twice daily to termination. For the radiosensitivity
study, backside tumors (control) and right hind limb tumors were
directly injected with DMSO (dissolved in PBS), and left hind limb
tumors were directly injected with 5 µM SP600125 (dissolved in DMSO
and PBS) 6 h before localized irradiation. The drugs were injected
twice daily to termination. Both hind limbs were irradiated with a
fractionated schedule (3×3.5 Gy, total 8 Gy). Tumors were measured
with calipers and the volume was calculated with the formula:
Volume (V) = length (a) × width (b) × width (b) × 0.5. Radiation
was delivered with a 6-MV X-ray linear accelerator (Clinac 21EX;
Varian Inc., Palo Alto, CA, USA).
Immunohistochemistry
Tumor tissues were removed 10 days after
radiotherapy. The paraffin-embedded specimens were dewaxed in
xylene and subjected to heat-mediated antigen retrieval in target
retrieval solution (pH 9.0; Dako, Carpinteria, CA, USA). Endogenous
peroxidase activity was blocked by 3% H2O2,
and the sections were incubated with 3% bovine serum albumin
(Sigma) to block any non-specific binding. Anti-cleaved caspase-3
(Cell Signaling Technology) was added at 4°C overnight, and then
the secondary antibody (Dako) was added, and samples were incubated
at room temperature for 1 h. The tissue sections were detected
using DAB (Dako). Nuclei were stained with Harris hematoxylin
(ScyTek, Logan, UT, USA).
Statistical analyses
Data are presented as mean ± SD. Between-group
comparisons were performed using the Student's t-test. The paired
t-test was used to compare the volume of two tumors in one
individual. A value of P<0.05 was considered to indicate a
statistically significant result.
Results
Responses of the LLC and 4T1 cell
lines to γ-irradiation and SP600125
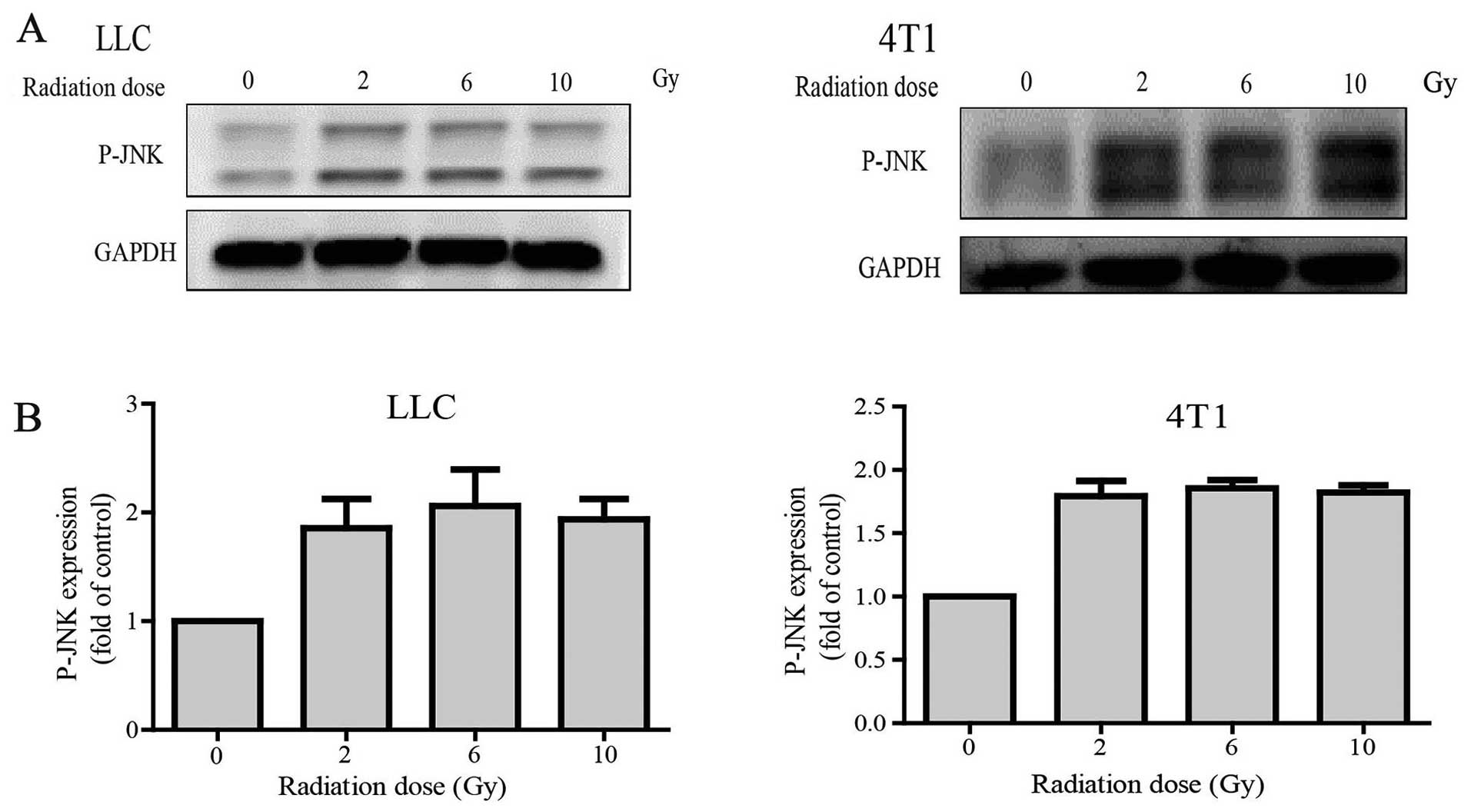
Western blot data showed that P-JNK expression was
significantly increased in the radiation group compared with that
noted in the control group in both the LLC and 4T1 cell lines.
Nevertheless, P-JNK expression was independent of the dose of
γ-irradiation (2, 6 and 10 Gy) (Fig. 1A
and B). In the presence of the JNK inhibitor SP600125, JNK
signaling was found to be specifically blocked in other cells
(11,12). SP600125 was used in the present
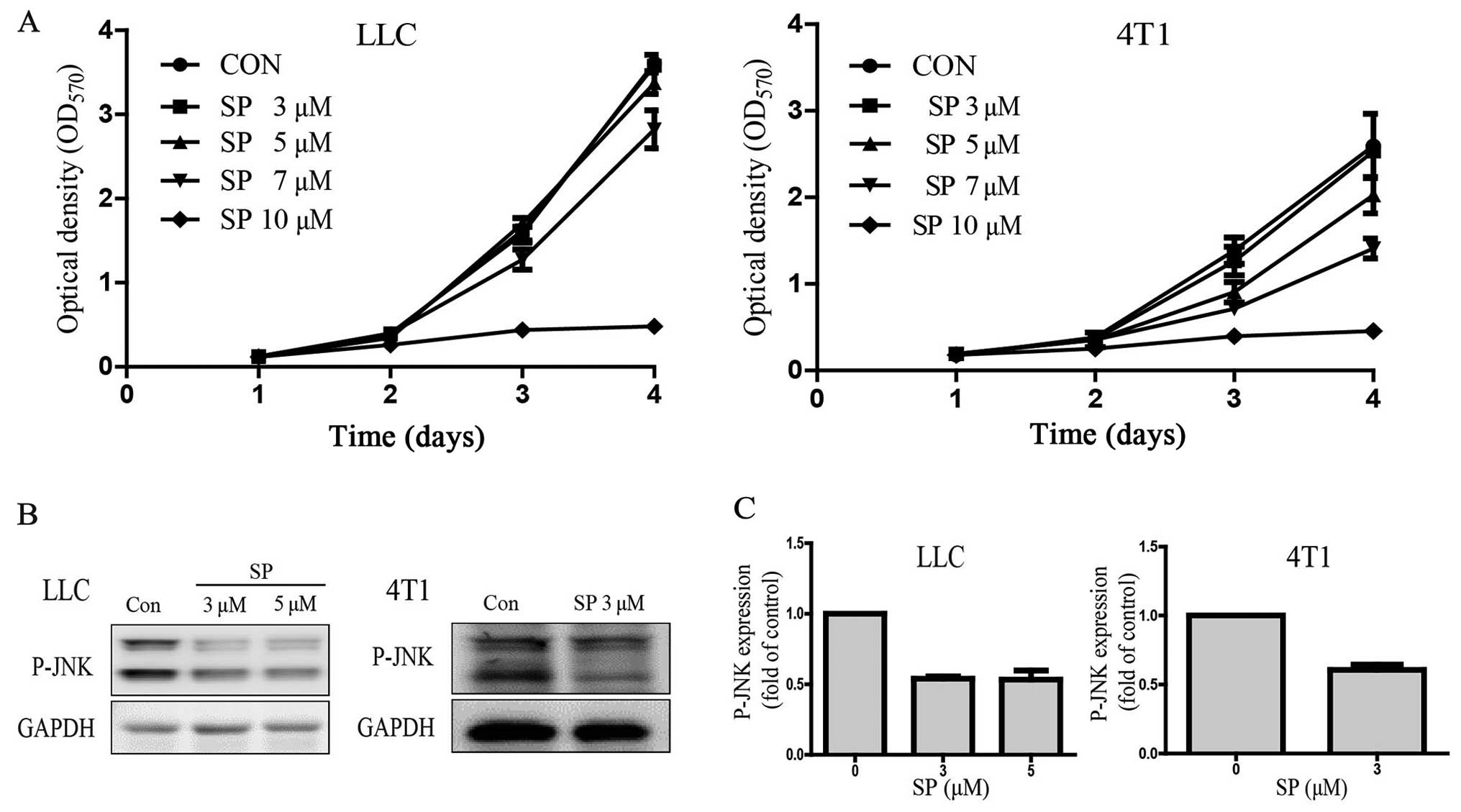
study. MTT assay results showed that optical density
dose-dependently decreased after SP600125 treatment in the LLC and
4T1 cells. The cells barely grew in the culture medium containing
10 µM SP600125 (Fig. 2A). The
IC5 of SP600125 was obtained by statistical analysis.
The IC50 was 8.14 µM and the IC5 was 5.10 µM
for LLC, and the IC50 was 7.37 µM and the IC5
was 3.04 µM for 4T1. For the subsequent clonogenic assay, doses of
SP600125 that caused toxicity below the IC5 were used:
LLC, 5 µM and 4T1, 3 µM. The results confirmed that SP600125
effectively suppressed JNK signaling at 5 µM in LLC and 3 µM in 4T1
cells (Fig. 2B and C).
Blocking of JNK signaling sensitizes
LLC and 4T1 cells to γ-irradiation
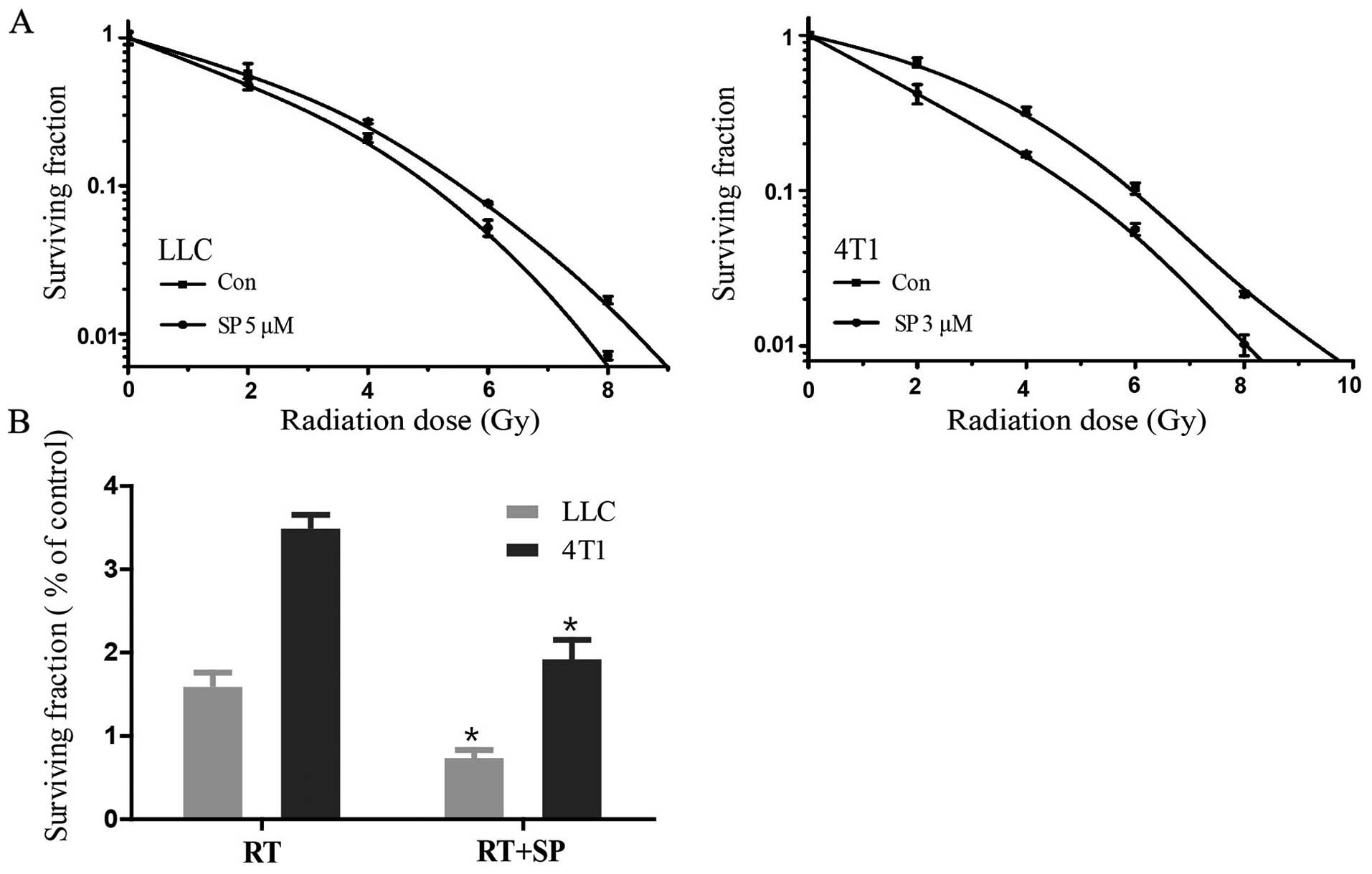
LLC and 4T1 cells were treated with different
concentrations of SP600125, 5 and 3 µM, respectively (Fig. 3A). The DEF, the ratio of the
radiation dose alone and in combination with SP600125, was
calculated by clonogenic survival at 10% (D0.1). D0.1 was defined
as the quotient of D0.1 RT/D0.1 RT + drug. A DEF >1 indicated
radiosensitization. SP600125 showed a radiosensitizing effect on
LLC and 4T1 cells with a DEF 0.1 of 1.11 and 1.21, respectively
(Table I). Treatment with a dose of
8 Gy γ-irradiation following exposure to SP600125 caused
significant decreases in the clonal formation of both cell lines
(Fig. 3B; P<0.05).
 | Table I.Dose enhancement factor (DEF) in LLC
and 4T1 cells. |
Table I.
Dose enhancement factor (DEF) in LLC
and 4T1 cells.
| Cells | Treatment | D0.1 (Gy) | DEF |
|---|
| LLC | RT | 5.56 |
|
|
| RT + SP | 5.02 | 1.11 |
| 4T1 | RT | 5.93 |
|
|
| RT + SP | 4.89 | 1.21 |
JNK regulates γH2AX expression after
γ-irradiation
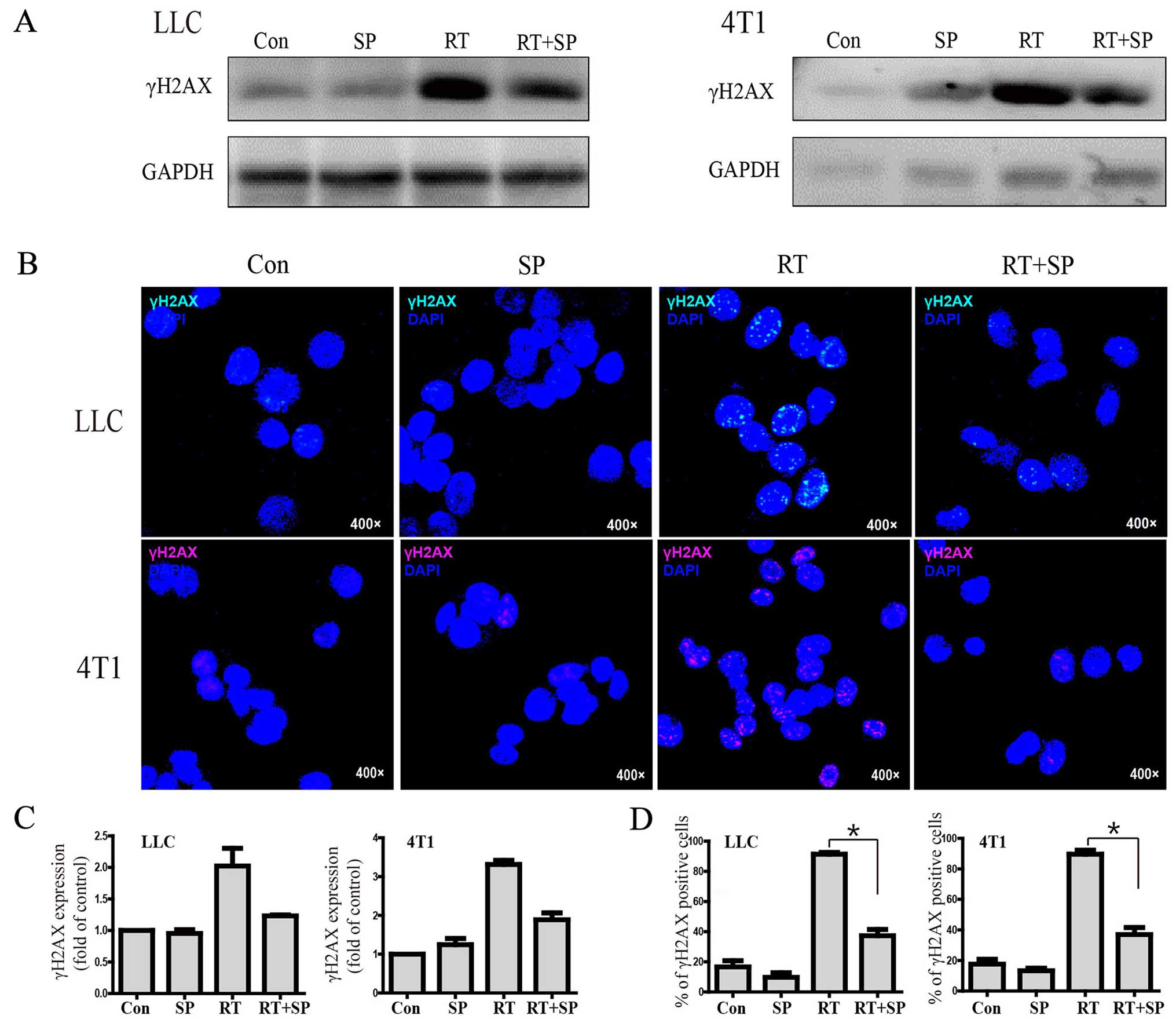
Western blot results showed that γH2AX expression
was significantly increased in the radiation treatment group
compared with the level in the control and SP600125 treatment
groups; however, it was decreased by SP600125 therapy in the LLC
and 4T1 cell lines. Similarly, immunohistochemistry results showed
that γH2AX focus formation was significantly increased in the
radiation treatment group compared with that noted in the control
and SP600125 treatment groups, however, it was decreased by
SP600125 treatment (Fig. 4A-D).
Inhibition of JNK signaling induces
apoptosis after γ-irradiation
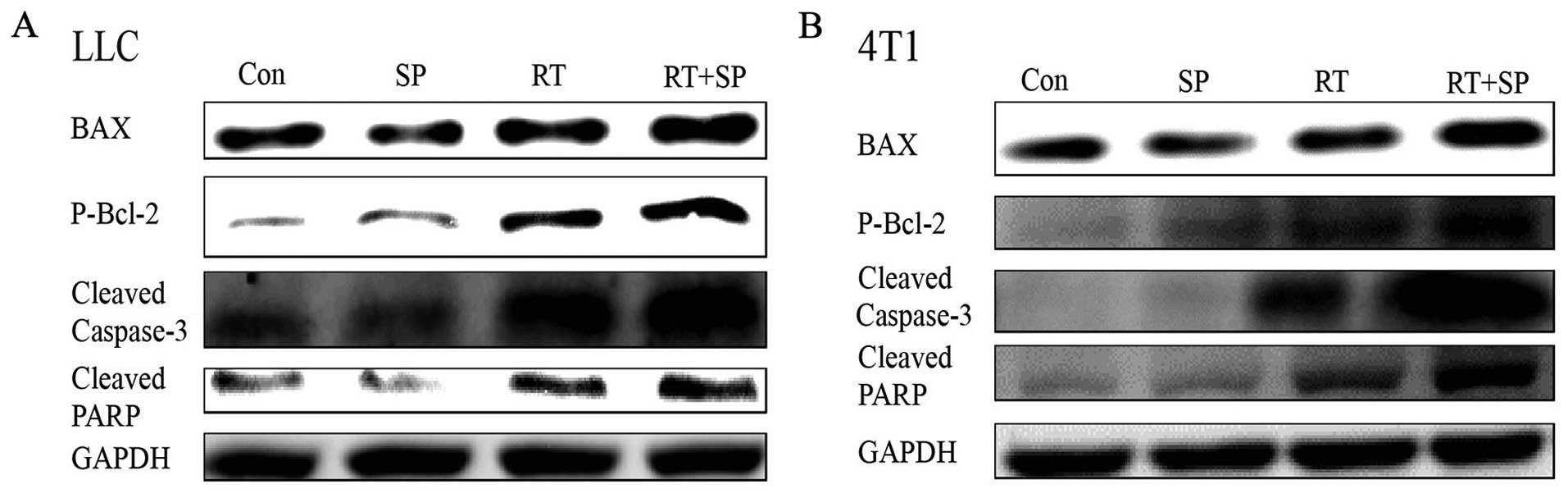
Western blot results showed that the levels of BAX,
P-Bcl-2, cleaved caspase-3 and cleaved PARP were significantly
increased in the radiation treatment group compared with these
levels in the control and SP600125 treatment groups. Moreover, this
effect was enhanced by SP600125 treatment in the LLC and 4T1 cells
(Fig. 5).
Combination of JNK inhibitor with
fractionated irradiation delays LLC tumor growth and promotes
apoptosis in vivo
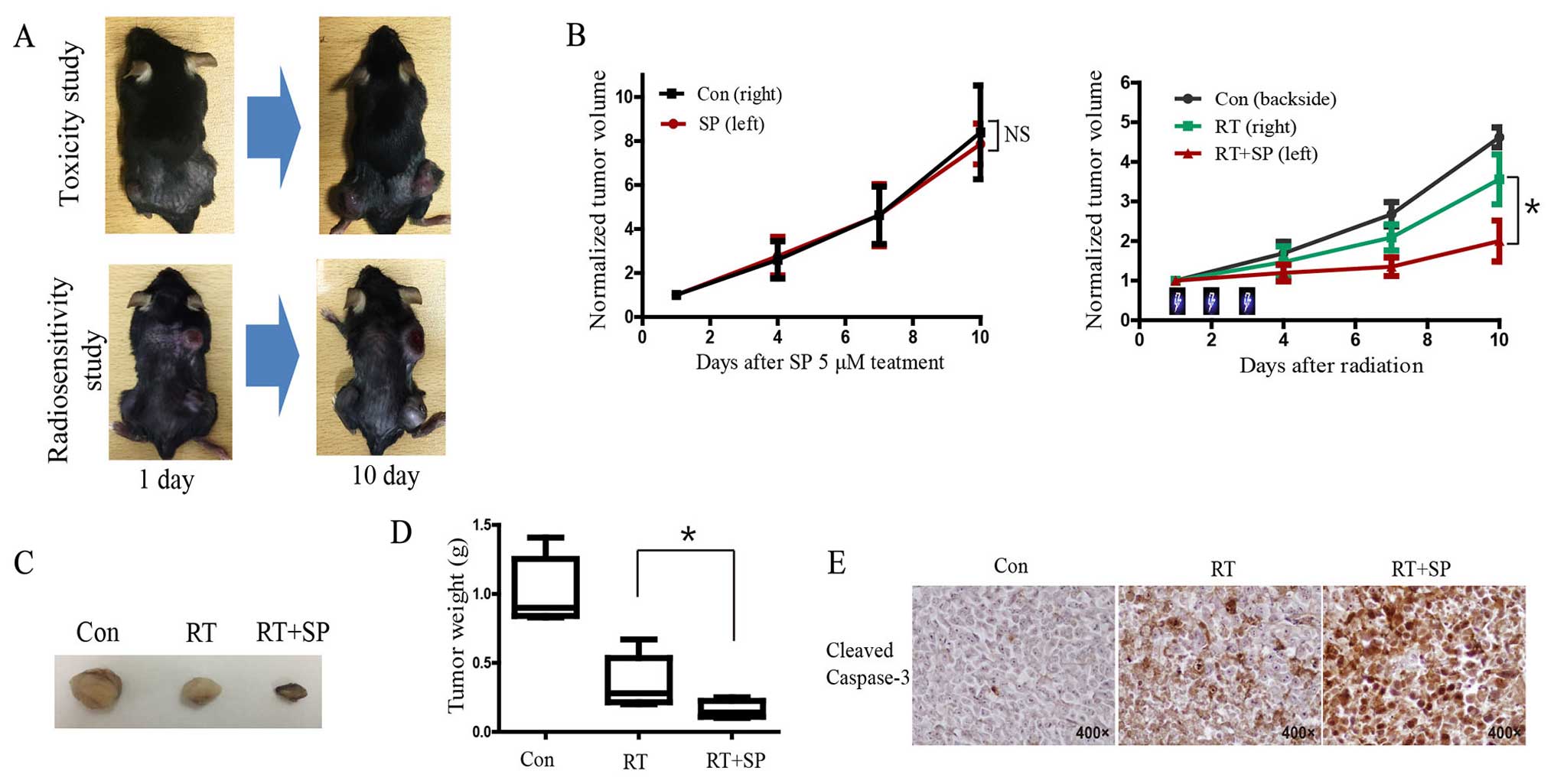
In the toxicity study on SP600125, tumor growth was
not significantly delayed in the 5 µM SP600125 treatment group
compared with the vehicle group in the hind limb LLC tumor model,
while there was significant delay in tumor growth in the SP600125
treatment group after fractionated radiotherapy in the
radiosensitivity study (Fig. 6A and
B). Tumor regrowth initiated ~6 days after RT was markedly
reduced in the SP600125-irradiated tumors. Imaging of tumor size
revealed that combined treatment with radiation and SP600125
produced a marked reduction in tumor volume at the end of the
experiment (day 10) (Fig. 6C).
Tumor weight was significantly decreased in the SP600125 treatment
group compared with that noted in the untreated group after
radiation (Fig. 6D; P<0.05).
Immunohistochemistry results showed that the level of cleaved
caspase-3 was significantly increased in the group receiving
combined SP600125 and radiation compared with the other groups
(Fig. 6E).
Discussion
It has been suggested that various drugs enhance the
radiosensitivity of metastatic brain tumor cells. Thus, more and
more research has focused on radiosensitizers to overcome the
limitations of radiation therapy. The most common sources of brain
metastases are lung (39%) and breast cancer (10–16%) (13,14).
Therefore, lung and breast cancer cell lines were used in the
present study.
Reducing the ability to repair DNA and increasing
the effects of radiotherapy are the objectives of a prolonged
endeavor in translational radiation research (15). Moreover, H2AX plays a key role in
the DNA repair system. γH2AX, which is the phosphorylated form of
H2AX, is recruited to various repair-related proteins such as p53
binding protein 1 (53BP1), breast cancer associated gene 1 (BRCA1),
Rad50 and NBS1 after DSB (16,17).
It is reported that ATM and Rad3-related protein (ATR), ataxia
telangiectasia mutated (ATM) and DNA-dependent protein kinase
(DNA-PK) play important roles in the phosphorylation of H2AX
(18–20). Moreover, Lu et al
demonstrated that JNK can phosphorylate H2AX in vitro
(4).
JNK protein is a subfamily of the mitogen activated
protein kinase (MAPK) superfamily (21). JNK consists of 10 isoforms, which
are derived from JNK1, JNK2 and JNK3 (22). JNK phosphorylation modifies the
activity of a number of proteins that reside in the mitochondria or
act in the nucleus, JNK1 and JNK2 are found in all cell and tissue
types, while JNK3 is mainly found in the brain, but also in the
heart and testes (2). JNK is
involved in cellular differentiation and proliferation,
neurodegeneration, inflammatory conditions and apoptosis mediated
by activation protein-1 (AP-1), RANTES, IL-8 and GM-CSF (23). Yue et al reported that
inhibition of JNK activity can enhance the radiosensitivity of
vestibular schwannoma cells (5).
In the present study, P-JNK was significantly
increased after radiation in the LLC and 4Tl cell lines. P-JNK may
play a vital role in H2AX phosphorylation. Unfortunately, P-JNK is
not dose-dependently increased after radiation. The JNK inhibitor
SP600125 is a small-molecule inhibitor that competes with ATP to
inhibit the phosphorylation of c-Jun (6,24). Our
results showed that SP600125 has dose-dependent cytotoxic effect on
the LLC and 4T1 cell lines. Previous studies have demonstrated that
phosphorylation of H2AX is time-dependently increased after
radiation (25). Thus, γH2AX was
measured 2 h after irradiation by western blotting and 6 h after
irradiation by immunofluorescence staining. Our results showed that
γH2AX expression was significantly increased after radiation.
However, it was decreased by SP600125 treatment in both the LLC and
4T1 cell lines. These data suggest that phosphorylation of H2AX was
suppressed by inhibition of JNK, and to the best of our knowledge,
this is the first study that γH2AX is inhibited by a JNK inhibitor
after radiation in LLC and 4T1 cell lines.
Radiosensitivity was measured by clonogenic
survival, the golden standard for measurement of radiation
sensitization. Our clonogenic assay results showed that the
necessary dose of radiation was significantly decreased in the
SP600125-treated group. This suggests that SP600125 increased the
radiosensitivity of the LLC and 4T1 cells. It has been reported
that P-JNK levels are increased after radiation and are associated
with apoptosis (26,27). Our results suggest that increased
P-JNK levels may contribute to increased phosphorylation of H2AX
after radiation. However, the mechanisms of action of the JNK
inhibitor in enhancing radiosensitivity has not yet been fully
understood.
Previous studies have demonstrated that reactive
oxygen species (ROS) levels are increased in the early (within
msec) and late (2–8 days) stages after radiation (28). ROS are formed as a natural byproduct
of oxygen metabolism and play a vital function in cell signaling
and homeostasis. ROS generally include the hydroxyl radicals (OH),
superoxide anion (O2−) and hydrogen peroxide
(H2O2), which are able to damage omnifarious
molecular targets including proteins, DNA and lipids (29,30).
Yue et al demonstrated that radiation enhanced ROS
generation, and ROS significantly increased in the radiation and
SP600125 treatment group compared with the only radiation group in
vestibular schwannoma cells (5,12). It
has been reported that mitochondrial and JNK pathways play vital
functions in ROS-induced cellular apoptosis (29,31).
The mitochondrial pathway is one of the typical apoptosis pathways.
BAX interacts with Bcl-2 and promotes the entry of cytochrome
c into the cytoplasm. Cytochrome c and caspase-9
combine to form an apoptotic body. Finally, caspase-3 is activated,
and the combination of activated caspase-3 and an apoptotic
substrate leads to apoptosis (30,32,33).
PARP is one of the enzymatic substrates of caspase-3 in the
nucleus. It plays an important role in DNA replication,
transcription, maintenance and adjustment of chromosomal structure,
stabilization of the genome and cellular apoptosis (34,35).
PARP can be degraded by caspase-3 and other caspases. Thus,
proteolysis is considered to be an early molecular marker of
apoptosis (36). Our results showed
that expression of BAX, P-Bcl-2, cleaved caspase-3 and cleaved PARP
were increased in the SP600125-treated group compared with these
levels in the untreated group after radiation. These findings
suggest that SP600125 has a radiosensitizing effect in LLC and 4T1
cells, and increases apoptosis after radiation.
Moreover, our in vivo study results showed
that there was no significant change after 5 µM SP600125 treatment
compared with vehicle treatment in the bilateral flank LLC tumor
model. However, tumor growth was significantly delayed in the
SP600125 treatment group after fractionated radiation therapy in
the backside of upper thoracic area and both flanks of the mouse
tumor model. These results suggest that SP600125 has antitumor
activity and suppressed tumor growth via inhibition of γH2AX and
DNA repair in the mouse model.
Normal brain cells, including neurons, are sensitive
to radiation; thus, the appropriate dose is limited in
radiotherapy. Therefore, a radiosensitizer provides a new method
for radiotherapy. JNK inhibitor increases radiosensitivity in
vestibular schwannoma and lung and breast cancer cells. Further
studies are needed to evaluate radiosensitivity in other cells.
In conclusion, blocking of JNK signaling decreased
γH2AX expression and increased apoptosis in lung and breast cancer
cells after radiotherapy. JNK plays an important role during
radiotherapy as a radiosensitizer via suppression of the DNA repair
system. In future, JNK inhibitors may be used as alternative
radiosensitizers to improve the outcomes of radiotherapy for
metastatic brain tumors.
References
|
1
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bode AM and Dong Z: The functional
contrariety of JNK. Mol Carcinog. 46:591–598. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weston CR and Davis RJ: The JNK signal
transduction pathway. Curr Opin Cell Biol. 19:142–149. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu C, Zhu F, Cho YY, Tang F, Zykova T, Ma
WY, Bode AM and Dong Z: Cell apoptosis: Requirement of H2AX in DNA
ladder formation, but not for the activation of caspase-3. Mol
Cell. 23:121–132. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yue WY, Clark JJ, Telisak M and Hansen MR:
Inhibition of c-Jun N-terminal kinase activity enhances vestibular
schwannoma cell sensitivity to gamma irradiation. Neurosurgery.
73:506–516. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bennett BL, Sasaki DT, Murray BW, O'Leary
EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et
al: SP600125, an anthrapyrazolone inhibitor of Jun N-terminal
kinase. Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guan QH, Pei DS, Zhang QG, Hao ZB, Xu TL
and Zhang GY: The neuroprotective action of SP600125, a new
inhibitor of JNK, on transient brain ischemia/reperfusion-induced
neuronal death in rat hippocampal CA1 via nuclear and non-nuclear
pathways. Brain Res. 1035:51–59. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li S, Li C, Ryu HH, Lim SH, Jang WY and
Jung S: Bacitracin inhibits the migration of U87-MG glioma cells
via interferences of the integrin outside-in signaling pathway. J
Korean Neurosurg Soc. 59:106–116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jin SG, Ryu HH, Li SY, Li CH, Lim SH, Jang
WY and Jung S: Nogo-A inhibits the migration and invasion of human
malignant glioma U87MG cells. Oncol Rep. 35:3395–3402.
2016.PubMed/NCBI
|
|
10
|
Wen M, Jung S, Moon KS, Jiang SN, Li SY
and Min JJ: Targeting orthotopic glioma in mice with genetically
engineered Salmonella typhimurium. J Korean Neurosurg Soc.
55:131–135. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang W, Shi L, Xie Y, Ma C, Li W, Su X,
Huang S, Chen R, Zhu Z, Mao Z, et al: SP600125, a new JNK
inhibitor, protects dopaminergic neurons in the MPTP model of
Parkinson's disease. Neurosci Res. 48:195–202. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yue WY, Clark JJ, Fernando A, Domann F and
Hansen MR: Contribution of persistent C-Jun N-terminal kinase
activity to the survival of human vestibular schwannoma cells by
suppression of accumulation of mitochondrial superoxides. Neuro
Oncol. 13:961–973. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin NU, Bellon JR and Winer EP: CNS
metastases in breast cancer. J Clin Oncol. 22:3608–3617. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shaw MG and Ball DL: Treatment of brain
metastases in lung cancer: Strategies to avoid/reduce late
complications of whole brain radiation therapy. Curr Treat Options
Oncol. 14:553–567. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fernandez-Capetillo O, Chen HT, Celeste A,
Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero
RD, Motoyama N, et al: DNA damage-induced G2-M
checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol.
4:993–997. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paull TT, Rogakou EP, Yamazaki V,
Kirchgessner CU, Gellert M and Bonner WM: A critical role for
histone H2AX in recruitment of repair factors to nuclear foci after
DNA damage. Curr Biol. 10:886–895. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Burma S, Chen BP, Murphy M, Kurimasa A and
Chen DJ: ATM phosphorylates histone H2AX in response to DNA
double-strand breaks. J Biol Chem. 276:42462–42467. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang H, Wang M, Wang H, Böcker W and
Iliakis G: Complex H2AX phosphorylation patterns by multiple
kinases including ATM and DNA-PK in human cells exposed to ionizing
radiation and treated with kinase inhibitors. J Cell Physiol.
202:492–502. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ward IM and Chen J: Histone H2AX is
phosphorylated in an ATR-dependent manner in response to
replicational stress. J Biol Chem. 276:47759–47762. 2001.PubMed/NCBI
|
|
21
|
Hibi M, Lin A, Smeal T, Minden A and Karin
M: Identification of an oncoprotein- and UV-responsive protein
kinase that binds and potentiates the c-Jun activation domain.
Genes Dev. 7:2135–2148. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Waetzig V and Herdegen T: Context-specific
inhibition of JNKs: Overcoming the dilemma of protection and
damage. Trends Pharmacol Sci. 26:455–461. 2005.PubMed/NCBI
|
|
23
|
Oltmanns U, Issa R, Sukkar MB, John M and
Chung KF: Role of c-jun N-terminal kinase in the induced release of
GM-CSF, RANTES and IL-8 from human airway smooth muscle cells. Br J
Pharmacol. 139:1228–1234. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barr RK, Kendrick TS and Bogoyevitch MA:
Identification of the critical features of a small peptide
inhibitor of JNK activity. J Biol Chem. 277:10987–10997. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
An J, Huang YC, Xu QZ, Zhou LJ, Shang ZF,
Huang B, Wang Y, Liu XD, Wu DC and Zhou PK: DNA-PKcs plays a
dominant role in the regulation of H2AX phosphorylation in response
to DNA damage and cell cycle progression. BMC Mol Biol. 11:182010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim CH, Won M, Choi CH, Ahn J, Kim BK,
Song KB, Kang CM and Chung KS: Increase of RhoB in
gamma-radiation-induced apoptosis is regulated by c-Jun N-terminal
kinase in Jurkat T cells. Biochem Biophys Res Commun.
391:1182–1186. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han Y, Wang Y, Xu HT, Yang LH, Wei Q, Liu
Y, Zhang Y, Zhao Y, Dai SD, Miao Y, et al: X-radiation induces
non-small-cell lung cancer apoptosis by upregulation of Axin
expression. Int J Radiat Oncol Biol Phys. 75:518–526. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao Z, Sarsour EH, Kalen AL, Li L, Kumar
MG and Goswami PC: Late ROS accumulation and radiosensitivity in
SOD1-overexpressing human glioma cells. Free Radic Biol Med.
45:1501–1509. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shen HM and Liu ZG: JNK signaling pathway
is a key modulator in cell death mediated by reactive oxygen and
nitrogen species. Free Radic Biol Med. 40:928–939. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jena NR: DNA damage by reactive species:
Mechanisms, mutation and repair. J Biosci. 37:503–517. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu CC, Ko FY, Yu CS, Lin CC, Huang YP,
Yang JS, Lin JP and Chung JG: Norcantharidin triggers cell death
and DNA damage through S-phase arrest and ROS-modulated apoptotic
pathways in TSGH 8301 human urinary bladder carcinoma cells. Int J
Oncol. 41:1050–1060. 2012.PubMed/NCBI
|
|
32
|
Zeng H, Kong X, Peng H, Chen Y, Cai S, Luo
H and Chen P: Apoptosis and Bcl-2 family proteins, taken to chronic
obstructive pulmonary disease. Eur Rev Med Pharmacol Sci.
16:711–727. 2012.PubMed/NCBI
|
|
33
|
Tang S, Hu J, Meng Q, Dong X, Wang K, Qi
Y, Chu C, Zhang X and Hou L: Daidzein induced apoptosis via
down-regulation of Bcl-2/Bax and triggering of the mitochondrial
pathway in BGC-823 cells. Cell Biochem Biophys. 65:197–202. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miwa M and Masutani M:
PolyADP-ribosylation and cancer. Cancer Sci. 98:1528–1535. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wei L, Nakajima S, Hsieh CL, Kanno S,
Masutani M, Levine AS, Yasui A and Lan L: Damage response of XRCC1
at sites of DNA single strand breaks is regulated by
phosphorylation and ubiquitylation after degradation of
poly(ADP-ribose). J Cell Sci. 126:4414–4423. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Germain M, Affar EB, D'Amours D, Dixit VM,
Salvesen GS and Poirier GG: Cleavage of automodified
poly(ADP-ribose) polymerase during apoptosis. Evidence for
involvement of caspase-7. J Biol Chem. 274:28379–28384. 1999.
View Article : Google Scholar : PubMed/NCBI
|