Introduction
Chronic myeloid leukemia (CML) is a clonal disease
of hematopoietic stem cells characterized by the Philadelphia
chromosome (Ph) generated by the reciprocal translocation
t(9;22)(q34;q11) resulting in the BCR-ABL1 fusion gene. A
constitutively upregulated tyrosine kinase activity of BCR-ABL1
contributes to malignant transformation of cells (1). The disease is usually recognized in a
chronic phase (CP) that may continue for several years. However,
the progression of CP may lead to an advanced stage that includes
an accelerated phase (AP) and blast crisis (BC). In most cases, CP
is suppressed with imatinib mesylate (IM), a tyrosine kinase
inhibitor (TKI) of the BCR-ABL1 protein. The resistance to IM that
develops in a portion of patients in CP is mostly caused by a
mutation in the kinase domain abolishing binding of IM to BCR-ABL1
or by amplification of the BCR-ABL1 gene (2).
Current molecular diagnosis of CML and evaluation of
the disease response to therapy are based on the detection of the
BCR-ABL1 transcript, monitoring of its level, and mutation analysis
of the BCR-ABL1 kinase domain. Increase in the BCR-ABL1 mRNA level
in CP is a marker of resistance to therapy, which suggests the risk
of a relapse and the necessity of treatment modification (3,4).
However, reliable markers of CML progression to predict disease
prognosis and thus to modify the treatment protocol accordingly are
still needed.
During CML progression, genomic instability in
leukemic cells leads to the accumulation of mutations including
chromosomal abnormalities, which are found in ~80% of CML patients
(5). These genetic changes result
in the generation of cells independent of the transformation
potential of the BCR-ABL1 protein. Moreover, in BC, the second type
of leukemia stem cells has been identified, granulocyte-macrophage
progenitors responsible for rapid expansion of immature blast cells
(6).
Centrosomes are cellular organelles that play an
essential role in the separation of chromosomes into daughter
cells. The development of malignant diseases is associated with
centrosome aberrations that lead to genetic instability and
chromosomal aberrations. In CML, the extent of centrosome
abnormalities is correlated with disease stage and karyotype
alterations (7).
Increased production of transcripts originating from
various genes encoding centrosomal proteins has been confirmed as a
negative prognostic marker in a variety of malignant diseases, but,
with the exception of the hyaluronan-mediated motility receptor
(HMMR) gene, their expression has not been tested in CML. Using
RT-PCR, Schmitt et al (8)
found HMMR mRNA expression in 75% of CML patients in CP and in 100%
of advanced CML patients. They also found a correlation of HMMR and
BCR-ABL1 expression in a patient with remission followed by relapse
and demonstrated an HMMR-specific T cell response in 80% of
patients. HMMR has been identified as a tumor-associated antigen
(TAA) in acute myeloid leukemia (AML) and CML (9). Induction of HMMR-specific T cell
responses has been found in patients with chronic lymphocytic
leukemia (CLL) after immunization with a peptide vaccine (10).
Aurora kinase A (AURKA) that regulates centrosome
duplication and separation is another centrosomal protein
overexpressed in CML and a candidate vaccine target (11). As the AURKA gene is overexpressed or
amplified in a variety of human tumors and kinase activity of the
produced protein is involved in tumorigenesis, inhibitors of this
kinase have been developed and tested in clinical trials (12).
Polo-like kinase 1 (PLK1) is implicated in numerous
mitotic events including centrosome maturation. Similar to AURKA,
it also has oncogenic potential and inhibitors of its kinase
activity have been developed (13).
In CML, the inhibition of PLK1 activity suppressed the
proliferation of both IM-sensitive and IM-resistant leukemic cells
(14).
The endopeptidase separase, the product of the extra
spindle poles-like 1 (ESPL1) gene, plays a vital role in the
separation of chromatids in mitosis. Its overexpression induced
aneuploidy and tumorigenesis (15).
Increased ESPL1 transcription in solid tumors is correlated with a
high incidence of relapse and metastasis and with a lower survival
rate (16). Moreover, a
meta-selection of genomic datasets obtained from solid tumors
identified ESPL1 as one of the four genes with the highest
prognostic impact on survival outcome (17). In CML cells treated with IM, the
separase level was reduced but its proteolytic activity was
increased (18).
Overexpression of TAAs can be accompanied by
serologic responses that show evidence of TAA immunogenicity.
Simultaneously, if immunity contributes to cancer elimination, the
generation of anti-TAA antibodies can be a positive prognostic
marker. For instance, in a study of WT1 in myelodysplastic
syndrome, an increased level of mRNA predicted worse survival,
while a high level of antibodies was associated with longer
survival (19).
In the present study, we measured the expression of
four genes (AURKA, ESPL1, HMMR and PLK1) encoding centrosomal
proteins and determined the production of antibodies against these
proteins in CML patients treated with IM. We obtained data at
diagnosis and during treatment and evaluated their significance for
disease prognosis.
Materials and methods
Patients and samples
Patients diagnosed with CML and treated at the
Institute of Hematology and Blood Transfusion, Prague, between 2010
and 2015 were included in the present study for prospective
follow-up. In total, 36 patients newly diagnosed with CML in CP
were enrolled. All but 2 patients were treated with 400 mg IM/day
for at least 12 months. In one patient, this dose was reduced to
300 mg for two months due to side-effects and in one patient, IM
was applied in a 600 mg dose from the seventh month of therapy.
Thirty-three patients were followed up for at least 24 months and
the remaining 3 patients for only 9, 12 and 13 months due to their
death. Altogether, 5 patients died during the study, 2 of them due
to progression of CML. Peripheral blood samples were taken at
diagnosis and regular trimonthly monitoring for BCR-ABL1 was
conducted during IM therapy. Blood samples of age- and
gender-matched healthy controls were also collected. The
characteristics of the study cohort are summarized in Table I. The response to IM treatment
(optimal, warning or failure) was evaluated according to the
European LeukemiaNet (ELN) recommendations based on the BCR-ABL1
transcript levels (20). The
present study was approved by the Institutional Ethics Committee of
the Institute of Hematology and Blood Transfusion, Prague and
conducted according to the Declaration of Helsinki. All subjects
signed the informed consent to participate in the present
study.
 | Table I.Baseline characteristics of the 36
patients newly diagnosed with CML in the chronic phase. |
Table I.
Baseline characteristics of the 36
patients newly diagnosed with CML in the chronic phase.
| No. of patients | Total |
|---|
| Age (years) |
|
| Median
(range) | 53.5 (19–81) |
| Gender, n (%) |
|
|
Males/females | 15/21
(41.7/58.3) |
| Months of
follow-up |
|
| Median
(range) | 26.6 (9.0–39.3) |
| Sokal risk score, n
(%) |
|
| Low | 15 (41.7) |
|
Intermediate | 14 (38.9) |
|
High | 7
(19.4) |
| EUTOS risk score, n
(%) |
|
|
Low | 29 (80.6) |
|
High | 7
(19.4) |
| Transcript type
(%) |
|
| b2a2
(e13b2) | 15 (41.7%) |
| b3a2
(e14b2) | 19 (52.8%) |
| b2a2
and b3a2 | 2
(5.5%) |
| WBC count
(×109/l) |
|
| Median
(range) | 63.25
(8.21–365.77) |
| Platelet count
(×109/l) |
|
| Median
(range) | 394.00
(108.00–1,376.00) |
| Hg level (g/l) |
|
| Median
(range) | 119.50
(85.00–154.00) |
| Basophils in blood
(%) |
|
| Median
(range) | 6.70
(1.70–14.70) |
| Blast cells in
blood (%)a |
|
| Median
(range) | 0.70 (0–23.0) |
Gene expression analysis
Total RNA was prepared from peripheral blood
leukocytes using TRIzol reagent, and cDNA was synthesized by M-MLV
reverse transcriptase (Promega, Madison, WI, USA) using random
hexamer primers (21). The level of
mRNA expression of BCR-ABL1 and genes encoding centrosomal proteins
(called centrosomal genes in the present study) were analyzed
trimonthly since the start of IM treatment by reverse-transcriptase
quantitative real-time polymerase chain reaction (RT-qPCR). The
β-glucuronidase gene (GUSB) that is used for standardized BCR-ABL1
monitoring in international scale (IS) units served as a reference
gene also for expression analysis of centrosomal genes. BCR-ABL1
quantification was standardized within the European Treatment and
Outcome Study (EUTOS) for the CML project of ELN and data were
reported in the IS units. Primers and probes for BCR-ABL1 and GUSB
were applied according to the Europe Against Cancer
recommendations, and commercial plasmid standards were used to
obtain calibration curves (Ipsogen, Marseille, France). The
transcripts of centrosomal genes were amplified in the following
TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA,
USA): Hs01582073_m1 for AURKA, Hs00234864_m1 for HMMR,
Hs00901772_m1 for ESPL1 and Hs00153444_m1 for PLK1. The qPCR was
performed on Rotor-Gene 3000A (Qiagen, Hilden, Germany). Briefly,
10 µl of the PCR mixture contained 5 µl of TaqMan Universal Master
Mix II, without UNG (Applied Biosystems), 0.5 µl of TaqMan Gene
Expression Assay, and 2 µl of 3-fold diluted cDNA. After initial
incubation at 50°C for 2 min and polymerase activation at 95°C for
10 min, each of the 40 cycles consisted of denaturation at 94°C for
15 sec, and primer annealing and elongation at 60°C for 60 sec.
After amplification, analyses were performed using the Rotor-Gene
software. The relative quantification of mRNA expression was
evaluated by the ΔΔCq method (22).
The mean gene expression in the healthy controls was used as a
calibrator.
Recombinant proteins
Antigens for the detection of antibodies in an
enzyme-linked immunosorbent assay (ELISA) were produced using a
glutathione S-transferase (GST) gene fusion system in E.
coli. Due to the size limitation of this system, partial
sequences of the human genes AURKA (accession no. BC001280;
nucleotides 600–1212 of the coding sequence), HMMR (BC108904;
1551–2178), PLK1 (BC014846; 4–670), and ESPL1 (NM_012291; 4–2106)
were amplified from plasmids obtained from the Harvard Medical
School (Boston, MA, USA) by Phusion High-Fidelity DNA polymerase
(Finnzymes, Espoo, Finland) using the following primers containing
XhoI restriction sites (underlined sequences): AURKA,
5′-TTAGCTCGAGGTCCATGATGCTACCAGAGTCTAC-3′
(forward) and 5′-TTAGCTCGAGAGACTGTTTGCTAGCTGATTCTTTG-3′
(reverse); PLK1, 5′-TGTACTCGAGAGTGCTGCAGTGACTGCAGGG-3′
(forward) and 5′-TTAGCTCGAGGCTCAGCACCTCGGGAGCTATG-3′
(reverse); ESPL1, 5′-TGTACTCGAGAGGAGCTT
CAAAAGAGTCAACTTTGG-3′ (forward) and 5′-TTAGCTCGAGTCTCCGATCCCGCTCGATAC-3′
(reverse); HMMR, 5′-TGTGCTCGAGACCAAGTCAGCACTAAAGG
(forward) and 5′-TTAGCTCGAGCTTCCATGATTCTTGACACTCCATAG-3′
(reverse).
After digestion with XhoI (New England
Biolabs, Ipswich, MA, USA), the PCR products were cloned into the
pGEX-5X-1 vector carrying the GST gene (GE Healthcare, Little
Chalfont, UK) and the cloned sequences were verified using the
BigDye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems).
For the production of recombinant GST-fusion proteins, E.
coli BL21-CodonPlus (DE3)-RIPL competent cells (Agilent
Technologies, Santa Clara, CA, USA) were transformed with the
constructed plasmids and incubated at 37°C in Luria-Bertani medium
(Duchefa, Haarlem, The Netherlands) with 100 µg/ml ampicillin and
2% glucose (Sigma-Aldrich, St. Louis, MO, USA). At an
OD600 of 1.2–1.6, the production of recombinant proteins
was induced by adding 0.3 mM isopropyl-β-D-thio-galactoside (IPTG;
Duchefa) and the cells were incubated at 30°C for 6 h. The
bacterial pellets were resuspended in lysis buffer: 20 mM Tris-HCl
(pH 8.0), 200 mM NaCl, 2 mM DTT, 200 mM protease inhibitor
phenylmethylsulfonyl fluoride (PMSF; Serva, Heidelberg, Germany),
and 0.5% N-lauroylsarcosine (Sigma-Aldrich) in the case of soluble
GST-fusion proteins, HMMR, PLK1 and ESPL1. Since the GST-fusion
protein AURKA was insoluble in this lysis buffer it was dissolved
in a buffer with 2% N-lauroylsarcosine [10 mM Tris-HCl (pH 8.0),
150 mM NaCl, 1 mM EDTA, 7 mM DTT, 200 mM PMSF and 2%
N-lauroylsarcosine] (23). The
resuspended cells were sonicated three times for 10–15 sec on ice
and then lysed three times in the EmulsiFlex-C5 high pressure
homogenizer (Avestin, Ottawa, Canada). The lysates were
centrifugated (4°C, 30 min, 16,000 × g) and the supernatants were
mixed 1:1 with glycerol (Sigma-Aldrich) and stored at −20°C. The
production of recombinant proteins was verified by sodium dodecyl
sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and
immunoblotting.
Detection of antibodies
Polysorb 96-well plastic plates (Nunc, Roskilde,
Denmark) were coated overnight at 4°C with glutathione casein (200
ng/well) in 50 mM carbonate buffer (pH 9.6) as previously described
(24). Human sera were diluted 1:50
in blocking buffer [0.2% casein (Sigma-Aldrich) and 0.05% Tween-20
(Serva) in phosphate-buffered saline (PBS)] and incubated with the
lysate containing the fusion antigen (Ag) or only the GST tag at
room temperature for 3 h. Control samples were incubated without
lysates (without Ag). The wells of the coated plates were incubated
with 200 µl of blocking buffer at 37°C for 1 h and then with 50 µl
of the bacterial lysates containing the GST fusion proteins diluted
in blocking buffer (1 µg/µl total proteins) at 37°C for 1 h. The
plates were washed five times with washing buffer (0.05% Tween-20
in PBS) and the pre-incubated human sera were added and incubated
at 37°C for 1 h. After washing, the plates were loaded with donkey
anti-human IgG polyclonal antibodies conjugated to horseradish
peroxidase (Jackson ImmunoResearch Laboratories, West Grove, PA,
USA) diluted 1:7,500 in blocking buffer, and incubated at 37°C for
1 h. The plates were washed and stained with tetramethylbenzidine
substrate (Sigma-Aldrich). The absorbance was measured at 450 and
630 nm. The specificity of the antibody reactions was determined by
the inhibition of the reactions with the homologous antigens in
comparison with the effect of the control GST protein using the
formula: (Awithout Ag - Awith
Ag)/(Awithout Ag - Awith GST).
Statistical analysis
Comparison of groups was performed using the
Mann-Whitney test. The Spearman correlation coefficient was
calculated to evaluate the relationship between independent
variances. Survival outcomes were compared using the log-rank test.
Failure was defined as death, progression to AP or BC, failure to
achieve BCR-ABL1 transcript levels <10% at 6 months or <1% at
12 months, or major molecular response (MMR) loss. Results were
considered significantly different at P<0.05. Calculations were
performed using GraphPad Prism 5.02 software (GraphPad Software,
San Diego, CA, USA).
Results
In our previous study (25), we analyzed the dataset from gene
expression profiling of three subpopulations of CML stem/progenitor
cells (Lin−CD34−,
Lin−CD34+ and
Lin+CD34+) (4). Genes with at least twice increased
expression in all three subpopulations of leukemic cells in
comparison to healthy controls were identified and analyzed by the
Database for Annotation, Visualization and Integrated Discovery
(DAVID) v6.7 bioinformatics tool. Twelve upregulated genes encoding
centrosomal proteins (called centrosomal genes in the present
study) are listed in Table II. The
expression of four of them with relevance for CML as described in
the Introduction (AURKA, HMMR, PLK1 and ESPL1) was analyzed in the
present study.
| Table II.Centrosomal genes upregulated in
CMLa. |
Table II.
Centrosomal genes upregulated in
CMLa.
| Symbol | Official full
name |
|---|
| AURKA | Aurora kinase
A |
| BRCA2 | Breast cancer 2,
early onset |
| CALM3 | Calmodulin 3
(phosphorylase kinase, δ) |
| ESPL1 | Extra spindle pole
bodies homolog 1 (S. cerevisiae) |
| FEZ1 | Fasciculation and
elongation protein ζ1 (zygin I) |
| HMMR | Hyaluronan-mediated
motility receptor (RHAMM) |
| MARCKS | Myristoylated
alanine-rich protein kinase C substrate |
| NUDT21 | Nudix (nucleoside
diphosphate linked moiety X)-type motif 21 |
| PLK1 | Polo-like kinase
1 |
| PLK4 | Polo-like kinase
4 |
| RPGRIP1L | RPGRIP1-like |
| TOP2A | Topoisomerase (DNA)
IIα 170 kDa |
Expression of centrosomal genes is
significantly increased at diagnosis
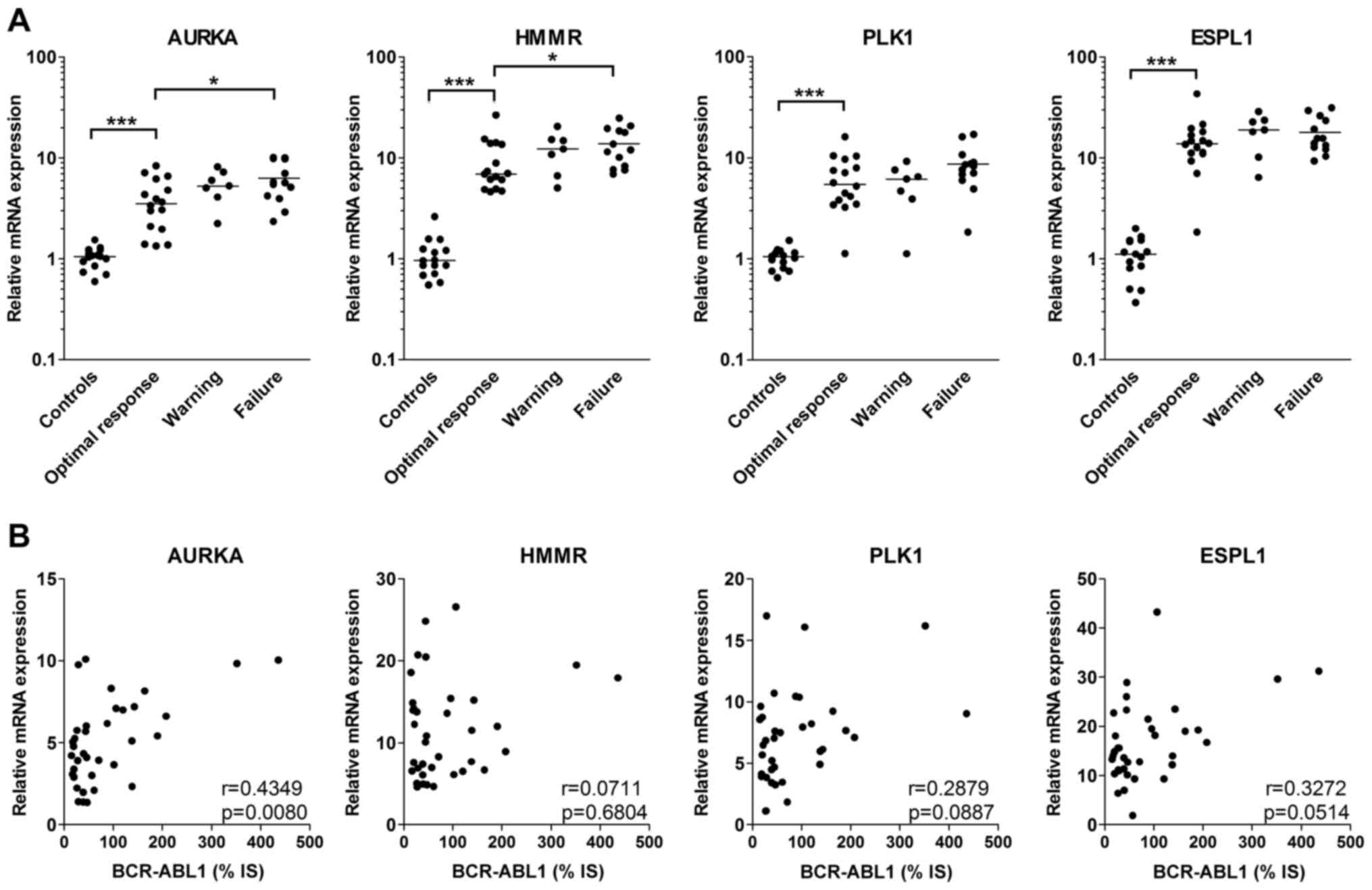
Significantly enhanced expression of all four
evaluated centrosomal genes in CML patients was found in total
peripheral blood leukocytes at diagnosis in comparison with the
level in the healthy controls (Fig.
1A). When patients were stratified based on the response to
one-year treatment with IM, patients with optimal response had
lower expression of the centrosomal genes at diagnosis than
patients with warning or failure, but this difference was
significant only for AURKA and HMMR in the patients with treatment
failure (Fig. 1A).
As the expression of all studied centrosomal genes
has been suggested to be upregulated by BCR-ABL1, the correlation
of mRNA expression at diagnosis between centrosomal genes and
BCR-ABL1 was also analyzed. However, a statistically significant
correlation is shown only for AURKA (Fig. 1B).
Delayed reduction in centrosomal-gene
expression is associated with worse overall survival
During IM treatment, peripheral blood samples were
regularly taken from patients and the mRNA expression of BCR-ABL1
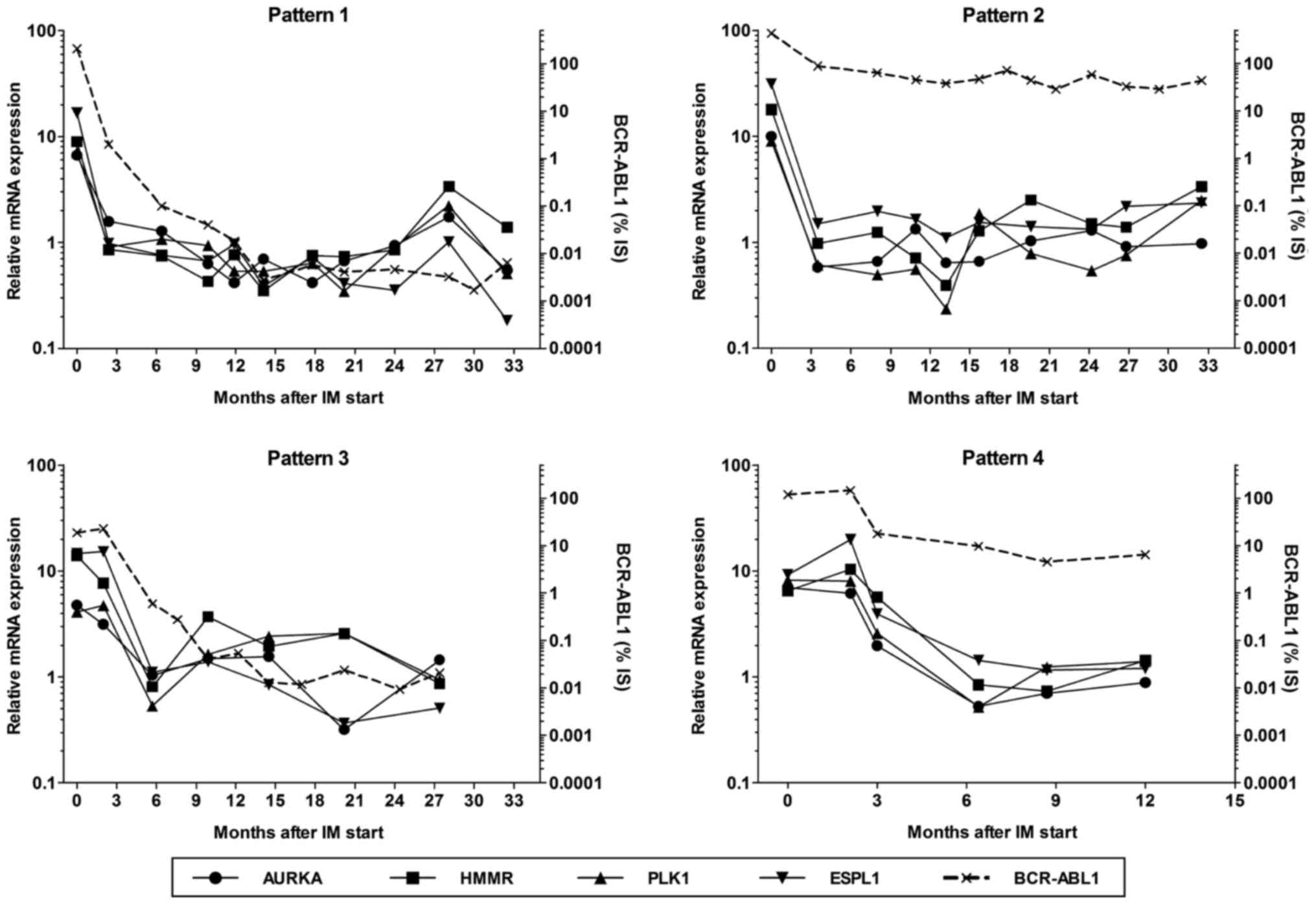
and centrosomal genes was quantified. We found four patterns of
gene expression kinetics (Fig. 2):
in pattern 1 (15 patients), the expression of centrosomal genes
reached the basal level (defined by a decrease in relative
expression below 2 for all genes; i.e. the mRNA levels were not
twice as high as those in the controls) after three months of IM
treatment and MMR was achieved within 12 months. In pattern 2 (15
patients), the expression of centrosomal genes was also quickly
reduced, but MMR was not recorded within 12 months of treatment.
Pattern 3, characterized by the delayed decrease in centrosomal
gene expression (i.e. the basal level was not achieved within three
months after IM start) and by MMR within 12 months, was found in
one patient who showed MMR three months after an increase in IM
dose to 600 mg/day. In 5 patients with pattern 4, the basal level
of transcripts of the centrosomal genes was detected with a delay
(5–9 months after IM start) and MMR was not achieved within 12
months. Both patients who died of CML showed this expression
pattern.
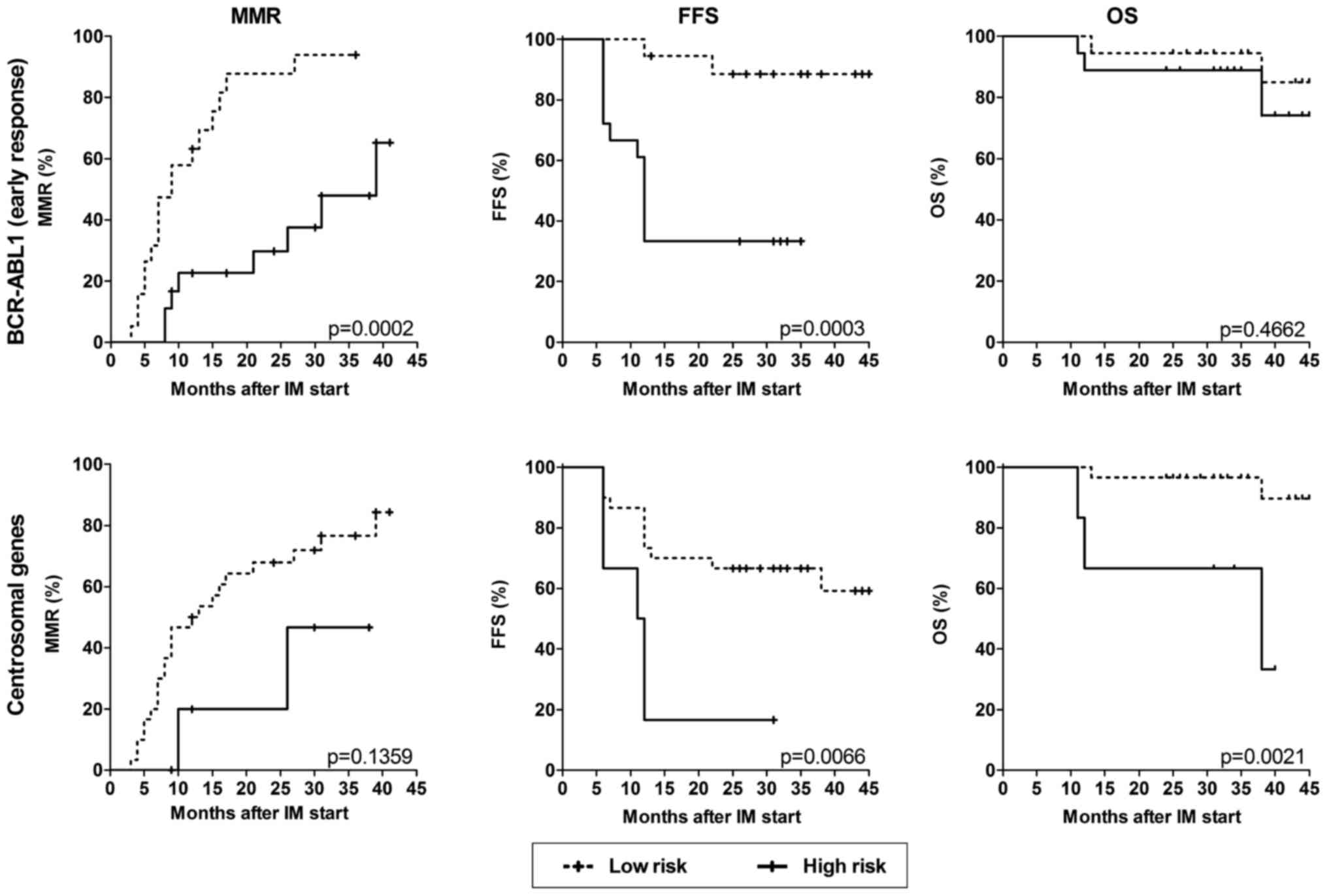
To analyze the association of the expression of
centrosomal genes with the response to IM treatment, patients were
stratified into a low-risk group with a decrease in centrosomal
gene expression at the first follow-up after IM start (patterns 1
and 2, 30 patients) and a high-risk group with a delayed decrease
(patterns 3 and 4, 6 patients). For comparison, early molecular
response (EMR) at three months was evaluated and patients were
divided into a low-risk group (BCR-ABL1 ≤10%, 19 patients) and a
high-risk group (BCR-ABL1 >10%, 17 patients). While EMR
predicted better cumulative incidence of MMR and failure-free
survival (FFS), the delayed reduction in centrosomal gene
expression in the course of IM treatment was significantly
associated with worse overall survival (OS) (Fig. 3).
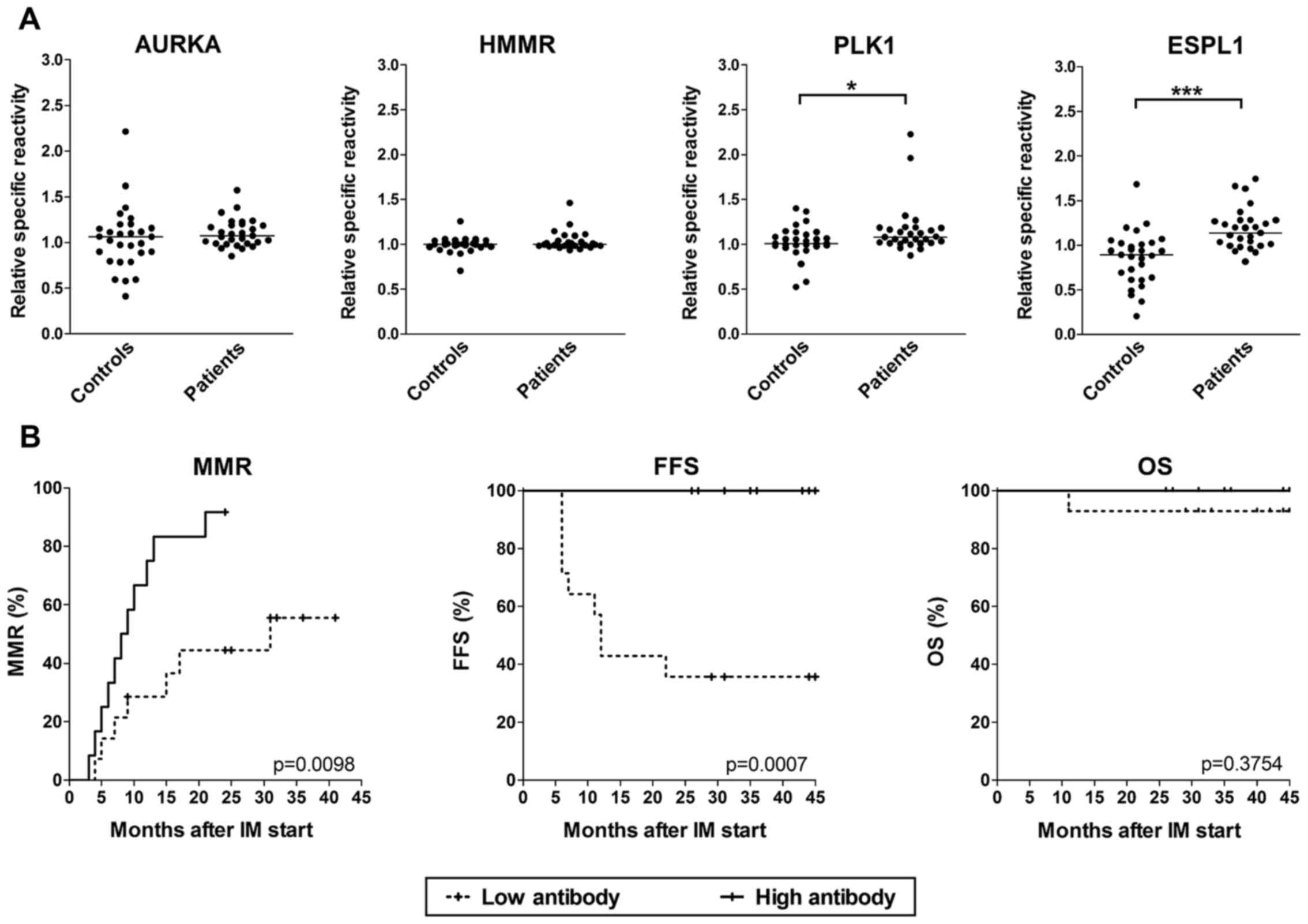
High overall antibody production is
associated with better FFS
In enrollment sera of 29 patients, the antibodies
against AURKA, HMMR, PLK1 and ESPL1 antigens were determined by
ELISA using GST-fusion recombinant proteins produced in E.
coli. Due to nonspecific reactions, we did not quantify
antibodies by sera titration, but the sera were incubated with
antigen-carrying fusion proteins or control GST and the relative
inhibition of specific reactions was calculated. For ESPL1 and
PLK1, a statistically significant increase was found in comparison
with sera from blood donors (Fig.
4A). After stratification of sera into low and high antibody
groups based on median production, no significant differences were
found for individual antigens (data not shown), but when the
overall antibody production against all four antigens was evaluated
(means were calculated from the values of all four antigens),
significantly higher cumulative incidence of MMR and FFS were shown
for patients with high production of antibodies (Fig. 4B).
Discussion
As centrosomal proteins play essential roles in cell
division, their expression is often increased in malignant cells.
This increase can be associated with centrosome aberration leading
to reduced genome stability, thus contributing to tumor
progression. Our analysis (25) of
the microarray dataset published by Lemoli et al (4) showed that the expression of numerous
centrosomal genes was upregulated in CML progenitor/stem cells.
Furthermore, a prognostic value of the HMMR overexpression has been
shown for multiple myeloma (26)
and B-cell chronic lymphocytic leukemia (27). Therefore, in the present study, we
tested the expression of four centrosomal genes. We found markedly
enhanced expression of all analyzed genes, particularly of ESPL1,
at diagnosis. However, when patients with optimal response and
failure were compared, only AURKA and HMMR showed a slightly
significant difference in gene expression. Moreover, the
probability of MMR achievement was significantly higher only in
patients with low AURKA expression, and no significant difference
was found for FFS (data not shown). Based on these data, we suggest
that the expression of centrosomal genes in total leukocytes at
diagnosis cannot be used as a prognostic marker in CML.
The follow-up of the expression of centrosomal genes
during IM treatment showed that all patients achieved the basal
expression. The majority of them (83%) did so in three months. As
in most of the remaining patients (5/6), the delayed decrease in
centrosomal gene expression was associated with treatment failure,
we assigned these patients to the high-risk group. The survival
analysis showed significantly reduced OS in these patients.
Notably, while the three patients from this group who died during
the study (two of them due to CML progression) had the BCR-ABL1
transcript level at the first follow-up after IM start reduced
below 20% of that at diagnosis, the remaining three patients who
survived had this level increased to >110%.
The four tested centrosomal genes have been
suggested to be induced by the BCR-ABL1 protein (8,14,18,28).
For AURKA, AKT signaling has been shown to be implicated in this
induction (28). However, at
diagnosis, we found a significant correlation with BCR-ABL1 at the
mRNA level only for AURKA. Surprisingly, in all patients with
therapy failure, centrosomal gene expression decreased to the basal
level during IM treatment which could suggest that centrosomal gene
expression can be normalized in differentiated cells in spite of
resistance to IM therapy.
When humoral response to centrosomal proteins was
investigated, we found that patients at diagnosis produced
significantly more IgG antibodies against the ESPL1 and PLK1
antigens than healthy controls. The antibody levels did not
correlate with mRNA expression of the particular gene (data not
shown). Survival analysis of antibodies against individual antigens
did not show significant differences, but that with the overall
antibodies against all four antigens revealed a higher probability
of MMR and FFS in patients with a high antibody production. IgM
antibodies were not found in any of the analyzed samples and IgG
antibodies did not significantly change after one-year IM treatment
(data not shown).
It has been shown that IM treatment has
immunosuppressive effects in CML patients that particularly
influence effector T cells and dendritic cells (29). Nevertheless, immune responses during
IM treatment contribute to its efficacy and can be enhanced by
immunization (30). Various target
antigens, including HMMR, have been identified by serological
analysis (31). In the present
study, the level of detected antibodies against HMMR was low,
possibly due to the fact that we were able to produce only one half
of the HMMR antigen for ELISA which could lack immunodominant
epitope(s).
Despite the limited number of patients enrolled in
the present study, our data suggest that the increased production
of transcripts of centrosomal genes at CML diagnosis cannot be used
in disease prognosis, but the lack of early response of centrosomal
gene expression to IM therapy could be considered as a risk factor
for CML progression. Furthermore, we suppose that immune responses
against centrosomal proteins may contribute to the antitumor effect
of IM treatment.
Acknowledgements
We thank P. Vesela and K. Vlcanova for their
technical assistance and M. Duskova, I. Polakova, V. Ludvikova, K.
Kernova and P. Ptackova for their assistance with antibody
detection. The present study was supported by a grant
(NT13862-4/2012) from the Czech Ministry of Health.
References
|
1
|
Quintás-Cardama A and Cortes J: Molecular
biology of bcr-abl1-positive chronic myeloid leukemia. Blood.
113:1619–1630. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bixby D and Talpaz M: Seeking the causes
and solutions to imatinib-resistance in chronic myeloid leukemia.
Leukemia. 25:7–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ou J, Vergilio JA and Bagg A: Molecular
diagnosis and monitoring in the clinical management of patients
with chronic myelogenous leukemia treated with tyrosine kinase
inhibitors. Am J Hematol. 83:296–302. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lemoli RM, Salvestrini V, Bianchi E,
Bertolini F, Fogli M, Amabile M, Tafuri A, Salati S, Zini R,
Testoni N, et al: Molecular and functional analysis of the stem
cell compartment of chronic myelogenous leukemia reveals the
presence of a CD34− cell population with intrinsic
resistance to imatinib. Blood. 114:5191–5200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Johansson B, Fioretos T and Mitelman F:
Cytogenetic and molecular genetic evolution of chronic myeloid
leukemia. Acta Haematol. 107:76–94. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jamieson CH, Ailles LE, Dylla SJ,
Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating
A, et al: Granulocyte-macrophage progenitors as candidate leukemic
stem cells in blast-crisis CML. N Engl J Med. 351:657–667. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Giehl M, Fabarius A, Frank O, Hochhaus A,
Hafner M, Hehlmann R and Seifarth W: Centrosome aberrations in
chronic myeloid leukemia correlate with stage of disease and
chromosomal instability. Leukemia. 19:1192–1197. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schmitt M, Li L, Giannopoulos K, Chen J,
Brunner C, Barth T, Schmitt A, Wiesneth M, Döhner K, Döhner H, et
al: Chronic myeloid leukemia cells express tumor-associated
antigens eliciting specific CD8+ T-cell responses and
are lacking costimulatory molecules. Exp Hematol. 34:1709–1719.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Greiner J, Ringhoffer M, Taniguchi M,
Schmitt A, Kirchner D, Krähn G, Heilmann V, Gschwend J, Bergmann L,
Döhner H, et al: Receptor for hyaluronan acid-mediated motility
(RHAMM) is a new immunogenic leukemia-associated antigen in acute
and chronic myeloid leukemia. Exp Hematol. 30:1029–1035. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Giannopoulos K, Dmoszynska A, Kowal M,
Rolinski J, Gostick E, Price DA, Greiner J, Rojewski M,
Stilgenbauer S, Döhner H, et al: Peptide vaccination elicits
leukemia-associated antigen-specific cytotoxic CD8+
T-cell responses in patients with chronic lymphocytic leukemia.
Leukemia. 24:798–805. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ochi T, Fujiwara H, Suemori K, Azuma T,
Yakushijin Y, Hato T, Kuzushima K and Yasukawa M: Aurora-A kinase:
A novel target of cellular immunotherapy for leukemia. Blood.
113:66–74. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Karthigeyan D, Prasad SB, Shandilya J,
Agrawal S and Kundu TK: Biology of Aurora A kinase: Implications in
cancer manifestation and therapy. Med Res Rev. 31:757–793.
2011.PubMed/NCBI
|
|
13
|
McInnes C and Wyatt MD: PLK1 as an
oncology target: Current status and future potential. Drug Discov
Today. 16:619–625. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gleixner KV, Ferenc V, Peter B, Gruze A,
Meyer RA, Hadzijusufovic E, Cerny-Reiterer S, Mayerhofer M, Pickl
WF, Sillaber C, et al: Polo-like kinase 1 (Plk1) as a novel drug
target in chronic myeloid leukemia: Overriding imatinib resistance
with the Plk1 inhibitor BI 2536. Cancer Res. 70:1513–1523. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang N, Ge G, Meyer R, Sethi S, Basu D,
Pradhan S, Zhao YJ, Li XN, Cai WW, El-Naggar AK, et al:
Overexpression of Separase induces aneuploidy and mammary
tumorigenesis. Proc Natl Acad Sci USA. 105:13033–13038. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meyer R, Fofanov V, Panigrahi A, Merchant
F, Zhang N and Pati D: Overexpression and mislocalization of the
chromosomal segregation protein separase in multiple human cancers.
Clin Cancer Res. 15:2703–2710. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rouam S, Moreau T and Broët P: Identifying
common prognostic factors in genomic cancer studies: A novel index
for censored outcomes. BMC Bioinformatics. 11:1502010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Haaß W, Stehle M, Nittka S, Giehl M,
Schrotz-King P, Fabarius A, Hofmann WK and Seifarth W: The
proteolytic activity of separase in BCR-ABL-positive cells is
increased by imatinib. PLoS One. 7:e428632012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tamura H, Dan K, Yokose N, Iwakiri R, Ohta
M, Sakamaki H, Tohyama K, Kondo A, Hyodo H, Nakamura K, et al:
Prognostic significance of WT1 mRNA and anti-WT1 antibody levels in
peripheral blood in patients with myelodysplastic syndromes. Leuk
Res. 34:986–990. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Baccarani M, Deininger MW, Rosti G,
Hochhaus A, Soverini S, Apperley JF, Cervantes F, Clark RE, Cortes
JE, Guilhot F, et al: European LeukemiaNet recommendations for the
management of chronic myeloid leukemia: 2013. Blood. 122:872–884.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Poláková KM, Polívková V, Rulcová J,
Klamová H, Jurcek T, Dvoráková D, Zácková D, Pospísil Z, Mayer J
and Moravcová J: Constant BCR-ABL transcript level ≥=0.1% (IS) in
patients with CML responding to imatinib with complete cytogenetic
remission may indicate mutation analysis. Exp Hematol. 38:20–26.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Park DW, Kim SS, Nam MK, Kim GY, Kim J and
Rhim H: Improved recovery of active GST-fusion proteins from
insoluble aggregates: Solubilization and purification conditions
using PKM2 and HtrA2 as model proteins. BMB Rep. 44:279–284. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sehr P, Zumbach K and Pawlita M: A generic
capture ELISA for recombinant proteins fused to glutathione
S-transferase: Validation for HPV serology. J Immunol Methods.
253:153–162. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smahel M: Antigens in chronic myeloid
leukemia: Implications for vaccine development. Cancer Immunol
Immunother. 60:1655–1668. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maxwell CA, Rasmussen E, Zhan F, Keats JJ,
Adamia S, Strachan E, Crainie M, Walker R, Belch AR, Pilarski LM,
et al: RHAMM expression and isoform balance predict aggressive
disease and poor survival in multiple myeloma. Blood.
104:1151–1158. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Giannopoulos K, Mertens D, Bühler A, Barth
TF, Idler I, Möller P, Kröber A, Greiner J, Chocholska S,
Dmoszyñska A, et al: The candidate immunotherapeutical target, the
receptor for hyaluronic acid-mediated motility, is associated with
proliferation and shows prognostic value in B-cell chronic
lymphocytic leukemia. Leukemia. 23:519–527. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang J, Ikezoe T, Nishioka C, Udaka K and
Yokoyama A: Bcr-Abl activates AURKA and AURKB in chronic myeloid
leukemia cells via AKT signaling. Int J Cancer. 134:1183–1194.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Krusch M and Salih HR: Effects of BCR-ABL
inhibitors on anti-tumor immunity. Curr Med Chem. 18:5174–5184.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smith BD, Kasamon YL, Kowalski J, Gocke C,
Murphy K, Miller CB, Garrett-Mayer E, Tsai HL, Qin L, Chia C, et
al: K562/GM-CSF immunotherapy reduces tumor burden in chronic
myeloid leukemia patients with residual disease on imatinib
mesylate. Clin Cancer Res. 16:338–347. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qin L, Smith BD, Tsai HL, Yaghi NK, Neela
PH, Moake M, Fu J, Kasamon YL, Prince GT, Goswami M, et al:
Induction of high-titer IgG antibodies against multiple
leukemia-associated antigens in CML patients with clinical
responses to K562/GVAX immunotherapy. Blood Cancer J. 3:e1452013.
View Article : Google Scholar : PubMed/NCBI
|