Introduction
The B lymphoma Mo-MLV insertion region 1 homolog
(BMI-1) protein is associated with cancer development and tumor
progression; its roles include tumor invasion, metastasis and
repression of apoptotic cell death or cell senescence (1–4). The
BMI-1 gene is an important member of the polycomb group protein
(PcG) family, which plays oncogenic roles in several types of
tumors. Structurally, BMI-1 has a conserved RING finger domain at
the N-terminus and a central helix-turn-helix motif (5). Upregulation of BMI-1 was found in
various human cancers including ovarian, breast, cervical (6–8),
glioblastoma multiforme (9), and
colorectal cancers (10,11), as well as high-grade B-cell
non-Hodgkin lymphomas (NHLs) (12),
skin cancer (13), neuroblastomas
(14), and pancreatic cancer
(15). BMI-1 overexpression has
been associated with poor survival. Previous studies have
discovered that BMI-1 interacts with a number of tumor-related
signaling pathways, specifically, NF-κB activation and p16/Rb and
p19Arf/MDM2/p53 tumor suppressive signaling pathway
inhibition (16–18). It has also been reported that BMI-1
controls the expression of several genes, including the PTEN tumor
suppressors, HOX and WWOX (19–21).
Also, Cao et al (22)
validated that compounds from one of our top series suppressed
tumor growth and significantly decreased intratumoral levels of
BMI-1. Recently, BMI-1 was considered as a promising drug target
for lung adenocarcinomas with low CEBPα expression (23), pancreatic cancer with gemcitabine
resistance (24), and MDR1 mediated
chemoresistance (25). However, the
precise roles of BMI-1 in tumor invasion and metastasis are largely
unknown.
The suppressor of MEK1 (sMEK1) tumor suppressor
protein, defined as protein phosphatase 4 regulatory subunit 3
(PP4R3), is a vital modulator involved in cellular biological and
physiological functions, such as apoptotic cell death, microtubule
organization, cell cycle arrest, DNA damage checkpoints, and
PI3K/Akt/mTOR signaling pathways. It is a highly conserved member
of the phosphatase family of serine/threonine phosphatases
associated with sensitivity to traditional chemotherapeutic drugs,
such as cisplatin, gemcitabine and paclitaxel (26–30).
sMEK1 functionally binds with several intracellular proteins that
cooperate with biological and physiological processes, as well as
apoptotic cell death, microtubule growth and cell cycle arrest,
including target of rapamycin (TOR) (31), insulin receptor substrate 4 (IRS4)
(32), adenosine triphosphate
(ATP)-dependent chaperonin (33),
and histone deacetylase 3 (HDAC3) (28,34).
Previous studies have validated that ectopic expression of sMEK
upregulates hepatic gluconeogenesis, whereas knockdown of sMEK
decreases blood glucose levels while enhancing hepatic
CREB-regulated transcriptional coactivator (CRTC) phosphorylation.
The sMEK null protein is an important regulator of hepatic
gluconeogenesis (35). Byun et
al (30) reported that sMEK1
additively promotes the proapoptotic activity of gemcitabine by
activating p53 expression. In addition, the expression of sMEK1 is
remarkably decreased in ovarian and cervical cancer patient
tissues, as well as in cancer cell lines, and is hypermethylated
(36). Recently, we reported that
sMEK1 inhibits endothelial cell proliferation and angiogenesis by
suppressing VEGFR-2-mediated PI3K/Akt/eNOS signaling pathway in
ovarian tumors (37). More
specifically, protein phosphorylation/dephosphorylation plays an
important role in various cellular conditions involved in the
molecular basis for diseases such as diabetes, stroke,
hypertension, and unusual movements within the cardiovascular
system.
In this study, a yeast two-hybrid assay system was
utilized to screen a human cDNA library for novel BMI-1 interacting
protein partners in order to start the characterization of
BMI-1-dependent signaling pathways. Furthermore, we showed that
BMI-1 and the sMEK1 complex regulate the cellular physiological
function of the binding protein. We also demonstrated that the
signaling pathways were affected by BMI-1, and that the pattern of
expression for the apoptotic-regulatory proteins was mediated by
BMI-1 expression under siRNA (specific to BMI-1) of the
sMEK1-stimulated apoptotic features observed in ovarian carcinoma
cells. These observations suggest a role for BMI-1 in ovarian
tumorigenesis and it may be a potential target for antitumor
therapy of ovarian cancer.
Materials and methods
Cell lines, cell culture and
antibodies
Human ovarian cancer cell lines (SKOV-3 and OVCAR-3)
and human embryonic kidney 293T (HEK293T) cells were purchased from
the American Type Culture Collection (ATCC, Manassas, VA, USA).
Cells were grown according to the manufacturer's instructions.
Wortmannin and LY294002 chemicals were obtained from Sigma (St.
Louis, MO, USA). The primary antibodies used in this study were as
follows: anti-BMI-1, anti-cyclin D1, anti-CDK4, anti-Mcl-1,
anti-p53, anti-phospho-PI3K (Tyr508), anti-PI3K, anti-phospho-Akt
(Ser473), anti-Akt (Santa Cruz Biotechnology, Santa Cruz, CA, USA),
anti-sMEK1 (Abcam, Cambridge, UK), anti-p21, anti-p27, anti-NF-κB
(Ab-1; Oncogene, Cambridge, MA, USA), anti-Bax, anti-Bcl-xL,
anti-Bcl-2, anti-phospho-PDK1, anti-PDK1, anti-phospho-mTOR,
anti-mTOR, anti-phospho-4E-BP1, anti-4E-BP1 (Cell Signaling,
Beverly, MA, USA), and β-actin (Sigma).
Western blot analysis
For whole protein extraction, cultured ovarian
cancer cells were harvested, rinsed with PBS, centrifuged, and
disrupted by adding cell lysis buffer containing protease inhibitor
cocktail (50 mM Tris, pH 7.2, 150 mM NaCl, 1% Triton X-100, 1 µg/ml
leupeptin, 1 µg/ml pepstatin, 2 µg/ml aprotinin, and 200 µg/ml
phenylmethylsulfonyl fluoride) at 4°C for 1 h. Proteins were
subsequently separated by 8–12% SDS-PAGE and transferred to
Immobilon P membranes (Millipore Corp., Billerica, MA, USA). After
blocking, the membranes were incubated with the indicated primary
specific antibodies at 4°C overnight. The membranes were rinsed
three times in TBST washing buffer and incubated with horseradish
peroxidase-conjugated secondary antibodies. Protein bands were
developed using the ECL detection system (GE Healthcare, Little
Chalfont, Buckinghamshire, UK).
Co-immunoprecipitation analysis
For co-immunoprecipitation, cells were rinsed in
phosphate-buffered saline (PBS) and dissolved in cell lysis buffer
including 50 mM Tris-HCl, pH 7.2, 150 mM NaCl, 1% Triton X-100 and
a protease inhibitor cocktail (Sigma). Lysates were then incubated
with anti-Flag antibody (Santa Cruz) and precipitated using protein
A-agarose (Invitrogen, Carlsbad, CA, USA). Precipitated proteins
were separated by 10% sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE), transferred to an Immobilon-P membrane
(GE Healthcare, Piscataway, NJ, USA), and subjected to immunoblot
analysis using either anti-BMI-1 or anti-sMEK1 antibodies. Enhanced
chemiluminescent (ECL) western blotting detection reagent (Pierce,
Rockford, IL, USA) was used to visualize the gels. β-actin served
as an internal control.
Gene silencing by small interference
RNA (siRNA)
Oligonucleotides containing the BMI-1 siRNA sequence
5′-ATAT GAAGAGAAGAAGGGATT-3′ were synthesized using an RNAi
construction kit (Ambion, Austin, TX, USA) and transfected into
cells using Lipofectamine 2000 reagent (Invitrogen) according to
the manufacturer's instructions to silence endogenous BMI-1
expression. Cells were seeded for 24 h followed by transfection
with 150 nM siBMI-1. At 24 h after transfection, cells were
prepared for MTT assays and immunoblot analysis.
Apoptotic cell death analysis
Cells were seeded on 6-well plates at a density of
1×105 per well and transfected. Next, cells were
incubated with FITC-labeled Annexin V and propidium iodide (PI) for
15 min according to the manufacturer's instructions (BD Pharmingen,
Mississauga, ON, Canada) and analyzed using a fluorescence
activated cell sorting (FACS) Vantage BD FACSCalibur flow
cytometer. To determine cell viability, the relative rate of cell
viability was evaluated via MTT assay. Cells were grown at a
density of 4.5×103 per well in 96-well plates. Three
days after transfection, fresh medium including 10% fetal bovine
serum (FBS) and 20 µl of
3-(4,5-dimethylthiazol-2-yl)-2.5-diphenyl-2H-tetrazolium
bromide (MTT) solution (Sigma, 5 µg/ml) was added to each well and
incubated for an additional 4 h at 37°C. After centrifugation at
500 g for 10 min, the supernatant was removed from the wells, and
the formazan product was dissolved in dimethyl sulfoxide (DMSO).
The amount of MTT-formazan added was determined by the absorbance,
calculated using a microplate reader at 540–550 nm.
Luciferase reporter gene analysis
BMI-1 luciferase activity in vitro was
carried out as reported previously (38). In brief, cells at 85% confluency
were transfected using a BMI-1 reporter plasmid vector. After lysis
using RIPA buffer, lysates were cleared by centrifugation at 14,000
rpm for 15 min, and cell extracts were incubated with a luciferase
substrate reagent at room temperature for 30 min according to the
manufacturer's instructions. Five-microliter aliquots of each
sample were measured using a MicroLumat Plus LB96V luminometer
(Berthold Technologies, Bad Wildbad, Germany).
Data and statistical analysis
All results are presented as the means ± standard
deviations (SD) from at least three independent experiments.
Statistical comparisons between the different groups were analyzed
using the Student's t-test. Significance was set at P<0.05.
Results
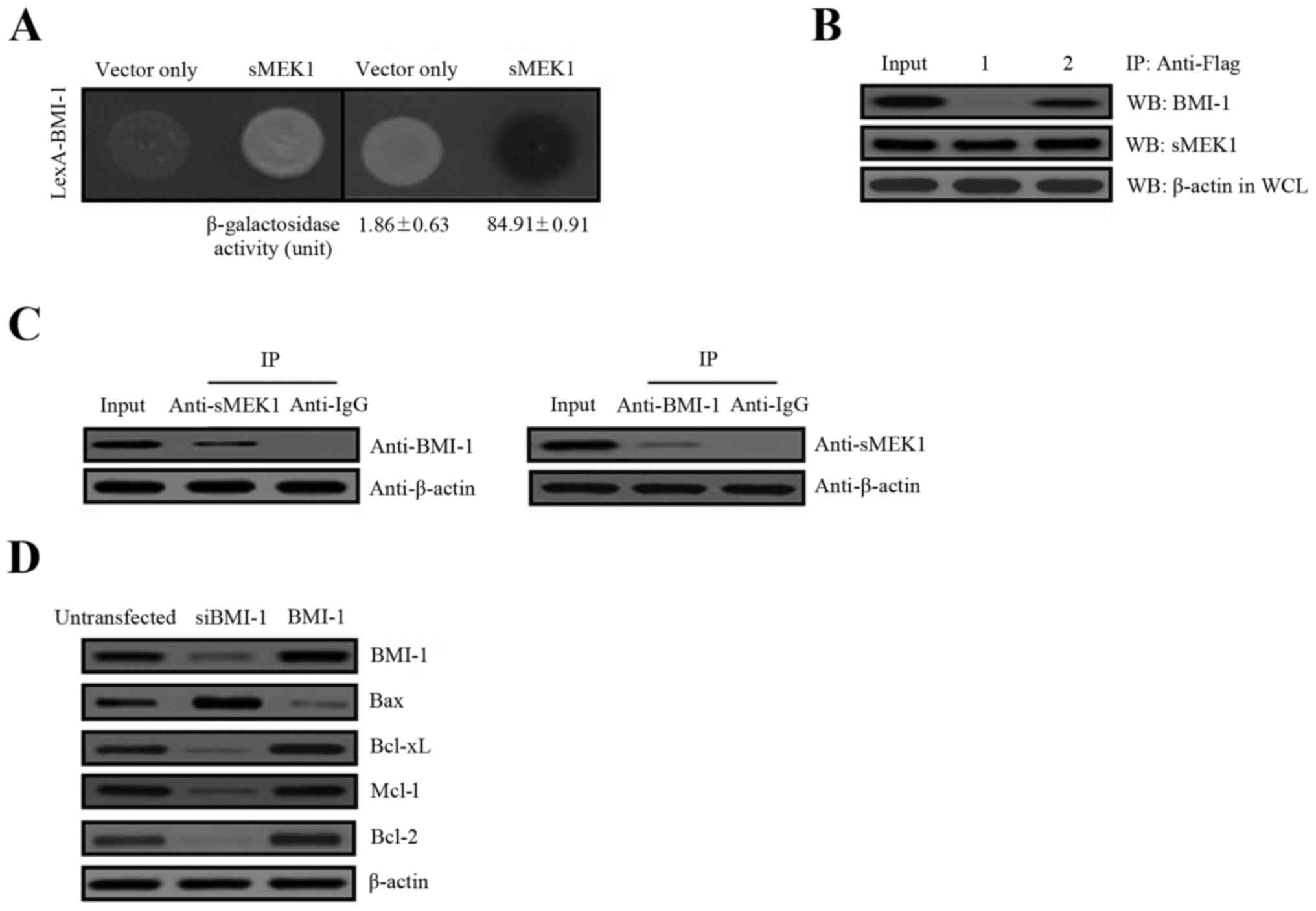
Physical interaction between BMI-1 and
sMEK1
To explore the cellular biological and physiological
relevance of BMI-1 on the effects of tumor metastasis, we performed
yeast two-hybrid and co-immunoprecipitation assays to identify
potential BMI-1-interacting partners. The human cDNA library fused
to the transcriptional activator pJG4-5/B42 gene was introduced
into yeast cells containing the pGilda/LexA-BMI-1 as bait.
Approximately 6.0×106 independent transformants were
pooled. Five positive colonies were obtained after respreading on
selection media (Ura-, His-, Trp-, and Leu-). Of the five sequenced
clones, all encoded sMEK1 (accession no. NM_001284280), indicating
that BMI-1 interacted with sMEK1. To confirm these results,
positive interaction was monitored by both cell growth on
leucine-deficient plates and ONPG β-galactosidase activity. An
empty plasmid (vector only) served as a negative control. As shown
in Fig. 1A, β-galactosidase
activity was fully activated, indicating an interaction between
BMI-1 and sMEK1 (84.91±0.91), but such was not observed with the
empty plasmid (vector only, 1.86±0.63). To further confirm a direct
interaction between BMI-1 and sMEK1 as revealed in the yeast
two-hybrid assay, co-immunoprecipitation experiments were used.
Gene constructs of sMEK1 (pcDNA3.1/Flag-sMEK1) and BMI-1
(pcDNA3.1/BMI-1) or sMEK1 (pcDNA3.1/Flag-sMEK1) and vector only
(pcDNA3.1), were co-transfected into ovarian carcinoma cells. Next,
immunoprecipitation was performed using an anti-Flag antibody in
lysates from both transfected cell lines. After
immunoprecipitation, precipitated proteins were subjected to
immunoblotting using anti-BMI-1 or anti-sMEK1 antibodies. As shown
in Fig. 1B, pcDNA3.1-BMI-1
co-immunoprecipitated with pcDNA3.1/Flag-sMEK1 (lane 2, upper
panel), but not with pcDNA3.1 (vector only) (lane 1, upper panel).
Subsequently, cellular interaction between these two proteins was
also demonstrated by co-immunoprecipitation of endogenous BMI-1 and
sMEK1. As shown in Fig. 1C,
endogenous BMI-1 co-immunoprecipitated directly with sMEK1. We next
evaluated BMI-1-induced cell growth by determining the expression
of the apoptosis-related proteins Bax, Bcl-xL, Mcl-1 and Bcl-2.
These proteins are well-known pivotal regulators of apoptotic cell
death and growth. As presented in Fig.
1D, Bax expression was decreased, while Bcl-xL, Mcl-1 and Bcl-2
were increased by BMI-1, compared with the non-transfectants.
Consistent with these results, cells transfected with a specific
BMI-1 RNAi (siBMI-1) showed enhanced Bax expression, whereas
Bcl-xL, Mcl-1 and Bcl-2 were decreased remarkably by siBMI-1.
Collectively, these results strongly indicate that BMI-1 directly
interacts with sMEK1 under biologic conditions.
sMEK1 controls cell cycle- and
apoptosis-associated proteins
Cell cycle control is a fundamental process in
cellular homeostasis and involves DNA repair, DNA replication and
cell division. This regulation is generally mediated by cyclin and
cyclin-dependent kinases (CDKs). In particular, CDKs are
serine/threonine kinases that play an important role in the complex
feedback regulation of cell cycle progression. Therefore, we
examined expression levels of cell cycle- and apoptotic cell
death-associated proteins via western blot analysis. As shown in
Fig. 2A, overexpression of BMI-1
increased the expression of cyclin D1 and CDK4 remarkably, but
decreased the expression of p21 and p27, acting as a CDK inhibitor.
These results demonstrate that overexpression of BMI-1 strikingly
interrupts sMEK1-induced apoptosis (Fig. 2B). Nuclear factor kappa B (NF-κB)
and Bcl-2 family genes, as well as the p53 tumor suppressor, are
major modulators of cell growth and apoptosis. To explore the
functional effects of BMI-1 on sMEK1-induced apoptotic cell death,
cells were transfected with either a BMI-1 expression plasmid or
siBMI-1, and promoter activity was then measured using a dual
luciferase reporter gene assay. As presented in Fig. 2B, the promoter activities of p21 and
p53 were significantly reduced, whereas Bcl-2 and NF-κB activity
were noticeably enhanced by BMI-1 overexpression. Taken together,
these results demonstrate clearly that BMI-1 controls apoptosis
modulator protein levels through downregulation of p53 and
upregulation of NF-κB activity during sMEK1-induced apoptosis in
tumor cells.
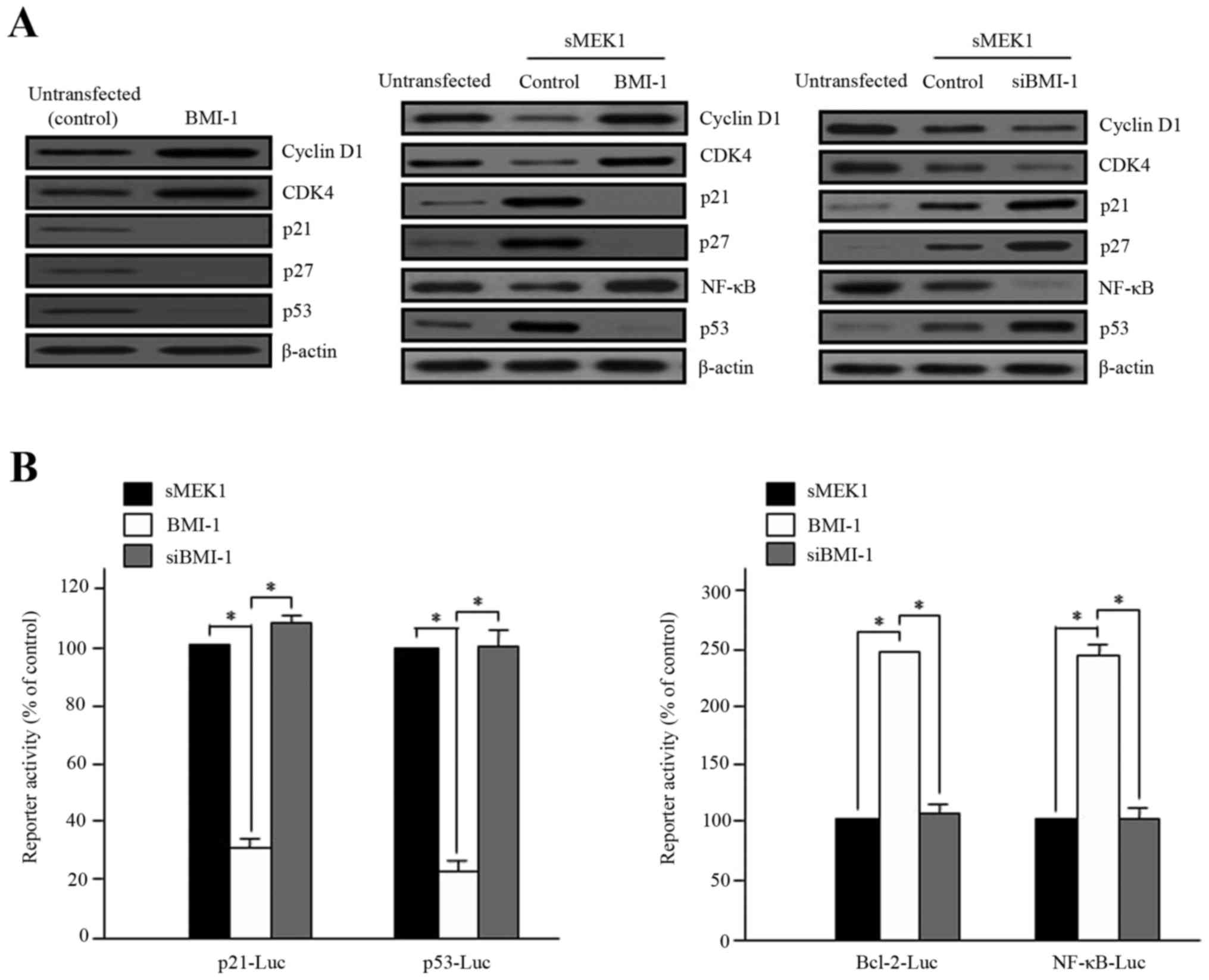 | Figure 2.Effects of BMI-1 on sMEK1-induced
apoptosis, p21 and NF-κB activity. (A) Cells were transfected with
BMI-1 expression plasmid (pcDNA3.1/BMI-1) (left) and sMEK1 for 24
h, and sMEK1-expressing cells were then transiently transfected
with a control, BMI-1 (middle) or siBMI-1 (right), respectively.
After 48 h, cells were collected and treated with lysis buffer.
Cell lysates were subsequently subjected to immunoblot analysis.
Protein expression levels of cell cycle- and apoptosis-related
genes were evaluated. Protein expression was assessed via
immunoblotting using specific antibodies. (B) Promoter activities
of p21, p53, Bcl-2 and NF-κB were measured using a luciferase
reporter-gene assay system with a p21 promoter reporter (p21-Luc),
p53 (p53-Luc), Bcl-2 (Bcl-2-Luc) and NF-κB (NF-κB-Luc),
respectively. For example, the p53-luciferase reporter gene
constructs or promoter-less plasmid vector only were transiently
transfected into sMEK1-induced ovarian carcinoma cells. Cells were
incubated for 24 h and then added to lysis buffer. After collection
by centrifugation, cells were lysed, mixed with luciferase reaction
substrate, and assayed for luciferase activity. Experiments were
performed in triplicate, and error bars represent the means ± SD.
Differences between groups were considered significant at the level
of *P<0.05. |
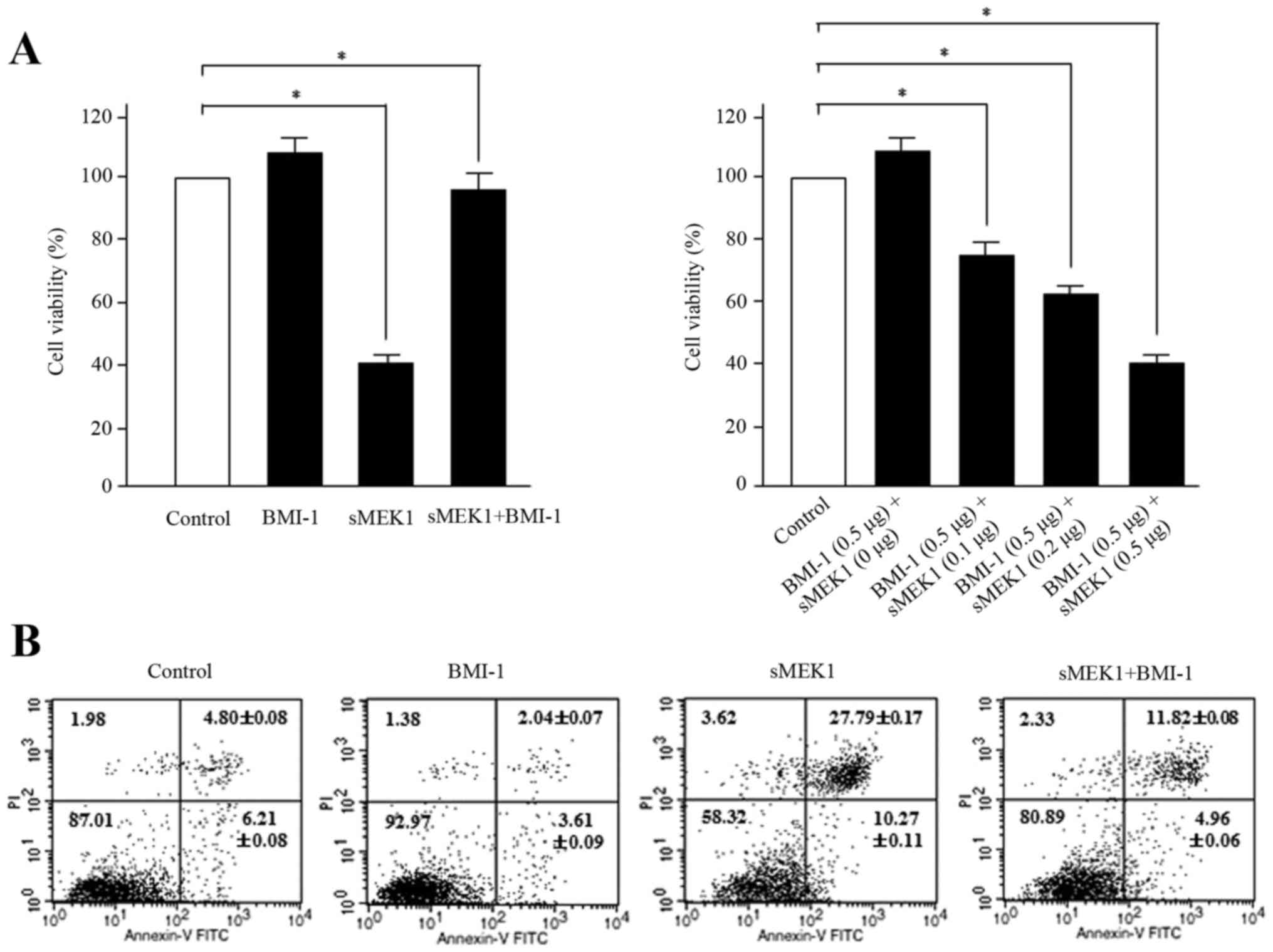
BMI-1 inhibits sMEK1-stimulated
apoptosis in ovarian carcinoma cells
To explore the biological function of BMI-1 during
sMEK1-induced apoptosis, ovarian carcinoma cells were transfected
with BMI-1, sMEK1, or sMEK1 plus BMI-1. Control transfectant
contained the expression vector only. According to the reduced
number of cells, which is indicative of cell viability,
sMEK1-transfected cells were suppressed to ~60% compared with
control cells. In contrast, enhanced viability of the BMI-1 and
sMEK1 plus BMI-1 transfectants was observed (Fig. 3A, left panel). Furthermore, after
cotransfection with BMI-1 (0.5 µg) and sMEK1 (0–0.5 µg), apoptotic
cell death increased gradually in a dose-dependent manner (Fig. 3A, right panel). Subsequently, flow
cytometric analysis confirmed that the loss of viability was in
fact due to cell death. Cells were transfected with a control
vector (expression plasmid vector only), BMI-1, sMEK1, or a
combination of sMEK1 and BMI-1. As shown in Fig. 3B, cell growth of ovarian cancer
cells was inhibited by sMEK1 transfection compared with that of
control transfectants. Interestingly, cell viability was recovered
in sMEK1-expressing BMI-1 transfectants (sMEK1 plus BMI-1) compared
with sMEK1-expressing only transfectants, whereas sMEK1-expressing
siBMI-1 transfectants (sMEK1 plus siBMI-1) exhibited suppression of
cell viability by 20% compared with siBMI-1-expressing cells
(siBMI-1 plasmid only; data not shown). These results strongly
indicate that BMI-1 has an antagonistic effect on sMEK1-mediated
apoptotic cell death.
sMEK1 causes a decrease in
PI3K/mTOR/4E-BP1 phosphorylation
To explore the underlying molecular mechanism by
which BMI-1 promotes PI3K activation, we examined the involvement
of PDK1, Akt, mTOR, and 4E-BP1, all of which are downstream
signaling cascade components of the PI3K pathway. Cell lysates from
sMEK1-expressing cells (control) and BMI-1 transfected cells were
examined by western blot analysis. BMI-1-induced phosphorylation of
PI3K, Akt and mTOR plays a major role in BMI-1-stimulated
tumorigenesis, migration, invasion, and tumor metastasis. As
presented in Fig. 4, sMEK1-induced
PI3K and Akt dephosphorylation was significantly restored by BMI-1.
Ectopic BMI-1 expression also enhanced Akt phosphorylation in a
dose-dependent manner (Fig. 4B).
These data were comparable to those of wortmannin and LY294002,
well-known PI3K inhibitors. Wortmannin and LY294002 were shown to
additively decrease (by 40–45%) the PI3 kinase activity induced by
sMEK1 (Fig. 4A). We next
investigated the phosphorylation of essential components of the
mTOR/4E-BP1 signaling pathways. As presented in Fig. 4C, sMEK1 significantly reduced PDK1
phosphorylation, as well as the phosphorylation of mTOR and 4E-BP1.
Collectively, our results indicate that BMI-1 recovers
sMEK1-stimulated apoptotic cell death through activation of the
PI3K/mTOR/4E-BP1 signaling pathway in ovarian tumor cells.
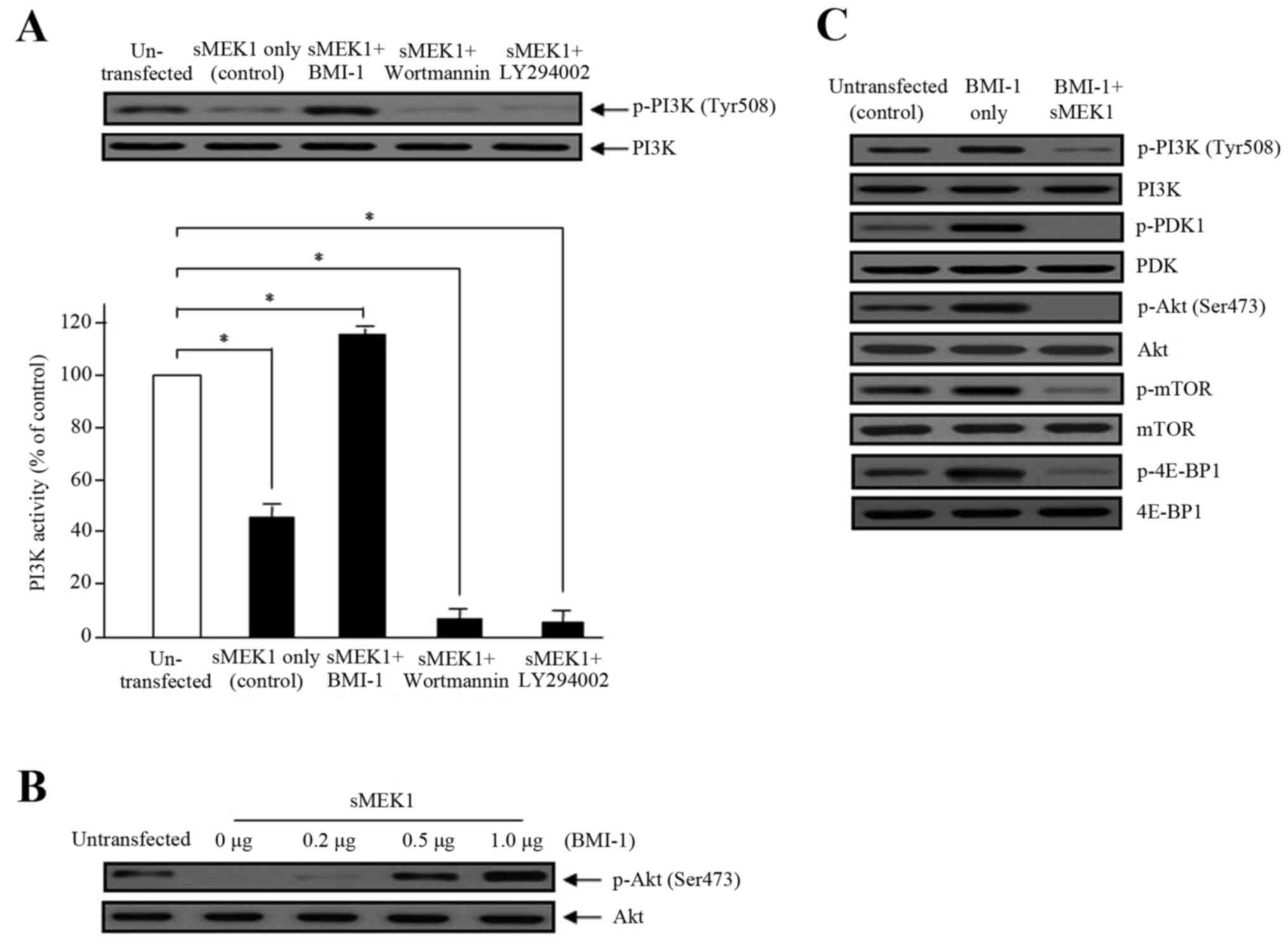 | Figure 4.Effects of BMI-1 on PI3 kinase
activity and phosphorylation of the Akt/mTOR signaling components
by sMEK1 transfection. (A) The effects of BMI-1 on PI3K activity,
in the presence of sMEK1, was measured using an in vitro PI3
kinase assay. Data represent the means ± SD from three independent
experiments with similar results. Differences between groups were
considered significant at the level of *P<0.05. Box, cells
transfected with sMEK1 were transfected/treated with the BMI-1
expression plasmid, wortmannin, or LY294002 for 24 h, collected,
and subjected to immunoblotting to identify the indicated proteins.
PI3K was used to verify equal sample loading. (B) sMEK1-expressing
cells were transfected with various concentrations of the BMI-1
expression plasmid. Akt phosphorylation was determined by western
blot analysis. Non-phosphorylated Akt served as a loading control.
(C) Cells were transfected with control, BMI-1, or BMI-1 plus
sMEK1, harvested, and treated with lysis buffer. After collection
by centrifugation, equal amounts of cellular protein (30 µg) were
separated by 10% SDS-PAGE, followed by immunoblotting using
specific antibodies (PI3K/phosphorylated PI3K, PDK1/phosphorylated
PDK1, Akt/phosphorylated Akt, mTOR/phosphorylated mTOR and
4E-BP1/phosphorylated-4E-BP1). All experiments were performed at
least three times with consistent and similar results. |
Discussion
Previous studies have reported that overexpression
of BMI-1 correlates with therapy failure in various cancer types,
including ovarian, breast, lung, and prostate tumor patients
(22,39–42).
BMI-1 is significantly overexpressed in ovarian cancer and is
correlated with a poor prognosis, indicating that this protein
participates in the development and progression of ovarian cancer.
Therefore, the BMI-1 protein may be a potential target for novel
antitumor therapies (7,43). Ovarian cancer is the fourth most
common cause of cancer-related death in women. Ovarian tumors
typically have a poor prognosis (44). In most cases, the exact cause of
ovarian cancer remains unknown. Carriers of certain BRCA mutations
are at significant risk. Germline mutations in the BRCA1 and
BRCA2 genes contribute to ~18% of hereditary ovarian tumors,
conferring an estimated lifetime risk of 15–50% (45,46).
The risk increases with age and decreases with the number of
pregnancies. Treatment usually involves chemotherapy, surgery, and
sometimes radiotherapy. The major limitation of standard treatment
using taxane and platinum analogues (cisplatin and carbo-platin) is
the development of chemoresistance (47). Inhibition of apoptotic cell death is
generally accepted as one of the major contributing factors to
chemoresistance. Despite high initial response rates to aggressive
primary therapy, most ovarian cancers develop drug resistance,
resulting in patient death (48).
Unfortunately, the 5-year survival rate for these patients has not
exceeded 20–25%. Hence, there is a critical need to develop better
therapeutic agents and strategies (49,50).
However, it is unknown whether BMI-1 directly affects metastasis
and progression in ovarian tumorigenesis. In this study, we provide
new evidence that BMI-1 ultimately enhances tumor metastasis by
increasing BMI-1-dependent PI3K/mTOR/4E-BP1 phosphorylation. First,
higher BMI-1 expression activated tumor metastasis by suppressing
sMEK1-stimulated apoptotic cell death via control of various cell
cycle- and apoptosis-associated proteins, such as p21, cyclin D1,
p53 and Bcl-2 family genes. Second, BMI-1 directly binds to sMEK1
and the bound sMEK1 inactivates BMI-1, causing overexpression of
sMEK1 in ovarian tumor cells. Third, induction of the
BMI-1-regulated PI3K/mTOR/4E-BP1 signaling pathway and tumor
metastasis was remarkably suppressed by transient overexpression of
sMEK1.
Cancer cell invasion and metastasis are regulated by
many factors that can induce or enhance cell motility, demolition
of the cellular matrix, angiogenesis, and various biological and
physiological events at the molecular level. High BMI-1 expression
is strongly associated with advanced stages of cancer and
carcinomas with serous histology. BMI-1 expression displayed a
significant inverse association with overall and mean patient
survival. For example, Wang et al (51) demonstrated in vitro and in
vivo that BMI-1 silencing synergizes with enhanced oxidative
stress, leading to accumulated DNA damage and apoptotic cell death.
Subsequently, knockdown of BMI-1 can effectively inhibit tumor cell
proliferation and tumorigenicity in several tumors. Silencing BMI-1
by RNA interference can promote senescence and effectively decrease
metastasis of gastric cancer cells (52–54).
In addition, Wu et al (55)
validated that BMI-1 functions as an oncogene in osteosarcoma and
enhances tumorigenicity and resistance to chemotherapy. BMI-1
knockdown sensitized cells to cisplatin-induced apoptotic cell
death via suppression of the PI3K/Akt signaling pathway. Bax, Mcl-1
and Bcl-2 proteins are well-known pivotal regulators of apoptotic
cell death and cell growth. As indicated in Fig. 1D, Bax expression was decreased
compared with non-transfectants, while Bcl-xL, Mcl-1 and Bcl-2
expression levels were increased by BMI-1. In support of these
results, in cells transfected with a specific BMI-1 RNAi (siBMI-1),
Bax expression was enhanced, whereas Bcl-xL, Mcl-1 and Bcl-2 were
remarkably decreased by siBMI-1. PI3K communicates with
3-phosphoinositide-dependent protein kinase-1 (PDK1) and regulates
cell death, cell growth, or possibly both through Akt/mTOR
phosphorylation. Our data demonstrate that sMEK1 suppresses PI3K
and Akt activation via BMI-1 but has no effect on PI3K inhibitors,
such as wortmannin and LY294002 (Fig.
4A). These results indicate that BMI-1 promotes key signaling
regulators during tumor cell progression.
In conclusion, our findings provide important new
insights in cancer-related cell metastasis and tumorigenesis.
Overall, we have shown that ectopic expression of sMEK1 contributes
to enhanced metastatic potential via disruption of the
PI3K/mTOR/4E-BP1 signaling pathways, which may be associated with
BMI-1-sMEK1 interaction and transcriptional regulation of the mTOR
signaling pathway. We postulate that targeting sMEK1 may provide a
new strategy for modulating cancer metastasis and may serve as a
novel therapeutic target for BMI-1-associated diseases, including
various solid tumors.
Acknowledgements
This study was supported by a grant from the
National Cancer Center, Korea (NCC-1410312-3).
Glossary
Abbreviations
Abbreviations:
|
BMI-1
|
B lymphoma Mo-MLV insertion region 1
homolog
|
|
PcG
|
polycomb group protein
|
|
sMEK1
|
suppressor of mek1
|
|
PP4R3
|
protein phosphatase 4 regulatory
subunit 3
|
|
CDK
|
cyclin-dependent kinases
|
|
HDAC3
|
histone deacetylase 3
|
|
CRTC
|
CREB regulated transcriptional
coactivator
|
|
ONPG
|
O-nitrophenyl
β-d-galactopyranoside
|
|
PDK1
|
3-phosphoinositide-dependent protein
kinase-1
|
|
mTOR
|
mammalian target of rapamycin
|
References
|
1
|
Jacobs JJ, Kieboom K, Marino S, DePinho RA
and van Lohuizen M: The oncogene and Polycomb-group gene bmi-1
regulates cell proliferation and senescence through the ink4a
locus. Nature. 397:164–168. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Song LB, Zeng MS, Liao WT, Zhang L, Mo HY,
Liu WL, Shao JY, Wu QL, Li MZ, Xia YF, et al: Bmi-1 is a novel
molecular marker of nasopharyngeal carcinoma progression and
immortalizes primary human nasopharyngeal epithelial cells. Cancer
Res. 66:6225–6232. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li J, Gong LY, Song LB, Jiang LL, Liu LP,
Wu J, Yuan J, Cai JC, He M, Wang L, et al: Oncoprotein Bmi-1
renders apoptotic resistance to glioma cells through activation of
the IKK-nuclear factor-kappaB pathway. Am J Pathol. 176:699–709.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guo BH, Feng Y, Zhang R, Xu LH, Li MZ,
Kung HF, Song LB and Zeng MS: Bmi-1 promotes invasion and
metastasis, and its elevated expression is correlated with an
advanced stage of breast cancer. Mol Cancer. 10:102011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Itahana K, Zou Y, Itahana Y, Martinez JL,
Beausejour C, Jacobs JJ, Van Lohuizen M, Band V, Campisi J and
Dimri GP: Control of the replicative life span of human fibroblasts
by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 23:389–401.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bhattacharya R, Nicoloso M, Arvizo R, Wang
E, Cortez A, Rossi S, Calin GA and Mukherjee P: MiR-15a and MiR-16
control Bmi-1 expression in ovarian cancer. Cancer Res.
69:9090–9095. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Honig A, Weidler C, Häusler S,
Krockenberger M, Buchholz S, Köster F, Segerer SE, Dietl J and
Engel JB: Overexpression of polycomb protein BMI-1 in human
specimens of breast, ovarian, endometrial and cervical cancer.
Anticancer Res. 30:1559–1564. 2010.PubMed/NCBI
|
|
8
|
Tong YQ, Liu B, Zheng HY, He YJ, Gu J, Li
F and Li Y: Overexpression of BMI-1 is associated with poor
prognosis in cervical cancer. Asia Pac J Clin Oncol. 8:e55–e62.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abdouh M, Facchino S, Chatoo W, Balasingam
V, Ferreira J and Bernier G: BMI1 sustains human glioblastoma
multiforme stem cell renewal. J Neurosci. 29:8884–8896. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim JH, Yoon SY, Kim CN, Joo JH, Moon SK,
Choe IS, Choe YK and Kim JW: The Bmi-1 oncoprotein is overexpressed
in human colorectal cancer and correlates with the reduced
p16INK4a/p14ARF proteins. Cancer Lett. 203:217–224. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kreso A, van Galen P, Pedley NM,
Lima-Fernandes E, Frelin C, Davis T, Cao L, Baiazitov R, Du W,
Sydorenko N, et al: Self-renewal as a therapeutic target in human
colorectal cancer. Nat Med. 20:29–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van Kemenade FJ, Raaphorst FM, Blokzijl T,
Fieret E, Hamer KM, Satijn DP, Otte AP and Meijer CJ: Coexpression
of BMI-1 and EZH2 polycomb-group proteins is associated with
cycling cells and degree of malignancy in B-cell non-Hodgkin
lymphoma. Blood. 97:3896–3901. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Balasubramanian S, Lee K, Adhikary G,
Gopalakrishnan R, Rorke EA and Eckert RL: The Bmi-1 polycomb group
gene in skin cancer: Regulation of function by
(−)-epigallocatechin-3-gallate. Nutr Rev. 66:(Suppl 1). S65–S68.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cui H, Hu B, Li T, Ma J, Alam G, Gunning
WT and Ding HF: Bmi-1 is essential for the tumorigenicity of
neuroblastoma cells. Am J Pathol. 170:1370–1378. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yin T, Wei H, Leng Z, Yang Z, Gou S, Wu H,
Zhao G, Hu X and Wang C: Bmi-1 promotes the chemoresistance,
invasion and tumorigenesis of pancreatic cancer cells.
Chemotherapy. 57:488–496. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lindström MS, Klangby U and Wiman KG:
p14ARF homozygous deletion or MDM2 overexpression in Burkitt
lymphoma lines carrying wild type p53. Oncogene. 20:2171–2177.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dhawan S, Tschen SI and Bhushan A: Bmi-1
regulates the Ink4a/Arf locus to control pancreatic beta-cell
proliferation. Genes Dev. 23:906–911. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang L, Li J and Song L: Bmi-1, stem
cells and cancer. Acta Biochim Biophys Sin (Shanghai). 41:527–534.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bracken AP, Dietrich N, Pasini D, Hansen
KH and Helin K: Genome-wide mapping of Polycomb target genes
unravels their roles in cell fate transitions. Genes Dev.
20:1123–1136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fan C, He L, Kapoor A, Rybak AP, De Melo
J, Cutz JC and Tang D: PTEN inhibits BMI1 function independently of
its phosphatase activity. Mol Cancer. 8:982009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kimura M, Takenobu H, Akita N, Nakazawa A,
Ochiai H, Shimozato O, Fujimura Y, Koseki H, Yoshino I, Kimura H,
et al: Bmi1 regulates cell fate via tumor suppressor WWOX
repression in small-cell lung cancer cells. Cancer Sci.
102:983–990. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cao L, Bombard J, Cintron K, Sheedy J,
Weetall ML and Davis TW: BMI1 as a novel target for drug discovery
in cancer. J Cell Biochem. 112:2729–2741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yong KJ, Basseres DS, Welner RS, Zhang WC,
Yang H, Yan B, Alberich-Jorda M, Zhang J, de Figueiredo-Pontes LL,
Battelli C, et al: Targeted BMI1 inhibition impairs tumor growth in
lung adenocarcinomas with low CEBPα expression. Sci Transl Med.
8:350ra1042016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yin T, Zhang Z, Cao B, Duan Q, Shi P, Zhao
H, Camara SN, Shen Q and Wang C: Bmi1 inhibition enhances the
sensitivity of pancreatic cancer cells to gemcitabine. Oncotarget.
7:37192–37204. 2016.PubMed/NCBI
|
|
25
|
Mustafi S Banerjee, Chakraborty PK, Naz S,
Dwivedi SK, Street M, Basak R, Yang D, Ding K, Mukherjee P and
Bhattacharya R: MDR1 mediated chemoresistance: BMI1 and TIP60 in
action. Biochim Biophys Acta. 1859:983–993. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hastie CJ, Vázquez-Martin C, Philp A,
Stark MJ and Cohen PT: The Saccharomyces cerevisiae orthologue of
the human protein phosphatase 4 core regulatory subunit R2 confers
resistance to the anticancer drug cisplatin. FEBS J. 273:3322–3334.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen GI, Tisayakorn S, Jorgensen C,
D'Ambrosio LM, Goudreault M and Gingras AC: PP4R4/KIAA1622 forms a
novel stable cytosolic complex with phosphoprotein phosphatase 4. J
Biol Chem. 283:29273–29284. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chowdhury D, Xu X, Zhong X, Ahmed F, Zhong
J, Liao J, Dykxhoorn DM, Weinstock DM, Pfeifer GP and Lieberman J:
A PP4-phosphatase complex dephosphorylates gamma-H2AX generated
during DNA replication. Mol Cell. 31:33–46. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakada S, Chen GI, Gingras AC and Durocher
D: PP4 is a gamma H2AX phosphatase required for recovery from the
DNA damage checkpoint. EMBO Rep. 9:1019–1026. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Byun HJ, Kim BR, Yoo R, Park SY and Rho
SB: sMEK1 enhances gemcitabine anti-cancer activity through
inhibition of phosphorylation of Akt/mTOR. Apoptosis. 17:1095–1103.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bertram PG, Choi JH, Carvalho J, Ai W,
Zeng C, Chan TF and Zheng XF: Tripartite regulation of Gln3p by
TOR, Ure2p, and phosphatases. J Biol Chem. 275:35727–35733. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mihindukulasuriya KA, Zhou G, Qin J and
Tan TH: Protein phosphatase 4 interacts with and down-regulates
insulin receptor substrate 4 following tumor necrosis factor-alpha
stimulation. J Biol Chem. 279:46588–46594. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gingras AC, Caballero M, Zarske M, Sanchez
A, Hazbun TR, Fields S, Sonenberg N, Hafen E, Raught B and
Aebersold R: A novel, evolutionarily conserved protein phosphatase
complex involved in cisplatin sensitivity. Mol Cell Proteomics.
4:1725–1740. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang X, Ozawa Y, Lee H, Wen YD, Tan TH,
Wadzinski BE and Seto E: Histone deacetylase 3 (HDAC3) activity is
regulated by interaction with protein serine/threonine phosphatase
4. Genes Dev. 19:827–839. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoon YS, Lee MW, Ryu D, Kim JH, Ma H, Seo
WY, Kim YN, Kim SS, Lee CH, Hunter T, et al: Suppressor of MEK null
(SMEK)/protein phosphatase 4 catalytic subunit (PP4C) is a key
regulator of hepatic gluconeogenesis. Proc Natl Acad Sci USA.
107:17704–17709. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dong SM, Byun HJ, Kim BR, Lee SH, Trink B
and Rho SB: Tumor suppressor BLU enhances pro-apoptotic activity of
sMEK1 through physical interaction. Cell Signal. 24:1208–1214.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim BR, Seo SH, Park MS, Lee SH, Kwon Y
and Rho SB: sMEK1 inhibits endothelial cell proliferation by
attenuating VEGFR-2-dependent-Akt/eNOS/HIF-1α signaling pathways.
Oncotarget. 6:31830–31843. 2015.PubMed/NCBI
|
|
38
|
Rho SB, Song YJ, Lim MC, Lee SH, Kim BR
and Park SY: Programmed cell death 6 (PDCD6) inhibits angiogenesis
through PI3K/mTOR/p70S6K pathway by interacting of VEGFR-2. Cell
Signal. 24:131–139. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Glinsky GV: Stem cell origin of
death-from-cancer phenotypes of human prostate and breast cancers.
Stem Cell Rev. 3:79–93. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vrzalikova K, Skarda J, Ehrmann J, Murray
PG, Fridman E, Kopolovic J, Knizetova P, Hajduch M, Klein J, Kolek
V, et al: Prognostic value of Bmi-1 oncoprotein expression in NSCLC
patients: A tissue microarray study. J Cancer Res Clin Oncol.
134:1037–1042. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang H, Pan K, Zhang HK, Weng DS, Zhou J,
Li JJ, Huang W, Song HF, Chen MS and Xia JC: Increased
polycomb-group oncogene Bmi-1 expression correlates with poor
prognosis in hepatocellular carcinoma. J Cancer Res Clin Oncol.
134:535–541. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li DW, Tang HM, Fan JW, Yan DW, Zhou CZ,
Li SX, Wang XL and Peng ZH: Expression level of Bmi-1 oncoprotein
is associated with progression and prognosis in colon cancer. J
Cancer Res Clin Oncol. 136:997–1006. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gavrilescu MM, Todosi AM, Aniţei MG, Filip
B and Scripcariu V: Expression of bmi-1 protein in cervical, breast
and ovarian cancer. Rev Med Chir Soc Med Nat Iasi. 116:1112–1117.
2012.PubMed/NCBI
|
|
44
|
Chobanian N and Dietrich CS III: Ovarian
cancer. Surg Clin North Am. 88285–299. (vi)2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lakhani SR, Manek S, Penault-Llorca F,
Flanagan A, Arnout L, Merrett S, McGuffog L, Steele D, Devilee P,
Klijn JG, et al: Pathology of ovarian cancers in BRCA1 and BRCA2
carriers. Clin Cancer Res. 10:2473–2481. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Stavropoulou AV, Fostira F, Pertesi M,
Tsitlaidou M, Voutsinas GE, Triantafyllidou O, Bamias A, Dimopoulos
MA, Timotheadou E, Pectasides D, et al: Prevalence of BRCA1
mutations in familial and sporadic Greek ovarian cancer cases. PLoS
One. 8:e581822013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shih KK and Chi DS: Maximal cytoreductive
effort in epithelial ovarian cancer surgery. J Gynecol Oncol.
21:75–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Agarwal R and Kaye SB: Ovarian cancer:
Strategies for overcoming resistance to chemotherapy. Nat Rev
Cancer. 3:502–516. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ozols RF: Treatment goals in ovarian
cancer. Int J Gynecol Cancer 15 (Suppl 1). 3–11. 2005. View Article : Google Scholar
|
|
50
|
Spannuth WA, Nick AM, Jennings NB,
Armaiz-Pena GN, Mangala LS, Danes CG, Lin YG, Merritt WM, Thaker
PH, Kamat AA, et al: Functional significance of VEGFR-2 on ovarian
cancer cells. Int J Cancer. 124:1045–1053. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang E, Bhattacharyya S, Szabolcs A,
Rodriguez-Aguayo C, Jennings NB, Lopez-Berestein G, Mukherjee P,
Sood AK and Bhattacharya R: Enhancing chemotherapy response with
Bmi-1 silencing in ovarian cancer. PLoS One. 6:e179182011.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xin T, Zhang FB, Sui GJ and Jin XM: Bmi-1
siRNA inhibited ovarian cancer cell line growth and decreased
telomerase activity. Br J Biomed Sci. 69:62–66. 2012.PubMed/NCBI
|
|
53
|
Gao FL, Li WS, Liu CL and Zhao GQ:
Silencing Bmi-1 enhances the senescence and decreases the
metastasis of human gastric cancer cells. World J Gastroenterol.
19:8764–8769. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang Y, Zhang YL, Chen HM, Pu HW, Ma WJ,
Li XM, Ma H and Chen X: Expression of Bmi-1 and PAI-1 in esophageal
squamous cell carcinoma. World J Gastroenterol. 20:5533–5539. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wu Z, Min L, Chen D, Hao D, Duan Y, Qiu G
and Wang Y: Overexpression of BMI-1 promotes cell growth and
resistance to cisplatin treatment in osteosarcoma. PLoS One.
6:e146482011. View Article : Google Scholar : PubMed/NCBI
|