Introduction
Human cutaneous melanoma (CM) has been, until
recently, resistant to conventional chemotherapy. However, the
finding that approximately 50% of human CM harbor the driver
mutations V600E or V600K in the BRAF oncogene, a component of the
MAPK pathway (1), led to the
development of MAPK inhibitors (MAPKi), such as vemurafenib
(PLX4032) and dabrafenib directed either to the BRAF-mutated
protein (2–4), and cobimetinib (GDC-0973) and
trametinib targeting downstream kinase MEK (2,5,6). A
phase III clinical study of vemurafenib, alone or combined with
cobimetinib, demonstrated the superiority of the drug combination,
with progression-free survival increasing from 6.9 months for
PLX4032 alone to 9.9 months for the combination (7). The utility of dual BRAF V600E and MEK
inhibition was confirmed in a phase III trial in which dabrafenib
alone was compared to dabrafenib plus trametinib, with an increased
progression-free survival from 8.8 to 9.3 months (8). In both studies, although the overall
response rates were higher than 60%, the number of complete
responses was only 10%. Several resistance mechanisms, most of them
involving a reactivation of the MAPK pathway, have been described
(9–14), but no effective solution to drug
resistance has been found. Intriguingly, Long et al analyzed
biopsies from 15 patients obtained before, early after treatment
and on progression after treatment with vemurafenib or dabrafenib.
They observed that inhibition of proliferative markers was
universal yet unrelated to clinical response, but that cell death
markers were more prominent in responders (15). Therefore, the link between
inhibition of the MAPK pathway, triggered apoptosis, necrosis and
ensuing clinical responses remains to be established. A possible
explanation for clinical relapses could be the presence in tumors
of ‘persister’ cells, a subpopulation of cancer cells that survives
targeted therapy and that could be responsible for therapy failure
and tumor progression (16,17).
Another successful approach to CM therapy has been
the introduction of immune checkpoint inhibitors (18–20).
Although different lines of evidence suggest that the combination
of MAPK-targeted therapies with immunotherapy may offer additional
benefit to eliminate residual disease, treatment with BRAF-mutated
inhibitors apparently increases melanoma differentiation antigen
(MDA) expression (21,22) and T cell tumor infiltration
(23). At present it is not known
whether MAPK inhibition and immunotherapy may be successfully
combined in the clinic. Due to the difficulty in obtaining biopsies
from treated patients, we undertook such analysis using BRAF V600E
mutated cell lines. In this study we employed MAPKi to investigate
in vitro whether surviving populations exist after long-term
MAPKi treatment and, if that were the case, their sensitivity to
immune effectors. We report that after exposure to MAPKi for
several weeks, alone or in combination, a small number of cells
remained alive (SUR) and displayed a complex phenotype with
overlapping characteristics of cancer stem cells (CSCs) and
senescent cells. When released from drug inhibition, SUR cells
proliferated and regained their parental drug sensitivity. Most
importantly, we demonstrated that SUR cells were sensitive to
CD8+ effectors, thereby providing a useful system for
analyzing combination therapy.
Materials and methods
Cell lines
The MEL-XY3 cell line has already been described
(24). The MEL-XY13 cell line was
obtained from a lymph node amelanotic metastasis of an 82-year-old
male patient. Both cell lines are HLA-A*0201-positive and have the
BRAF V600E mutation, and c-kit (exons 11 and 17) and Nras (exons 2
and 3) sequencing revealed no additional mutations. Both cell lines
were grown in melanoma medium (MM) (25) plus 10% fetal bovine serum (FBS)
(Natocor, Carlos Paz, Córdoba, Argentina) at 37°C in
air:CO2 (95:5%) humid incubator.
MEL-XY3SUR and MEL-XY13SUR
were generated by exposing cancer cells to 10 µM PLX4032, 1 µM
GDC-0973 or combined treatment for 5 weeks. Media were changed
twice a week. PLX4032 and GDC-0973 were provided by Genentech
(South San Francisco, CA, USA).
DNA synthesis
DNA synthesis was assessed by measuring
3[H]-labeled thymidine incorporation. Ten thousand
cells/well were seeded in 96-well plates in 200 µl of MM. When
indicated, cells were incubated overnight and PLX4032 and/or
GDC-0973 were subsequently added for different periods. After
performing a 2-h pulse at 37°C with 1 µCi/ml
3[H]-labeled thymidine (Perkin-Elmer, Boston, MA, USA),
the cells were harvested with a NuncCell Harvester 8 (Nalge Nunc
International Corp., Rochester, NY, USA) and the incorporated
radioactivity was determined with a liquid scintillation counter
(Wallac 1214 RackBeta; Pharmacia, Turku, Finland).
MTT cell viability assay
Cells were seeded in 96-well flat-bottomed plates in
triplicate. Twenty-four hours later, serial dilutions of PLX4032
and/or GDC-0973 were added. After incubation for 72 h, 100 µl of 1
mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT; Sigma-Aldrich, St. Louis, MO, USA) diluted in MM were added
to each well and incubation was carried out for 90 min. The
supernatant was discarded and the crystal products were resuspended
with isopropyl alcohol and incubated for 1 h at 30°C. Colorimetric
evaluation was performed with a spectrophotometer at 570 nm. The
inhibition of proliferation induced by the drugs was shown as the
percentage of the growth of the untreated control cells.
IC50 was determined using GraphPad Prism 5.0
software.
Quantitative real-time reverse
transcriptase PCR (RT-qPCR)
Total RNA was purified using TRIzol reagent (Ambion,
Invitrogen, Carlsbad, CA, USA) according to the manufacturer's
instructions. Quantification was performed with a NanoDrop 2000
(Thermo Fisher Scientific, Inc., Wilmington, DE, USA). One
microgram of RNA was reverse-transcribed using SuperScript™ II
Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). The product
was used in subsequent RT-qPCR using KAPA SYBR FAST Universal
Master Mix (Applied Biosystems, Carlsbad, CA, USA) with the primers
listed in Table I. Relative
expression levels were determined by the ΔΔCq method (26), using β-actin gene expression to
normalize all samples. Α melting curve analysis and gel
electrophoresis assessment were used to confirm the specificity of
PCR reactions.
 | Table I.List of primers. |
Table I.
List of primers.
| Genes | Sequences |
|---|
| CD271 | F:
CTGGACAGCGTGACGTTCTCC |
|
| R:
CTGCCACCGTGCTGGCTATGA |
| CD133 | F:
GGACCCATTGGCATTCTC |
|
| R:
CAGGACACAGCATAGAATAATC |
| ABCB5 | F:
CCAAATCGGGGGCTGCGCATCTGTT |
|
| R:
AGCCGCTGCTCCCCACAAATGCTA |
| β-actin | F:
GCCATCTCTTGCTCGAAGTCCAG |
|
| R:
ATGTTTGAGACCTTCAACACCCC |
| Sox10 | F:
GACCAGTACCCGCACCTG |
|
| R:
CGCTTGTCACTTTCGTTCAG |
| Sox2 | F:
CAGTCTGCCGAGAATCCATG |
|
| R:
TTTTTTTTTTCAGTGTCCATATTTC |
| NanoG | F:
CAGCTGTGTGTACTCAATGATAGA |
|
| R:
ACACCATTGCTATTCTTCGGCCAG |
| Oct4 | F:
ACATCAAAGCTCTGCAGAAAGAACT |
|
| R:
CTGAATACCTTCCCAAATAGAACCC |
| NGF | F:
TCATCATCCCATCCCATCTT |
|
| R:
CTTGACAAAGGTGTGAGTCG |
| BDNF | F:
AGCCTCCTCTTCTCTTTCTGCTGGA |
|
| R:
CTTTGTCTATGCCCCTGCAGCCTT |
| NT-3 | F:
TTTCTCGCTTATCTCCGTGGCATCC |
|
| R:
GGCAGGGTGCTCTGGTAATTTTCCT |
| NT-4 | F:
ATGCTCCCTCTCCCCTCAT |
|
| R:
GCATGGGTCTCAGGCCCG |
| MART-1 | F:
GAGAAAAACTGTGAACCTGTGGT |
|
| R:
GACTGTTCTGCAGAGAGTTTCTCAT |
| gp100 | F:
GCTGATCGTGGGCATCTTG |
|
| R:
AGTGACTGCTGCTATGTGG |
| Tyr | F:
TCTGCCAACGATCCTATCT |
|
| R:
AATGGGTGCATTGGCTTCT |
| Trp-2 | F:
CGACTCTGATTAGTCGGAACTCA |
|
| R:
GGTGGTTGTAGTCATCCAAGC |
| MITF | F:
CGAGCTCATGGACTTTCCCTTA |
|
| R:
CTTGATGATCCGATTCACCAAA |
Western blot analysis
Cells were detached, centrifuged, washed with cold
phosphate-buffered saline (PBS) and resuspended in RIPA lysis
buffer and protease inhibitor cocktail (Sigma-Aldrich). Phosphatase
inhibitor cocktail 2 (Sigma-Aldrich) and NaF 10 mM were added for
phospho-protein determinations. Cell extracts were run on a 10%
SDS-PAGE gel and transferred to a nitrocellulose membrane
(Immobilon, 0.45-µm pore size, HATF 304 FO; Millipore, Bedford, MA,
USA). After blocking in 3% BSA with PBS, the blots were incubated
with mouse anti-human CD271 mAb (clone C40-1457; BD Pharmingen, San
Jose, CA, USA), rabbit anti-ERK1 polyclonal Ab (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), rabbit anti-p-ERK
polyclonal Ab (Cell Signaling Technology, Inc., Beverly, MA, USA)
or mouse anti-β-actin (clone AC-74; Sigma-Aldrich). After being
washed, blots were further incubated with 1/2,500 alkaline
phosphatase-AffiniPure F(ab')2 fragment goat anti-mouse
IgG (H+L) (Jackson ImmunoResearch Laboratories, Inc., West Grove,
PA, USA) and developed with nitro blue
tetrazolium/5-bromo-4-chloro-3-indolyl phosphate (NBT-BCIP)
(Promega, Madison, WI, USA). Alternatively, blots were incubated
with peroxidase anti-rabbit antibody (Vector Laboratories,
Burlingame, CA, USA), developed with SuperSignal West Dura Extended
Duration substrate (Thermo Fisher Scientific, Rockford, IL, USA),
and chemoluminescence was measured in an ImageQuant LAS 4000
instrument (GE Healthcare, Uppsala, Sweden).
Flow cytometry
Cells were incubated with human-specific CD271
(clone C40-1457; BD Pharmingen), MART-1 (2A9) (27), gp100 (clone Hmb45; Dako, Glostrup,
Denmark) and HLA-A2-PE (clone BB7.2; BD Pharmingen) antibodies for
1 h at 4°C. Before the use of MART-1 and gp100 antibodies a
permeabilization step with 0.1% saponin was performed. Washed cells
were resuspended in PBS and stored on ice or labeled with RPE
conjugated anti-mouse polyclonal Ab (1:50; Dako). Ag expression was
assessed using a FACSCalibur flow cytometer (BD Biosciences, San
Diego, CA, USA). Analysis was performed with the FlowJo 7.6
software. Isotype-matched mouse antibodies were used as
controls.
For the carboxyfluorescein succinimidyl ester (CFSE)
assay, cells were detached, washed with PBS and labeled with 10 µM
CFSE according to the manufacturer's indications (Invitrogen), and
then plated for 96 h in the presence or absence of PLX4032. Cells
were then detached and analyzed with a FACSCalibur flow
cytometer.
For cell cycle analysis, cells were detached and
fixed with 70% ethanol ON at 4°C. Cells were stained with propidium
iodide solution (50 µg/ml with 100 µg/ml RNase A in PBS) for 30 min
at 37°C. Cells were analyzed on a FACSCalibur flow cytometer using
the FlowJo 7.6 software for data analysis.
MACS
Magnetic-activated cell sorting (MACS) to separate
the CD271+ and CD271− subpopulations was
performed according to the manufacturer's instructions using
anti-human-specific CD271 (clone C40-1457; BD Pharmingen),
anti-mouse IgG microbeads and MS columns (Miltenyi Biotec, Bergisch
Gladbach, Germany). Sorting verification was performed by flow
cytometry.
SA-β-Gal staining
Cells were either left untreated or were treated
with 10 µM PLX4032 for 72 h or 5 weeks. Senescence-associated
β-galactosidase (SA-β-Gal) staining was performed according to the
chromogenic assay described by Debacq-Chainiaux et al
(28).
Reverse-phase protein array (RPPA)
analysis
Cell lysates were 2-fold serial diluted for five
dilutions (from undiluted to 1:16 dilutions) and arrayed on
nitrocellulose-coated slides. Samples were probed with antibodies
by a tyramide-based signal amplification approach and visualized by
DAB colorimetric reaction. Slides were scanned on a flatbed scanner
to produce 16-bit tiff images. Spots from tiff images were
identified and the density was quantified by an Array-Pro Analyzer.
Relative protein levels for each sample were determined by
interpolation of each dilution curve from the standard curve
(supercurve) of the slide (antibody). All data points were
normalized for protein loading and transformed into a linear value
and normalized linear values were transformed to log2
values.
In vivo tumorigenicity assay
NOD/LtSz-scid IL2Rγnull mice (NSG)
were subcutaneously injected with 500 MEL-XY3 parental or
MEL-XY3SUR-PLX cells in a suspension of Matrigel (BD
Matrigel™ Basement Membrane Matrix, BD Biosciences) with PBS in a
1:1 ratio. Tumor growth was monitored three times a week for the
entire period of the experiment. Tumor size was assessed using a
caliper to calculate tumor volume (V) by applying the formula: V =
[length (mm)] × [width (mm)]2/2. All animal procedures
were approved by the Institutional Animal Care Board of the Leloir
Institute (Buenos Aires, Argentina).
In vitro lysis of melanoma cells by
cytotoxic T lymphocyte (CTL) clones
To determine lysis, effector CD8+
lymphocytes (E) specific for the HLA-A*0201 restricted MART-1
(M27:AAGIGILTV) or gp100 (G154:KTWGQYWQV) antigens (29) and target MEL-XY3 (control,
MEL-XY3SUR-PLX or MEL-XY3SUR-GDC) cells (T)
were incubated overnight at different E:T ratios (1:1, 5:1 and
10:1) in AIM V medium (Invitrogen, Grand Island, NY, USA). Cells
were recovered and plated in quadruplicate for a clonogenic assay.
For this, the cells were resuspended in MM + 10% FBS with 1.5%
methylcellulose (Sigma-Aldrich), plated at 1,000 cells/well on MW24
plates over a 0.5% agar MM + 10% FBS underlayer and incubated for
14 days. Colonies ≥30 cells were counted using an inverted Olympus
microscope.
ELISA IFN-γ secretion
IFN-γ secretion into the supernatant by cytotoxic T
cells was determined with a BD OptEIA human IFN-γ ELISA kit (BD
Biosciences), following the manufacturer's recommendations. A
calibration curve was constructed for each experiment and the
sample concentration was calculated by regression analysis.
Controls for this experiment included CM cell lines
HLA-A*0201-negative that do not express MART-1 or gp100 and the
IIB-BRG breast cancer cell line HLA-A*0201-positive that does not
express MART-1 or gp100 (negative controls) and CTLs alone.
Results
Long-term treatment with MAPKi results
in a surviving population of nonproliferating cells
Since clinical resistance to MAPKi almost always
emerges after months of treatment, we sought to determine whether
some cells remained viable after long-term treatment of
BRAF-mutated cell lines with MAPKi and, if so, to characterize
them. Accordingly, we first investigated the effect of PLX4032 and
GDC-0973 on the MEL-XY3 CM cell line, heterozygous for the BRAF
V600E mutation. MEL-XY3 cells were sensitive to PLX4032
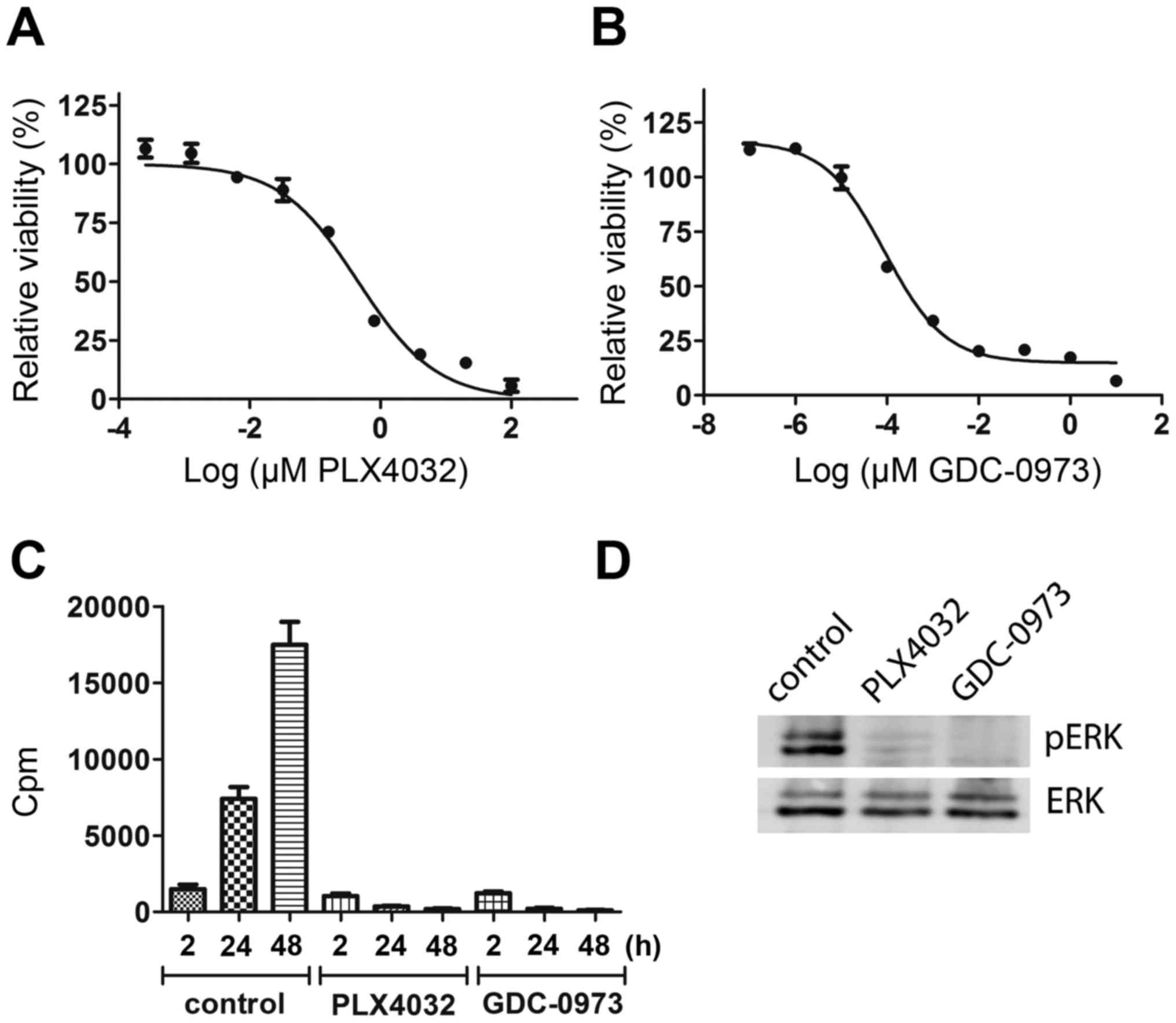
(IC50 0.45 µM) (Fig. 1A)
and to GDC-0973 (IC50 0.1 nM) (Fig. 1B). Inhibition of the MAPK pathway
was also confirmed by inhibition of DNA synthesis (Fig. 1C) and of ERK phosphorylation
(Fig. 1D). Though some DNA
synthesis occurred within 0–2 h of inhibitor treatment, after 24
and 48 h it was totally abolished. ERK phosphorylation was
inhibited 30 min after starting PLX4032 and GDC-0973 treatment. The
effect of short-term treatment of MEL-XY3 with both PLX4032 and
GDC-0973 was cytostatic, with cells starting to detach after a few
days. We next exposed MEL-XY3 cells to 10 µM PLX4032 for 5 weeks;
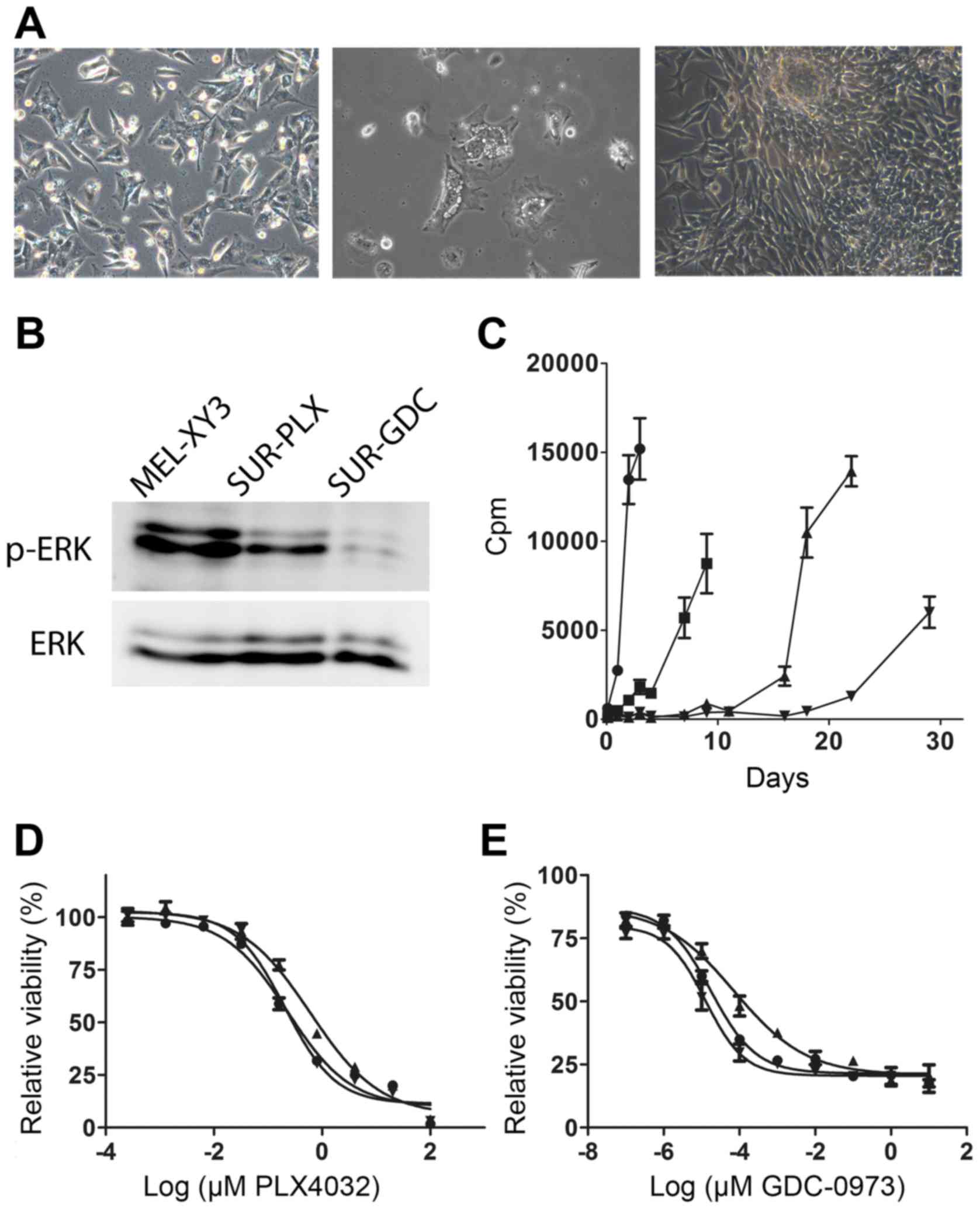
some cells remained attached and viable (Fig. 2A, left) and were detectable even
after five months of treatment. We referred to this subpopulation
of surviving cells as MEL-XY3SUR-PLX. We analyzed the
fate of MEL-XY3SUR-PLX cells when the inhibitor was
removed. After an initial period of several days, in which some
cells presented abundant cytoplasmic vesicles and some detached
from the plates (Fig. 2A, middle),
MEL-XY3SUR-PLX cells re-established vigorous growth
(Fig. 2A, right), and generated
cells with a similar phenotype to the parental cell line.
In order to determine whether such a quiescent
phenotype could also be generated by other MAPKi, MEL-XY3 cells
were maintained in the presence of 1 µM GDC-0973 for 5 weeks; a
small, viable subpopulation with analogous characteristics to
MEL-XY3SUR-PLX cells was also observed, and was named
MEL-XY3SUR-GDC. After combined treatment with 10 µM
PLX4032 and 1 µM GDC-0973 (MEL-XY3SUR-PLX-GDC),
surviving cells with a similar phenotype were observed. The status
of the MAPK pathway in MEL-XY3SUR-PLX and
MEL-XY3SUR-GDC cells while they were in the presence of
the inhibitors was investigated. MEL-XY3SUR-PLX cells
presented lower levels of p-ERK than parental cells, although
somewhat higher than MEL-XY3SUR-GDC cells (Fig. 2B).
When we studied DNA synthesis at different
time-points after drug removal, we observed that
MEL-XY3SUR-PLX cells started proliferation before
MEL-XY3SUR-GDC, while MEL-XY3SUR-PLX-GDC
cells were the last to resume growth (Fig. 2C). Notably, after a 5-week treatment
with PLX4032 or GDC-0973, and allowing treated cells to regrow in
the absence of the inhibitors, their sensitivity to PLX4032 or
GDC-0973 remained unaltered with respect to the parental cells
(Fig. 2D and E).
In order to evaluate whether the SUR phenotype also
arose in other melanoma cell lines, we studied MAPKi treatment in
the MEL-XY13 cell line. This cell line is heterozygous for the BRAF
V600E mutation and sensitive to PLX4032 (0.36 µM) and GDC-0973
(1.25 nM). When MEL-XY13 cells were treated for 5 weeks with
PLX4032 or GDC-0973, MEL-XY13SUR-PLX and
MEL-XY13SUR-GDC, respectively, were obtained.
Furthermore, when the drugs were removed, MEL-XY13SUR
cells restarted growth and their sensitivity to PLX4032 and
GDC-0973 was equal to the parental cell line (Fig. 3A and B).
Phenotypic characterization of
MEL-XY3SUR cells
Since MEL-XY3SUR cells were
non-proliferating and different reports associate drug resistance
with the CSC phenotype, we sought to determine whether they
displayed some CSC characteristics. After treatment with 10 µM
PLX4032 for 5 weeks, we analyzed the expression levels of putative
CSC markers, such as CD271, ABCB5 and CD133 (prominin-1) (30–32).
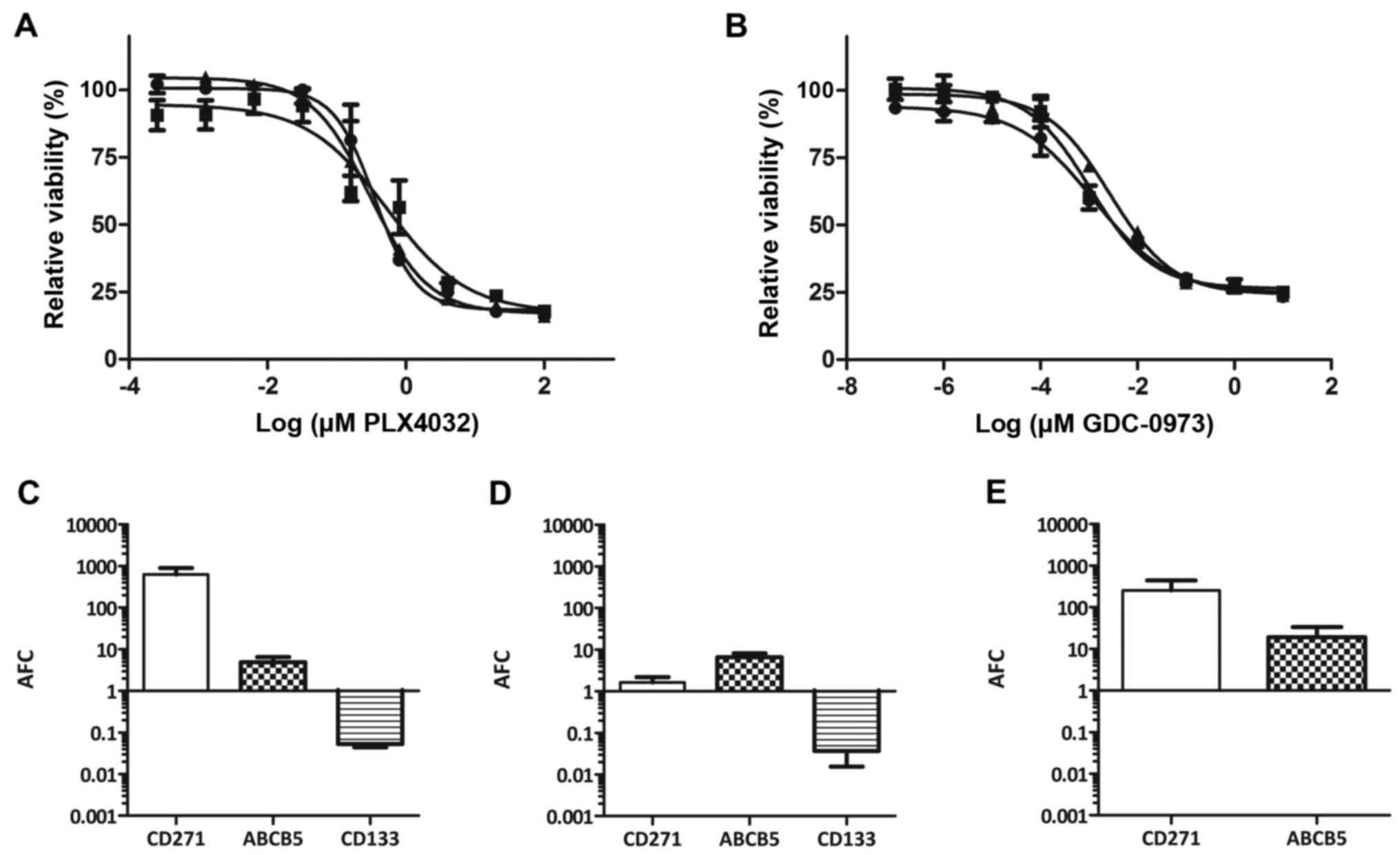
As compared to parental MEL-XY3, the mRNA levels for CD271 and
ABCB5 in MEL-XY3SUR-PLX cells increased 1,000- and
30-fold, respectively, whereas CD133 levels decreased (Fig. 4A). MEL-XY3SUR-GDC cells
obtained after 1 µM of GDC-0973 treatment (Fig. 4B) and MEL-XY3SUR-PLX-GDC
obtained after the combined treatment with both inhibitors
(Fig. 4C) also had increased CD271
and ABCB5 mRNA levels, although the increase in CD271 was smaller.
Similar results were obtained with MEL-XY13SUR, with the
exception that in MEL-XY13SUR-GDC cells, CD271 mRNA
levels did not change (Fig. 3C-E).
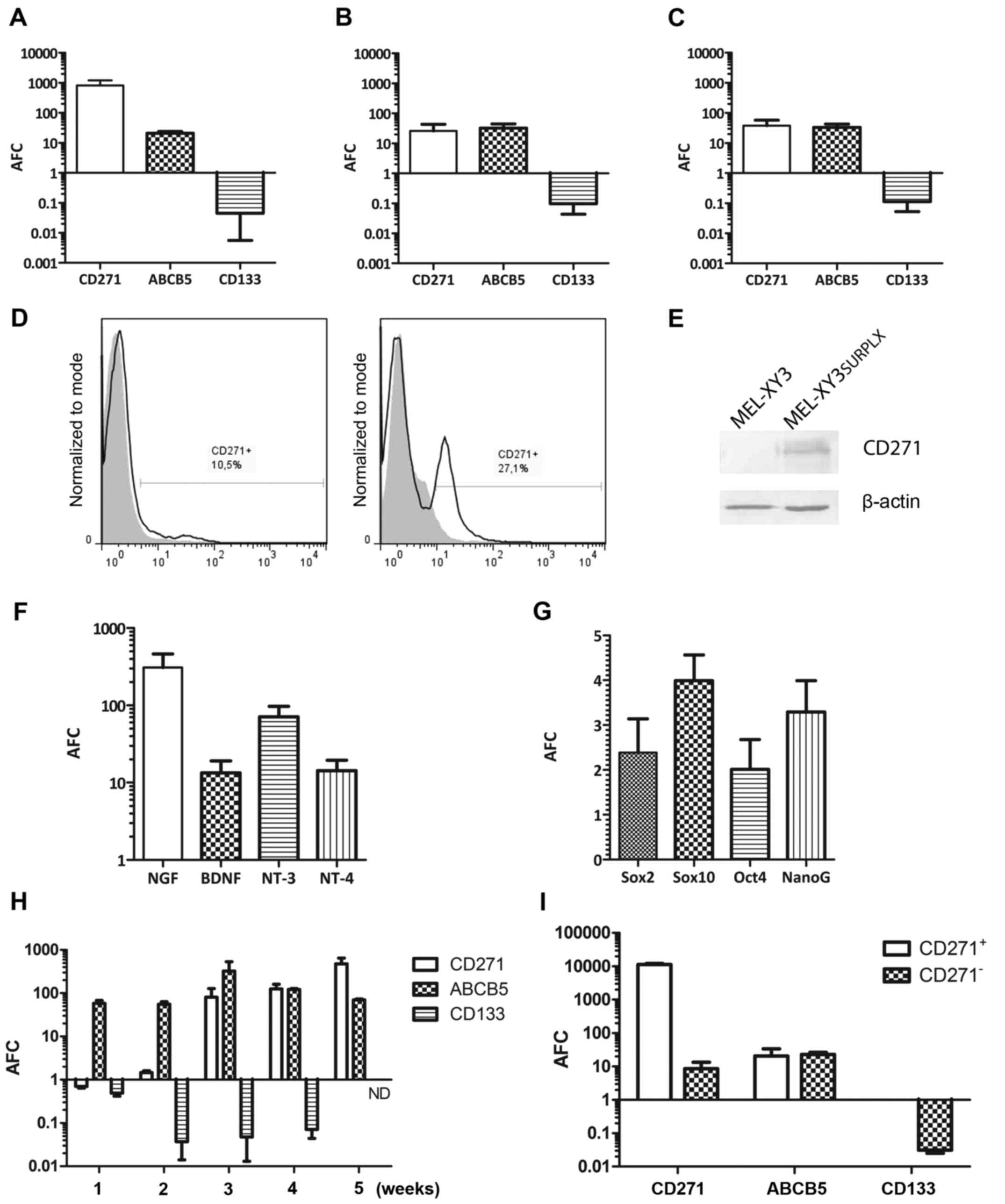
Increased CD271 expression in MEL-XY3SUR-PLX cells was
confirmed at the protein level by flow cytometry (Fig. 4D) and by western blot analysis
(Fig. 4E). In the flow cytometric
analysis of MEL-XY3SUR-PLX cells, two peaks were
observed, demonstrating the presence of two cell
subpopulations.
We also analyzed whether changes in CD271 ligand
expression also took place in the MEL-XY3SUR-PLX cells;
all the mRNA levels of neurotrophins that bind CD271 (NGF, BDNF,
NT-3, NT-4) increased 10- to −300-fold (Fig. 4F). Expression levels of the stem
cell associated transcription factors Sox10, Sox2, Oct4 and Nanog
were also determined, and a modest 2- to 4-fold increase was
observed in MEL-XY3SUR-PLX cells (Fig. 4G). We analyzed the expression time
course of ABCB5, CD271 and CD133 in MEL-XY3 cells exposed to 10 µM
PLX4032 at different time-points. ABCB5 expression rapidly
increased and remained at high levels during the 5-week treatment.
Instead, CD271 expression did not increase until the third week of
treatment and CD133 started decreasing after 2 weeks of treatment
(Fig. 4H). In order to determine
whether CD271 and ABCB5 were uniformly expressed in the
MEL-XY3SUR-PLX cell population, CD271+ and
CD271− cells were separated by magnetic sorting.
CD271− predominated over CD271+ cells but
both expressed ABCB5 (Fig. 4I),
suggesting that the expression of both markers was not related.
CD133 mRNA was not detected in CD271+
MEL-XY3SUR-PLX.
MEL-XY3SUR cells present
senescence-associated features
It has been previously shown that PLX4032 and
another mutant BRAF inhibitor, LGX818 (encorafenib), induce
senescent characteristics in melanoma cells (33,34).
It has also been shown that the expression of mutated BRAF in
normal melanocytes determines oncogene-induced senescence (35). Therefore, we decided to investigate
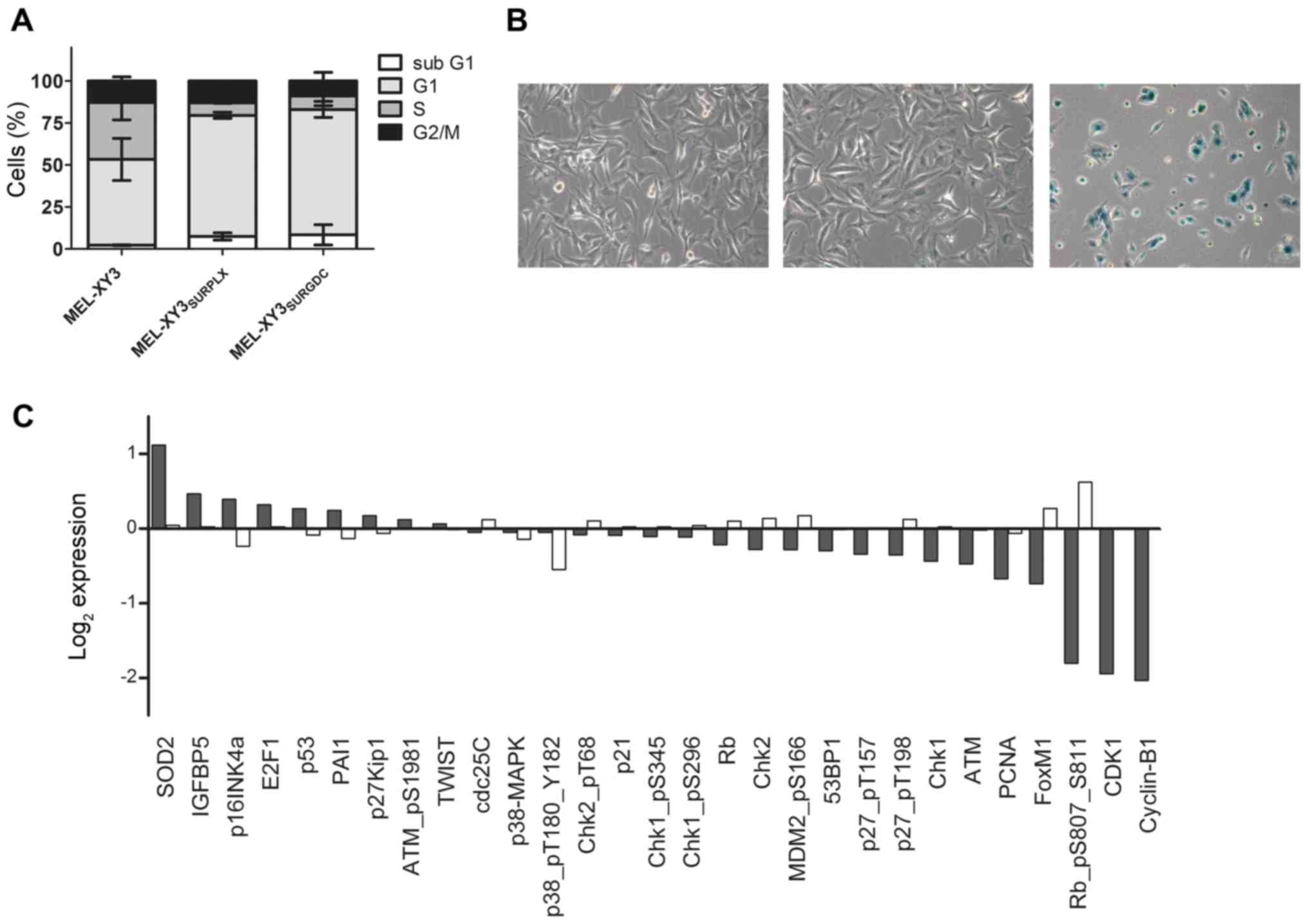
senescent characteristics in our SUR population. The cell cycle of
MEL-XY3SUR-PLX and MEL-XY3SUR-GDC was
analyzed by flow cytometry, and a decrease in the S phase and an
increase in the proportion of cells in the G0/G1 phase were
observed, consistent with the observations of Fig. 2 (Fig.
5A).
We then analyzed
senescence-associated-β-galactosidase (SA-β-gal) activity in
MEL-XY3SUR-PLX. Although MEL-XY3 cells treated with 10
µM PLX4032 for 72 h did not stain positive for SA-β-gal, most
MEL-XY3SUR-PLX cells did (Fig. 5B), thereby suggesting senescence. To
further investigate other senescence-related proteins, we performed
a RPPA (Fig. 5C). In relation to
the parental cell line, senescence-related proteins superoxide
dismutase 2 (SOD2), insulin-like growth factor binding protein-5
(IGFBP5) and p16INK4 were increased. On the other hand,
E2F1, p53, plasminogen activator inhibitor-1 (PAI1) and
p27kip1 displayed only small changes. In
MEL-XY3SUR-PLX cells, there was also a general decrease
in cell cycle-related proteins, with striking decreases in p-Rb,
CDK1 and cyclin B1.
Variable phenotype of
MEL-XY3SUR
We next investigated whether
MEL-XY3SUR-PLX and MEL-XY3SUR-GDC quiescent
cells coexisted in the cell line with proliferating cells before
MAPKi treatment. If that were the case, it could be assumed that
after drug removal and growth resumption, at least a portion of SUR
cells would remain quiescent, thereby confirming that they are a
‘reservoir’ for proliferating cells. On the other hand, if
quiescence is a transitory, variable state induced by chemotherapy,
every surviving cell would leave behind its quiescence and start
regrowth when the drugs were removed. We stained
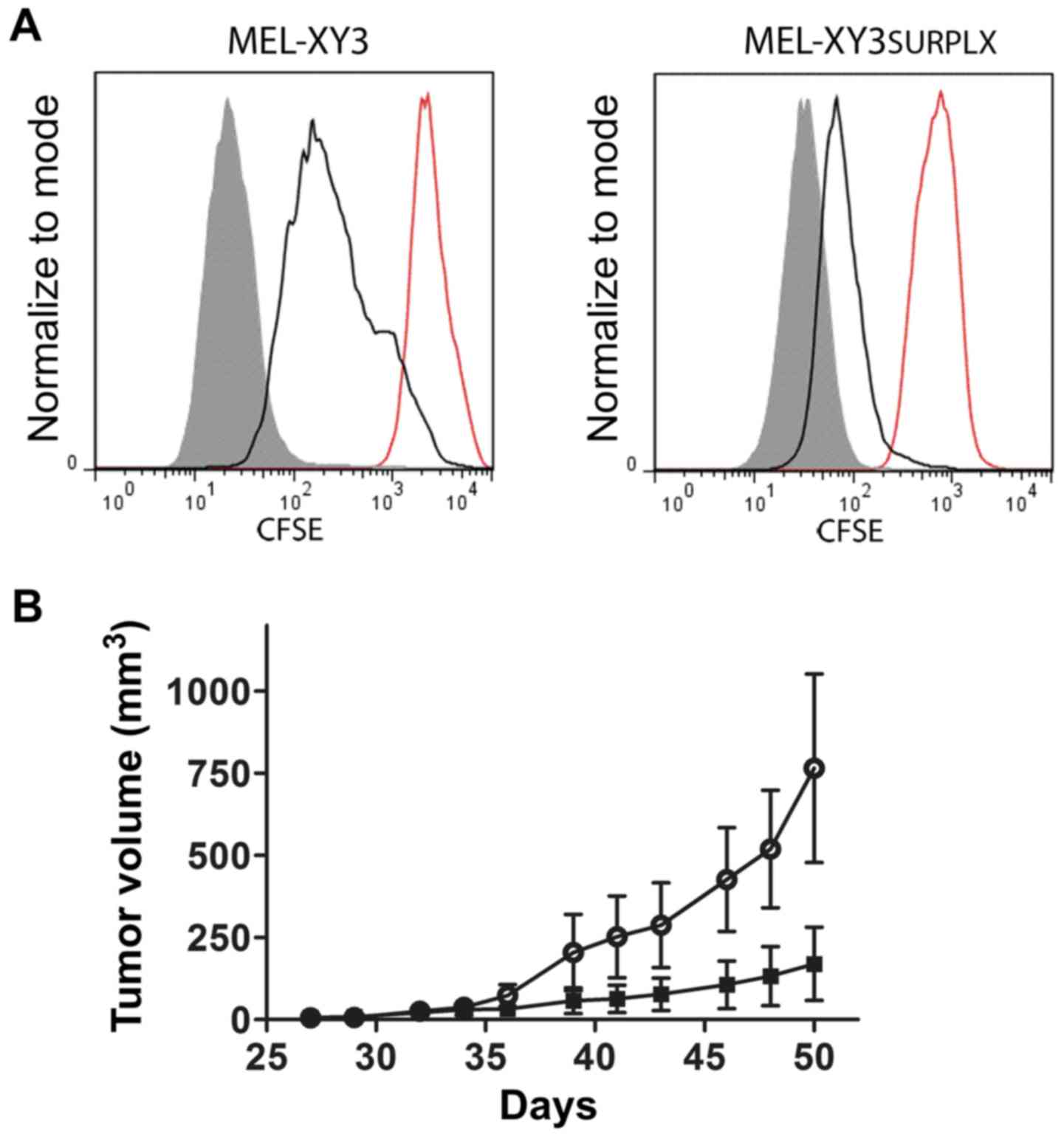
MEL-XY3SUR-PLX cells with CFSE, and they remained in
culture for 96 h in the absence or presence of the inhibitor;
retention of the label was then analyzed. During those 96 h,
MEL-XY3SUR-PLX cells underwent two rounds of cell
division without leaving behind a resting subpopulation. When a
similar experiment was performed with the parental cell line, three
rounds of cell division were achieved (Fig. 6A). This experiment suggests that
tumor cell plasticity and phenotypic switching would account for
the previously described acquisition of quiescence. When the same
experiment was performed with the MEL-XY3SUR-GDC cells,
no cell division was observed after 96 h, which is consistent with
the lagging DNA synthesis after drug removal (Fig. 2C).
Tumorigenicity of
MEL-XY3SUR-PLX cells
To determine whether MEL-XY3SUR-PLX cells
retained their tumorigenic potential, we injected 500 cells into
NSG mice and compared their growth rate with parental MEL-XY3
cells. Even though both cell types developed tumors,
MEL-XY3SUR-PLX grew faster than parental cells, although
the difference was not statistically significant (Fig. 6B).
Sensitivity of SUR cells to
MDA-directed CTL clones
It has been reported that MDA expression increases
after PLX4032 treatment, both in vitro and in patients
(21,22). We confirmed increased mRNA levels of
MDAs MART-1, gp100, TYR and Trp-2 when MEL-XY3 cells were treated
with 10 µM PLX4032 for 72 h (Fig.
7Α). We found that MEL-XY3SUR-PLX cells presented
MDA mRNA levels similar to or even higher than those of parental
cells (Fig. 7Β). Most importantly,
MART-1 and gp100 protein levels were similar in
MEL-XY3SUR-PLX and MEL-XY3SUR-GDC compared to
those of parental MEL-XY3 cells (Fig.
7Β), thereby revealing persistent MDA synthesis. MITF mRNA
expression was also analyzed in MEL-XY3SUR-PLX cells but
no significant changes in expression were found (Fig. 7Β).
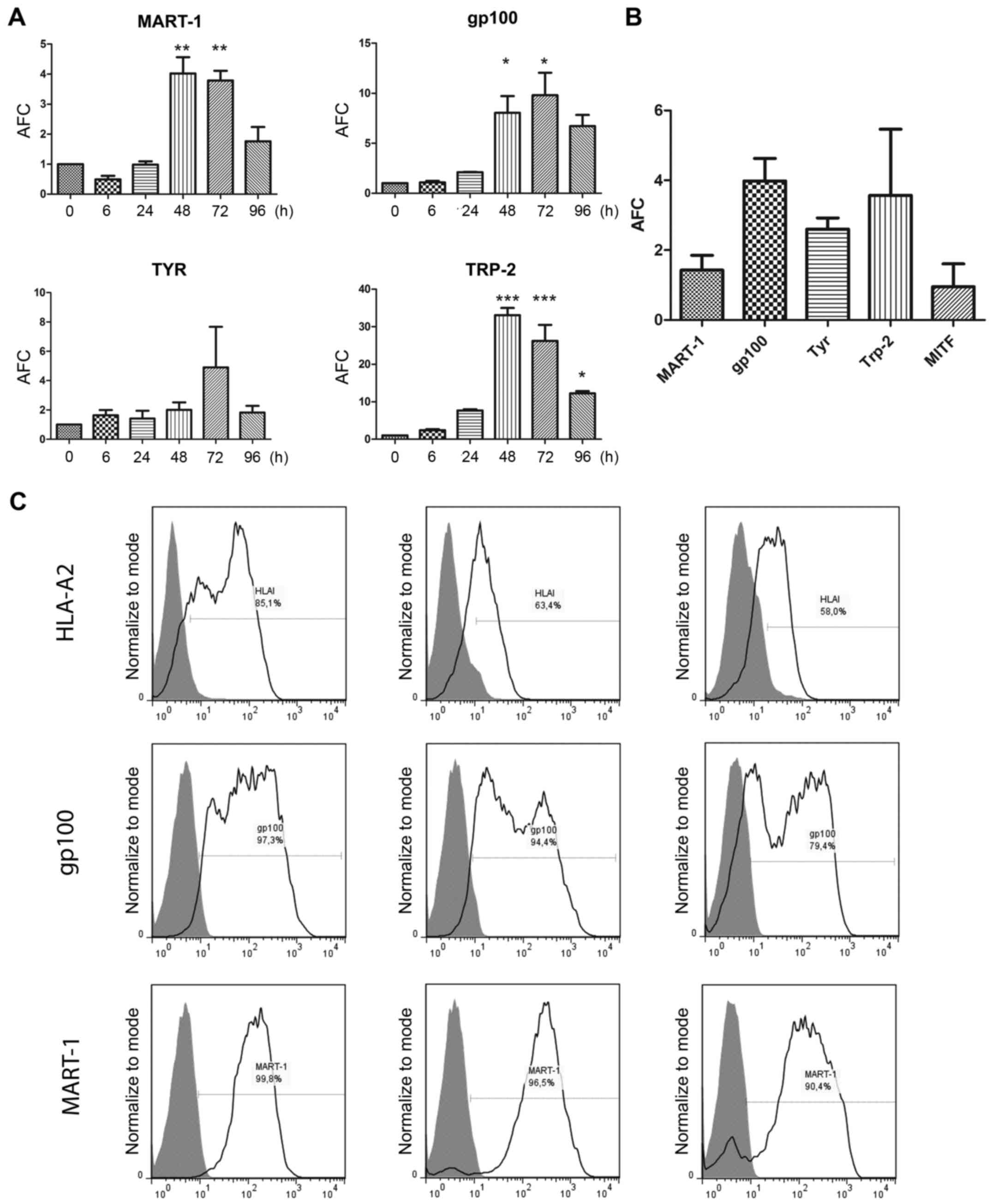 | Figure 7.MDA expression levels in
MEL-XY3-treated cells. (A) mRNA expression levels of MART-1, gp100,
Tyr and Trp-2 in MEL-XY3 cells treated with 10 µM PLX4032 for
different time-points. (B) mRNA expression levels of MART-1, gp100,
Tyr, Trp-2 and MITF in MEL-XY3SUR-PLX cells. mRNA levels
were determined by RT-qPCR. AFC with respect to parental cells; (C)
HLA-A2, MART-1 and gp100 protein expression levels were determined
by flow cytometry. Representative histograms for MEL-XY3 (left),
MEL-XY3SUR-PLX (middle) and MEL-XY3SUR-GDC
(right) are shown. Gray-filled histogram, isotype-matched control;
black line, HLA-A2 (upper panel), gp100 (middle panel) and MART-1
(lower panel). MDA, melanoma differentiation antigen; AFC, average
fold-change. *p<0.05, **p<0.01, ***p<0.001. |
It was therefore important to determine whether,
although resistant to chemotherapy, MEL-XY3SUR cells
could be killed by immunological effectors. First, we established
that MEL-XY3SUR cells expressed, although at lower
levels than the parental line, the HLA-A*0201 haplotype, in which
MART-1 and gp100 peptides are presented (Fig. 7C). We then studied the
susceptibility of MEL-XY3SUR cells to two CTL clones,
HLA-A*0201-restricted and specific for gp100 (G154) and MART-1
(M26) antigens (29). We had
previously shown that MEL-XY3 cells are lysed by specific CTL
(36). We now demonstrated that
MEL-XY3SUR-PLX cells were killed by specific CTL clones,
as determined by clonogenic assays. At a 1:1 effector:target ratio,
a 50% lytic effect was observed, as evidenced by the reduction of
colony formation; at higher ratios almost no cells survived.
MEL-XY3SUR-GDC cells were also sensitive to CTL killing,
although it is worth noting that MEL-XY3SUR-GDC cells
were less clonogenic than MEL-XY3SUR-PLX cells (Fig. 8A). As a surrogate measure confirming
that MEL-XY3SUR cells were sensitive to CTL action,
IFN-γ release after co-incubation of MEL-XY3SUR cells
with CTL clones was assessed. A strong cytokine release was
triggered by MEL-XY3SUR-PLX, indicating a potent
interaction between CTL and tumor cells, even stronger than with
parental cells. However, almost no CTL activation occurred after
incubation with MEL-XY3SUR-GDC cells, even though a
lytic effect was observed (Fig.
8B). Similar results were obtained with the CTL clone specific
for MART-1 (M26) (Fig. 8C and D).
Equivalent results were obtained with the CTL clone specific for
gp100 in MEL-XY13 and MEL-XY13SUR-PLX cells (Fig. 8E and F).
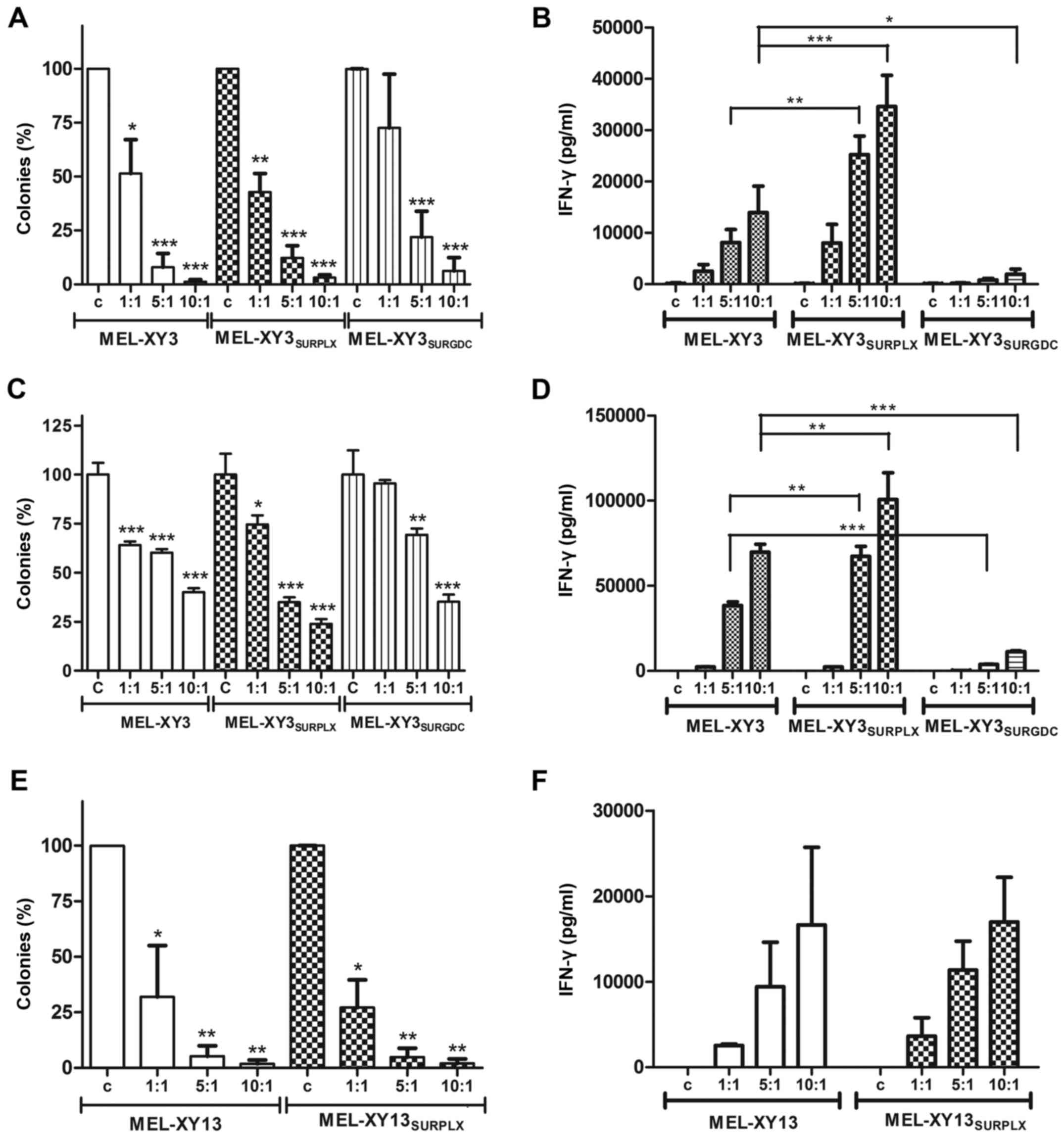 | Figure 8.Lysis of melanoma cells by MART-1 and
gp100-specific CTL. Target cells were incubated with effector (A,
B, E and F) gp100 CTL (C and D) MART-1 CTL. Effector:target cells
ratio 1:1, 5:1, 10:1; c, control without effector cells. (A, C and
E) Cell lysis was determined by inhibition of colony formation (%,
mean ± SD of three experiments). In every case, the number of
colonies formed in the absence of CTL was used to relativize the
colony growth of treated cells. (B, D and F) IFN-γ secretion by CTL
was determined by ELISA as a measure of CTL activation. Mean ± SEM
of three experiments. *p<0.05, **p<0.01, ***p<0.001. CTL,
cytotoxic T lymphocyte. |
Discussion
CM patients harboring BRAF V600E mutations and
treated with MAPKi almost invariably present with recurrence, as CM
cells acquire different resistance mechanisms, most of them
involving reactivation of the MAP kinase pathway (9–14), or
activation of other proliferative pathways such as PI3K signaling
(37,38). Through the study of two CM cell
lines, carrying BRAF V600E mutations, we provide evidence of an
alternative resistance mechanism: prolonged treatment of BRAF-V600E
CM cells with MAPKi induced surviving cell subpopulations, here
named SUR, whose main characteristics were as follows. i) They are
quiescent; ii) when the drugs are withdrawn SUR cells return to
proliferation, albeit remaining equally sensitive to the drugs as
the parental line; iii) they display a phenotype sharing features
of CSC and senescent cells; and iv) they are sensitive to specific
CD8 lymphocytes. In reference to quiescence, it was found that the
inhibition attained with GDC-0973 was more profound than that
attained with PLX4032. The time required to recover DNA synthesis
after drug removal was dramatically different when cells were
treated with PLX4032, GDC-0973 or their combination: a couple of
days after PLX4032 removal, two weeks after GDC-0973 removal, and
more than three weeks after PLX4032 and GDC-0973
combination-treatment removal, respectively. Coincidently, the
clonogenicity of the SUR-GDC cells was 7-fold less than that of the
SUR-PLX cells, which also pointed to a diminished vitality. In
addition, the p-ERK levels observed in the
MEL-XY3SUR-GDC cells were lower than these level in the
MEL-XY3SUR-PLX cells. With respect to the phenotype of
SUR cells, a mixture of protein expression normally attributed to
CSC markers and senescence was observed. With respect to CSC
markers, CD271 was greatly increased in the
MEL-XY3SUR-PLX and MEL-XY3SUR-GDC cells.
Redmer et al previously proposed that CD271 expression was
essential for the tumorigenicity of CM cells (39). However, Cheli et al described
CD271 as an imperfect CSC marker since only the slow growing cells
among the CD271+ subpopulation presented increased
tumorigenic potential (40). Our
results support those of Ravindran Menon et al (41), who demonstrated that PLX4032
increased CD271 expression in CM cells and induced the transition
into a slow cycling state. The same phenotype was also observed
when cells were exposed to different types of stress, such as
hypoxia or nutrient starvation, and the authors proposed that these
changes were part of an early innate response program. In regard to
the efflux pump ABCB5, MEL-XY3 expression increased within one week
after exposure to PLX4032 and two weeks before CD271 started to
increase. We suggest that an ABCB5 increase would allow some tumor
cells to transitorily resist PLX4032 and GDC-0973 treatment,
thereby allowing time to develop more stable survival mechanisms.
Our results confirm that ABCB5-expressing cells pre-exist in
melanoma as previously demonstrated (31,42),
and that such expression may be increased manifold in the SUR
population after MAPK inhibition. In addition, Chartrain et
al previously showed that treatment with PLX4032 leads to the
selection of ABCB5-positive cells (43). The observed downregulation of CD133
(prominin-1) in SUR cells suggests that, in our conditions, this
cell surface glycoprotein, expressed in CSC of different tumors
(44–46), is not essential to maintain SUR
conditions. Similar results were observed by Quintana et al
who did not find any correlation between the expression of CD133
and ABCB5 with cell tumorigenicity (47).
MEL-XY3SUR-PLX also presented
senescence-associated features, such as enhancement of SA-β-gal
activity and a 10-fold increase in SOD2 (mitochondrial Mn-SOD2).
Although some authors reported a diminution of SOD2 in senescent
cells (48), it was shown that
senescent drug-tolerant cells maintain low levels of reactive
oxygen species and present enhanced expression levels of
antioxidant enzymes (49).
Moreover, the acquisition of a senescent phenotype has been
proposed as an intermediate state that would allow escape from
chemotherapeutic-induced death and would lead to a CSC-like
phenotype (49). In an RPPA
analysis comparing SUR-PLX cells with the parental line, important
decreases in hyper-phosphorylated Rb, CDK1 and cyclin B1 were
observed, demonstrating that PLX4032 induces a blockage of cell
cycle and mitotic entry. The full reversibility of this blockage is
supported by: i) regrowth of cells after drug removal, both with
and without anchorage; and ii) the ability to generate tumors in
immunodeficient mice. A recent study also reported that
oncogene-induced senescence may not be irreversible and that the
senescent state could give rise to tumor-initiating cells (50).
Most importantly, we also established that SUR cells
are not below the radar of the immune system. Different studies
suggested that PLX4032 induces MDA expression, although diminished
MDA levels have also been reported after acquired resistance
(15,22). SUR cells express HLA-I molecules,
MDAs, and are susceptible to CTL killing, suggesting that the
combination of MAPKi with immunotherapy could eliminate cells that
present this type of variable resistance. This would be especially
important in patients who, after treatment with MAPKi, attain
complete or nearly complete responses; after that moment treatment
with anti-checkpoint inhibitors could trigger an attack of tumor
SUR cells when the tumor mass is at its lowest and before
resistance emerges.
In conclusion, we describe in this study the
acquisition of a variable, drug-tolerant, immunosensitive
phenotype, which would allow residual tumor cells to survive in the
presence of inhibitors of the MAPK pathway, but remain sensitive to
immune effectors. Further investigation will be required to
establish the in vivo presence and relevance of this
phenotype and its implication for targeted therapies.
Acknowledgements
This study was supported by the Agencia Nacional de
Promoción Científica y Tecnológica (ANPCyT), the Fundación Cáncer,
and the Fundación Sales and Fundación María Calderón de la Barca,
Argentina. J.M. is a member of the National Research Council of
Argentina (CONICET), and F.-P.M.R. and A.B. are fellows of the same
institution. The funders had no role in the manuscript design, data
collection and analysis, decision to publish, or preparation of the
manuscript. J.M. received a research grant from ROCHE.
Glossary
Abbreviations
Abbreviations:
|
CM
|
cutaneous melanoma
|
|
CSCs
|
cancer stem cells
|
|
CTLs
|
cytotoxic T lymphocytes
|
|
MAPKi
|
MAPK inhibitors
|
|
MDA
|
melanoma differentiation antigen
|
|
SA-β-Gal
|
senescence-associated
β-galactosidase
|
References
|
1
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bollag G, Hirth P, Tsai J, Zhang J,
Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, et al:
Clinical efficacy of a RAF inhibitor needs broad target blockade in
BRAF-mutant melanoma. Nature. 467:596–599. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et
al: BRIM-3 Study Group: Improved survival with vemurafenib in
melanoma with BRAF V600E mutation. N Engl J Med. 364:2507–2516.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hauschild A, Grob JJ, Demidov LV, Jouary
T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr,
Kaempgen E, et al: Dabrafenib in BRAF-mutated metastatic melanoma:
A multicentre, open-label, phase 3 randomised controlled trial.
Lancet. 380:358–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
King AJ, Arnone MR, Bleam MR, Moss KG,
Yang J, Fedorowicz KE, Smitheman KN, Erhardt JA, Hughes-Earle A,
Kane-Carson LS, et al: Dabrafenib; preclinical characterization,
increased efficacy when combined with trametinib, while BRAF/MEK
tool combination reduced skin lesions. PLoS One. 8:e675832013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hoeflich KP, Merchant M, Orr C, Chan J,
Den Otter D, Berry L, Kasman I, Koeppen H, Rice K, Yang NY, et al:
Intermittent administration of MEK inhibitor GDC-0973 plus PI3K
inhibitor GDC-0941 triggers robust apoptosis and tumor growth
inhibition. Cancer Res. 72:210–219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Larkin J, Ascierto PA, Dréno B, Atkinson
V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas
L, et al: Combined vemurafenib and cobimetinib in BRAF-mutated
melanoma. N Engl J Med. 371:1867–1876. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Long GV, Stroyakovskiy D, Gogas H,
Levchenko E, De Braud F, Larkin J, Garbe C, Jouary T, Hauschild A,
Grob JJ, et al: Combined BRAF and MEK inhibition versus BRAF
inhibition alone in melanoma. N Engl J Med. 371:1877–1888. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nazarian R, Shi H, Wang Q, Kong X, Koya
RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al: Melanomas
acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS
upregulation. Nature. 468:973–977. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Villanueva J, Vultur A, Lee JT,
Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu
X, Gimotty PA, Kee D, et al: Acquired resistance to BRAF inhibitors
mediated by a RAF kinase switch in melanoma can be overcome by
cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 18:683–695. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi H, Moriceau G, Kong X, Lee MK, Lee H,
Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, et al: Melanoma
whole-exome sequencing identifies V600EB-RAF amplification-mediated
acquired B-RAF inhibitor resistance. Nat Commun. 3:7242012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Johannessen CM, Boehm JS, Kim SY, Thomas
SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP,
Barretina J, et al: COT drives resistance to RAF inhibition through
MAP kinase pathway reactivation. Nature. 468:968–972. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Straussman R, Morikawa T, Shee K,
Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J,
Frederick DT, et al: Tumour micro-environment elicits innate
resistance to RAF inhibitors through HGF secretion. Nature.
487:500–504. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Poulikakos PI, Persaud Y, Janakiraman M,
Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al:
RAF inhibitor resistance is mediated by dimerization of aberrantly
spliced BRAF(V600E). Nature. 480:387–390. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Long GV, Wilmott JS, Haydu LE, Tembe V,
Sharma R, Rizos H, Thompson JF, Howle J, Scolyer RA and Kefford RF:
Effects of BRAF inhibitors on human melanoma tissue before
treatment, early during treatment, and on progression. Pigment Cell
Melanoma Res. 26:499–508. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bivona TG and Doebele RC: A framework for
understanding and targeting residual disease in oncogene-driven
solid cancers. Nat Med. 22:472–478. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sharma SV, Lee DY, Li B, Quinlan MP,
Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach
MA, et al: A chromatin-mediated reversible drug-tolerant state in
cancer cell subpopulations. Cell. 141:69–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hodi FS, O'Day SJ, McDermott DF, Weber RW,
Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel
JC, et al: Improved survival with ipilimumab in patients with
metastatic melanoma. N Engl J Med. 363:711–723. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Robert C, Long GV, Brady B, Dutriaux C,
Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C,
Kalinka-Warzocha E, et al: Nivolumab in previously untreated
melanoma without BRAF mutation. N Engl J Med. 372:320–330. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Robert C, Schachter J, Long GV, Arance A,
Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al:
KEYNOTE-006 investigators: Pembrolizumab versus Ipilimumab in
advanced melanoma. N Engl J Med. 372:2521–2532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boni A, Cogdill AP, Dang P, Udayakumar D,
Njauw CN, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher
DE, et al: Selective BRAFV600E inhibition enhances T-cell
recognition of melanoma without affecting lymphocyte function.
Cancer Res. 70:5213–5219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Frederick DT, Piris A, Cogdill AP, Cooper
ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et
al: BRAF inhibition is associated with enhanced melanoma antigen
expression and a more favorable tumor microenvironment in patients
with metastatic melanoma. Clin Cancer Res. 19:1225–1231. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wilmott JS, Long GV, Howle JR, Haydu LE,
Sharma RN, Thompson JF, Kefford RF, Hersey P and Scolyer RA:
Selective BRAF inhibitors induce marked T-cell infiltration into
human metastatic melanoma. Clin Cancer Res. 18:1386–1394. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
von Euw EM, Barrio MM, Furman D, Bianchini
M, Levy EM, Yee C, Li Y, Wainstok R and Mordoh J: Monocyte-derived
dendritic cells loaded with a mixture of apoptotic/necrotic
melanoma cells efficiently cross-present gp100 and MART-1 antigens
to specific CD8+ T lymphocytes. J Transl Med. 5:192007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barrio MM, De Motta PT, Kaplan J, von Euw
EM, Bravo AI, Chacón RD and Mordoh J: A phase I study of an
allogeneic cell vaccine (VACCIMEL) with GM-CSF in melanoma
patients. J Immunother. 29:444–454. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Barrio MM, Abes R, Colombo M, Pizzurro G,
Boix C, Roberti MP, Gélizé E, Rodriguez-Zubieta M, Mordoh J and
Teillaud JL: Human macrophages and dendritic cells can equally
present MART-1 antigen to CD8+ T cells after phagocytosis of
gamma-irradiated melanoma cells. PLoS One. 7:e403112012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Debacq-Chainiaux F, Erusalimsky JD,
Campisi J and Toussaint O: Protocols to detect
senescence-associated beta-galactosidase (SA-βgal) activity, a
biomarker of senescent cells in culture and in vivo. Nat Protoc.
4:1798–1806. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yee C, Thompson JA, Byrd D, Riddell SR,
Roche P, Celis E and Greenberg PD: Adoptive T cell therapy using
antigen-specific CD8+ T cell clones for the treatment of patients
with metastatic melanoma: In vivo persistence, migration, and
antitumor effect of transferred T cells. Proc Natl Acad Sci USA.
99:16168–16173. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boiko AD, Razorenova OV, van De Rijn M,
Swetter SM, Johnson DL, Ly DP, Butler PD, Yang GP, Joshua B, Kaplan
MJ, et al: Human melanoma-initiating cells express neural crest
nerve growth factor receptor CD271. Nature. 466:133–137. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schatton T, Murphy GF, Frank NY, Yamaura
K, Waaga-Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM,
Weishaupt C, et al: Identification of cells initiating human
melanomas. Nature. 451:345–349. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Frank NY, Margaryan A, Huang Y, Schatton
T, Waaga-Gasser AM, Gasser M, Sayegh MH, Sadee W and Frank MH:
ABCB5-mediated doxorubicin transport and chemoresistance in human
malignant melanoma. Cancer Res. 65:4320–4333. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Haferkamp S, Borst A, Adam C, Becker TM,
Motschenbacher S, Windhövel S, Hufnagel AL, Houben R and
Meierjohann S: Vemurafenib induces senescence features in melanoma
cells. J Invest Dermatol. 133:1601–1609. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Z, Jiang K, Zhu X, Lin G, Song F, Zhao
Y, Piao Y, Liu J, Cheng W, Bi X, et al: Encorafenib (LGX818), a
potent BRAF inhibitor, induces senescence accompanied by autophagy
in BRAFV600E melanoma cells. Cancer Lett. 370:332–344. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Michaloglou C, Vredeveld LC, Soengas MS,
Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi
WJ and Peeper DS: BRAFE600-associated senescence-like cell cycle
arrest of human naevi. Nature. 436:720–724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aris M, Zubieta MR, Colombo M, Arriaga JM,
Bianchini M, Alperovich M, Bravo AI, Barrio MM and Mordoh J:
MART-1- and gp100-expressing and -non-expressing melanoma cells are
equally proliferative in tumors and clonogenic in vitro. J Invest
Dermatol. 132:365–374. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Samatar AA and Poulikakos PI: Targeting
RAS-ERK signalling in cancer: Promises and challenges. Nat Rev Drug
Discov. 13:928–942. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Holderfield M, Deuker MM, McCormick F and
McMahon M: Targeting RAF kinases for cancer therapy: BRAF-mutated
melanoma and beyond. Nat Rev Cancer. 14:455–467. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Redmer T, Welte Y, Behrens D, Fichtner I,
Przybilla D, Wruck W, Yaspo ML, Lehrach H, Schäfer R and
Regenbrecht CR: The nerve growth factor receptor CD271 is crucial
to maintain tumorigenicity and stem-like properties of melanoma
cells. PLoS One. 9:e925962014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cheli Y, Bonnazi VF, Jacquel A, Allegra M,
De Donatis GM, Bahadoran P, Bertolotto C and Ballotti R: CD271 is
an imperfect marker for melanoma initiating cells. Oncotarget.
5:5272–5283. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Menon D Ravindran, Das S, Krepler C,
Vultur A, Rinner B, Schauer S, Kashofer K, Wagner K, Zhang G, Rad E
Bonyadi, et al: A stress-induced early innate response causes
multidrug tolerance in melanoma. Oncogene. 34:4448–4459. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sharma BK, Manglik V, O'Connell M,
Weeraratna A, McCarron EC, Broussard JN, Divito KA,
Simbulan-Rosenthal CM, Rosenthal DS and Zapas JL: Clonal dominance
of CD133+ subset population as risk factor in tumor progression and
disease recurrence of human cutaneous melanoma. Int J Oncol.
41:1570–1576. 2012.PubMed/NCBI
|
|
43
|
Chartrain M, Riond J, Stennevin A,
Vandenberghe I, Gomes B, Lamant L, Meyer N, Gairin JE, Guilbaud N
and Annereau JP: Melanoma chemotherapy leads to the selection of
ABCB5-expressing cells. PLoS One. 7:e367622012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Singh SK, Hawkins C, Clarke ID, Squire JA,
Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Singh SK, Clarke ID, Terasaki M, Bonn VE,
Hawkins C, Squire J and Dirks PB: Identification of a cancer stem
cell in human brain tumors. Cancer Res. 63:5821–5828.
2003.PubMed/NCBI
|
|
46
|
Ren F, Sheng WQ and Du X: CD133: A cancer
stem cells marker, is used in colorectal cancers. World J
Gastroenterol. 19:2603–2611. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Quintana E, Shackleton M, Sabel MS, Fullen
DR, Johnson TM and Morrison SJ: Efficient tumour formation by
single human melanoma cells. Nature. 456:593–598. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Velarde MC, Flynn JM, Day NU, Melov S and
Campisi J: Mitochondrial oxidative stress caused by Sod2 deficiency
promotes cellular senescence and aging phenotypes in the skin.
Aging (Albany NY). 4:3–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Achuthan S, Santhoshkumar TR, Prabhakar J,
Nair SA and Pillai MR: Drug-induced senescence generates
chemoresistant stemlike cells with low reactive oxygen species. J
Biol Chem. 286:37813–37829. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Leikam C, Hufnagel AL, Otto C, Murphy DJ,
Mühling B, Kneitz S, Nanda I, Schmid M, Wagner TU, Haferkamp S, et
al: In vitro evidence for senescent multinucleated melanocytes as a
source for tumor-initiating cells. Cell Death Dis. 6:e17112015.
View Article : Google Scholar : PubMed/NCBI
|