Introduction
Gastrointestinal stromal tumors (GISTs) are the most
common mesenchymal tumors and account for 18% of all sarcomas
(1).
GISTs develop from a small subset of interstitial
cells named Cajal cells and may arise anywhere in the
gastrointestinal tract (60–70% in the stomach and 20–30% in the
small intestine) and more rarely (less than 5%) in the omentum or
mesentery (2). GISTs usually occur
in adults with a median age of 55–60 years. The annual incidence of
GISTs worldwide is estimated to be between 11 and 19.6 per million
inhabitants, corresponding to 500–600 new cases per year in France
(3,4).
During the past decade, GISTs have emerged as a
distinct group of gastrointestinal tumors with the discovery of the
oncogenic role of the tyrosine kinase receptor KIT (also called
stem cell factor receptor) whose expression is observed by
immunohistochemical staining in more than 90% of GISTs (5). In 75% of GISTs expressing the
proto-oncogene KIT, a gain-of-function mutation in the
tyrosine kinase domain of KIT leads to its constitutive activation
(6). Alternatively, somatic
mutations in platelet-derived growth factor receptor α
(PDGFRA), another tyrosine kinase receptor encoding gene can
drive the development of GISTs in 15% of cases (7,8). In
approximately 85% of pediatric GISTs and in a small subset of adult
GISTs (10–15%), KIT and PDGFRA mutations have not
been identified (9). Mutations of
BRAF have been reported in 3.5–13% of
KIT/PDGFRA wild-type tumors but the pathogenic
significance of such mutations still remains unknown (10–12).
The vast majority of GISTs are sporadic but genetic
predispositions have also been described. Thus, 7% of patients with
neurofibromatosis type I develop GISTs, mostly multiple GISTs
without KIT mutations. More rarely, germline mutations in
succinate dehydrogenase complex, subunit B (SDHB),
KIT or PDGFRA have been observed in familial forms of
GIST (13–16).
The prognosis of GISTs varies widely. Since GISTs
generally evolve without symptoms, more than 10% are diagnosed at
the metastatic state. Complete surgical resection is the current
standard of care in most localized GISTs. After resection, the
estimated 15-year recurrence-free survival is 59.9%. Older age, a
tumor size larger than 10 cm, a high mitotic count, non-gastric
localization, presence of tumor rupture and male gender are
independent adverse prognostic factors (17,18).
For localized GISTs, the risk of relapse can be evaluated using
Armed Forces Institute of Pathology (AFIP) or National Institutes
of Health (NIH) classifications that are based on localization,
tumor size, mitotic index and presence of rupture of the primary
tumor (19,20). These classifications are critical
for the management of adjuvant treatment in patients with GISTs and
are likely to be enhanced by incorporating the mutational status of
GISTs (21).
Indeed, the rapid evolution in understanding the
oncogenesis of GISTs leads to the use of effective targeted
therapies. Most GISTs with KIT or PDGFRA mutations
respond to imatinib, a multikinase inhibitor (22–25).
Better responses are observed in GISTs with KIT exon 11
mutations than in patients with KIT exon 9 mutations,
PDGFRA mutations or without mutations (26). Unfortunately, around half of the
patients who initially respond to imatinib develop resistance after
a long period of treatment. Resistance to imatinib has been linked
to secondary mutations involving mostly the same gene as the
primary driver mutation (27–32).
Alternative tyrosine kinase inhibitors that target KIT and
PDGFRA such as sunitinib, nilotinib, sorafenib, regorafenib
as well as other investigational inhibitors are currently being
evaluated to treat imatinib-resistant GISTs (33–36).
The KIT and PDGFRA mutational
spectrums have been well characterized in population-based studies
in France (4), Norway (37) and Switzerland (38). These studies have shown that 50–60%
of primary GISTs present mutations in KIT exon 11 (encoding
the transmembrane domain), 5–10% in KIT exon 9
(extracellular domain), 1–3% in KIT exon 13 (tyrosine kinase
domain 1), <1% in KIT exon 17 (tyrosine kinase domain 2),
2–5% in PDGFRA exon 12 (transmembrane domain) and 2–12% in
PDGFRA exon 18 (tyrosine kinase domain 2). Mutations in
KIT exon 11 are the most heterogeneous mutations observed in
GISTs with about 50% deletions, 34% substitutions, 6%
duplications/insertions and 11% complex mutations.
We provide here a prospective study of the molecular
characteristics of a series of 104 GISTs in hospital-based French
adult patients.
Materials and methods
Study design and patients
All GIST cases diagnosed between August 2005 and
October 2014 in the University Hospital of Besançon, France (n=104)
were prospectively identified through the Department of Pathology
and the Regional Molecular Genetics Centre of Besançon.
All patients with GIST during this period had
routinely benefited from a molecular diagnosis according to the
French National Public Cancer Program managed by the National
Institute of Cancer (Institut National du Cancer, INCa) (39). All specimens used in the present
study were primary tumors except for 4 specimens corresponding to
metastasis.
Ethics statement
All procedures followed were in accordance with the
ethical standards of the committee responsible for human
experimentation (institutional and national) and with the Helsinki
Declaration of 1964 and later versions. According to the French
legislation (Public Health Code modified by the law no. 2004–806,
August 9, 2004 and the Huriet-Serusclat act 88–1138, December 20,
1988) and as this study only involved data extracted from medical
records and stored histological specimens, no informed consent from
the patients was necessary. Data collected from the Department of
Pathology were strictly anonymous. The collection of specimens and
their use for research were approved by the Ethics Committee of the
University Hospital of Besançon.
Histopathological evaluation
The diagnosis of GIST was based on histological
examination and confirmed using KIT/CD117 (clone 104D2, dilution
1/300; Dako, Les Ulis, France) and DOG-1 (clone SP31, dilution
1/150; Thermo Fisher Scientific, Villebon-sur-Yvette, France)
immunostaining when appropriate. In each case, the largest diameter
of the tumor was measured. Mitotic index was evaluated on 12.5
mm2 of tumor and then converted to the number of
mitoses/5 mm2. For localized GISTs, the potential risk
of relapse was evaluated according to Miettinen criteria (20).
DNA extraction
Tumor genomic DNA was extracted from formalin-fixed
and paraffin-embedded (FFPE) or frozen tissues using QIAmp DNA Mini
kit (Qiagen, Courtabeuf, France) according to the manufacturer's
instructions. Prior to DNA extraction, separate hematoxylin and
eosin stained slides were reviewed by a pathologist and manually
microdissected when appropriate to ensure tumor content greater
than 20%. Depending on the size of the fixed tissue, between 3 and
8 FFPE tissue sections of 10 µm thickness were processed for DNA
extraction. DNA and tissue samples were collected by the Biobank
BB-0033-00024 ‘Tumorothèque Régionale de Franche-Comté (TRFC)’.
KIT and PDGFRA mutations analysis by
direct sequencing
A sequential strategy analysis was adopted for the
screening of KIT and PDGFRA mutations by Sanger
sequencing. The most frequent sites of mutations (exons 9 and 11 of
KIT and exon 18 of PDGFRA) were first analyzed. When
no mutation was detected in the former exons, exons 13 and 17 of
KIT and exon 12 of PDGFRA were subsequently
sequenced. Genomic sequences of KIT (ENST00000288135) and
PDGFRA (ENST00000257290) were obtained from Ensembl database
(www.ensembl.org). Specific primers were designed
using the online Primer-BLAST software (www.ncbi.nlm.nih.gov/tools/primer-blast/)
(40). Table I shows the details of the primer
sequences and their annealing temperatures. Targeted sequences were
amplified by PCR using the Qiagen Multiplex PCR kit (Qiagen). PCR
conditions were as follows: 94°C for 15 min, 40 cycles of 92°C for
1 min, specific annealing temperature for 30 sec, 72°C for 45 sec
and finally 7 min at 72°C. PCR products were purified using the gel
extraction kit NucleoSpin Gel and PCR Clean-up (Macherey-Nagel,
Hoerdt, France). Bidirectional sequencing reaction was performed
using the DTCS Quick Start kit (SCIEX, Les Ulis, France). Reactions
were run according to the following protocol: one cycle at 96°C for
1 min; 15 cycles at 96°C for 10 sec, 50°C for 5 sec, 60°C for 1 min
15 sec; 5 cycles of 96°C for 10 sec, 50°C for 5 sec, 60°C for 1 min
30 sec; 5 cycles of 96°C for 10 sec, 50°C for 5 sec and 60°C for 2
min. After purification with a NucleoSEQ kit (Macherey-Nagel),
samples were run and analyzed on a CEQ 8000 sequencer (SCIEX).
Finally, the sequences obtained were compared with the reference
sequence of KIT or PDGFRA using CEQ 8000 analysis
software. Our procedure included a systematic double review by two
independent biologists.
 | Table I.PCR primers used for sequencing of
KIT and PDGFRA. |
Table I.
PCR primers used for sequencing of
KIT and PDGFRA.
| Gene/exon | Primer sequences
(5′→3′) | Annealing
temperature (°C) | Product size
(bp) |
|---|
| KIT 9 | F:
ATGCTCTGCTTCTGTACTG | 56 | 234 |
|
| R:
GCCTAAACATCCCCTTAAATTGG |
|
|
| KIT 11 | F:
CTCTCCAGAGTGCTCTAATGAC | 56 | 219 |
|
| R:
AGCCCCTGTTTCATACTGACC |
|
|
| KIT 13 | F:
GCTTGACATCAGTTTGCCAG | 56 | 294 |
|
| R:
GAGAACAACAGTCTGGGTAA |
|
|
| KIT 17 | F:
TCTCCTCCAACCTAATAGTGTAT | 56 | 173 |
|
| R:
GCAGGACTGTCAAGCAGAGAAT |
|
|
| PDGFRA
12 | F:
AAGCTCTGGTGCACTGGGACTT | 65 | 251 |
|
| R:
ATTGTAAAGTTGTGTGCAAGGGA |
|
|
| PDGFRA
18 | F:
TACAGATGGCTTGATCCTGAGT | 60 | 212 |
|
| R:
AGTGTGGGAGGATGAGCCTG |
|
|
KRAS and BRAF mutational analysis
Furthermore, all KIT/PDGFRA wild-type
samples (n=10) were tested for Kirsten rat sarcoma (KRAS)
codons 12 and 13 and BRAF codon 600 using a SNaPshot assay
as previously described (41). The
sensitivity of the SNaPshot assay that we developed was previously
evaluated using plasmid dilutions and ranged between 1–5% of mutant
alleles (Magnin et al, 2011; supplemental Figs. S1-S7)
(41). In comparison, the Sanger
assay that we used had a slightly higher level of detection that
ranged between 5 and 10% of mutant alleles.
Statistical analysis
Mean values and frequencies were used for the
description of continuous and categorical variables, respectively.
The proportions were compared using the Chi-squared test (or
Fisher's exact test, if appropriate). All statistical tests were
two-sided, and P-values <0.05 were considered as
significant.
Results
Clinicopathological
characteristics
Overall, samples from 104 GISTs corresponding to 103
patients including 60 males and 43 females were available for the
present study. The main clinical and pathological characteristics
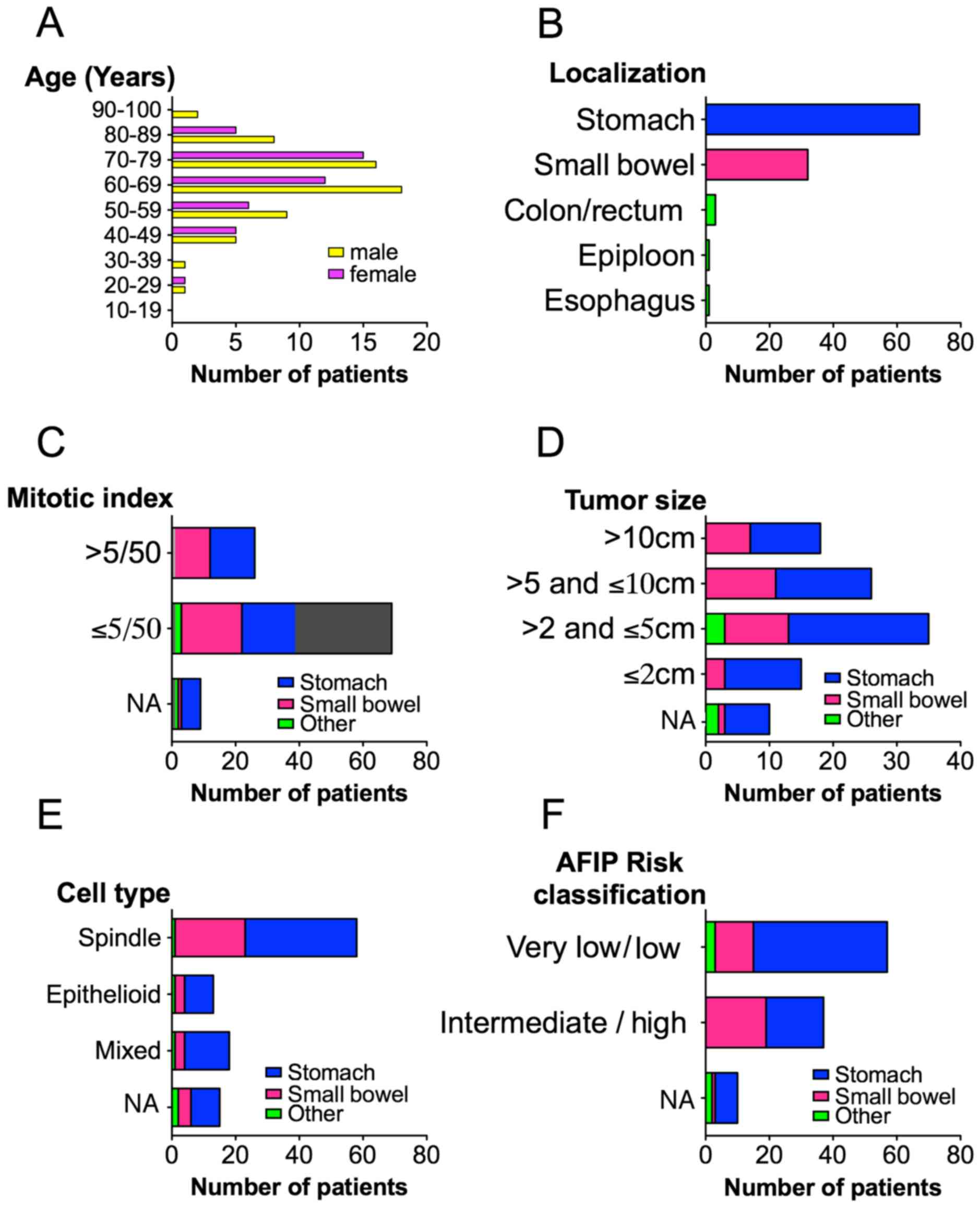
of the GISTs are shown in Fig. 1.
The mean age at the time of diagnosis was 66.2 years ranging from
29 to 92 years. Primary tumors were localized within the stomach
(66%), small bowel (29%), colon (2%), rectum (<1%), esophagus
(<1%) and epiploon (<1%). A majority of GISTs (65%) had a
tumor size between 2 and 10 cm and the mitotic index was <5/50
mm2 in the majority of cases (65%). Morphologically,
spindle cell type represented 56%, epithelioid 12.5% and mixed cell
type 17.5% of the GISTs. At the time of diagnosis, 11% of the GISTs
had synchronous metastasis. Thirty-six percent of localized GISTs
were intermediate to high risk according to AFIP
classification.
Mutational analysis
Characterization of the mutational status for
KIT and PDGFRA was performed in all GISTs. KIT
and PDGFRA mutations were detected in 90.4% cases, 71.2% in
KIT and 19.2% in PDGFRA while no mutation was found
in 9.6% specimens. A total of 43 different mutations were detected.
Among them 36 were localized in KIT exon 11 of which 13 were
not referenced in the COSMIC database (Table II).
| Table II.Novel KIT exon 11 mutations
observed in our series of GISTs. |
Table II.
Novel KIT exon 11 mutations
observed in our series of GISTs.
| Mutations |
|---|
| c.1649_1675del;
p.Lys550_Lys558delinsIle |
| c.1668_1692delinsA;
p.Trp557_Asn564del |
| c.1670_1720del;
p.Trp557_Thr574delinsSer |
| c.1676_1681del;
p.Val560_Glu561del |
| c.1676_1696del;
p.Val560_Asn566delinsAsp |
| c.1703_1726del;
p.Tyr568_Leu576delinsPhe |
| c.1708_1719dup;
p.Tyr570_Thr574dup |
| c.1709_1735dup;
p.Ile571_Asp579dup |
|
c.1717_1737dupinsCCA;
p.Asp572_Asp579dupinsPro |
| c.1718_1771dup;
p.Thr574_Phe590dupinsSer |
| c.1723_1758dup;
p.Gln575_Asn586dup |
| c.1726_1738delinsG;
p.Leu576_Asp579del |
| c.1711_1758dup;
p.Asp572_Asn586dup |
Altogether mutations in exon 9 and 11 of KIT
and exon 18 of PDGFRA accounted for 93% of all mutations.
Overall, the 9 most frequent mutations represented 55.2% of all
mutations (Table III).
| Table III.The 9 most frequent KIT and
PDGFRA mutations in our series of GISTs. |
Table III.
The 9 most frequent KIT and
PDGFRA mutations in our series of GISTs.
| Mutations | Percentage |
|---|
| 1. PDGFRA ex
18 p.Asp842Val | 13.95 |
| 2. KIT ex 9
p.Ala502_Tyr503dup | 9.60 |
| 3. KIT ex 11
p.Val560Asp | 7.69 |
| 4. KIT ex 11
p.Trp557Arg | 5.76 |
| 5. KIT ex 11
p.Leu576Pro | 4.80 |
| 6. KIT ex 11
p.Trp557_Lys558del | 4.80 |
| 7. KIT ex 11
p.Val559Asp | 2.88 |
| 8. KIT ex 11
p.Trp557Gly | 2.88 |
| 9. PDGFRA ex
18 p.Ile843_Asp846del | 2.88 |
In KIT exon 9, the classical duplication
(p.Ala502_Tyr503dup) was the only mutation identified.
As expected, KIT exon 11 mutations were more
heterogeneous. The most frequent types of KIT exon 11
mutations were substitutions in 44.4% cases followed by deletions
in 33.3% cases, complex mutations including insertions in 14.3%
cases and tandem duplications in 7.9% cases. The detailed frequency
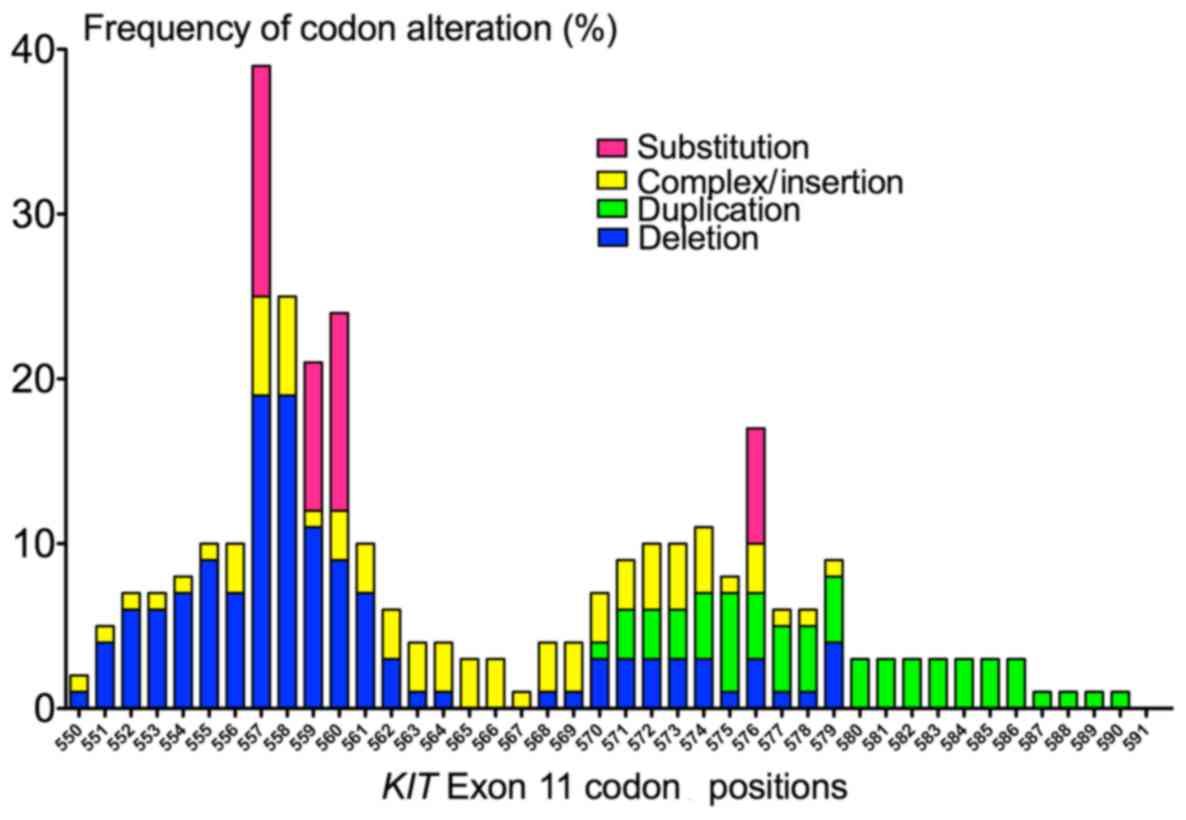
of codon alterations is shown in Fig.
2. KIT exon 11 deletions were predominantly clustered in
the 5′-end of exon 11. The most frequently mutated codons of
KIT exon 11 were 557 (in 39.6% of KIT exon 11
mutants), 558 and 560 (25.4% both). The most common deletion
p.Trp557_Lys558del was found in 5 cases (8% of KIT exon 11
mutants). By contrast, all tandem duplications (n=5) occurred in
the 3′-end of exon 11. The length of the duplications varied from 3
to 51 bp, mostly involving codons 573–579.
No mutation was found in KIT exon 17 and only
one mutation was found in KIT exon 13 (p.Lys642Glu).
Regarding PDGFRA, all mutations observed in
exon 18 corresponded to the classical p.Asp842Val, except for 2
deletions (p.Asp842_His845del and p.Ile843_Asp846del). Only one
mutation was found in exon 12 (p.Val561Asp). Of note, a patient was
diagnosed with double synchronous primary GISTs localized in the
stomach. Both tumors had the same histological characteristics but,
interestingly, they harbored 2 different mutations in PDGFRA
(p.Asp842Val and p.Val561Asp).
Distribution of patients in our series according to
the mutated exon was compared with that of patients from different
geographical origins included in population-based studies and
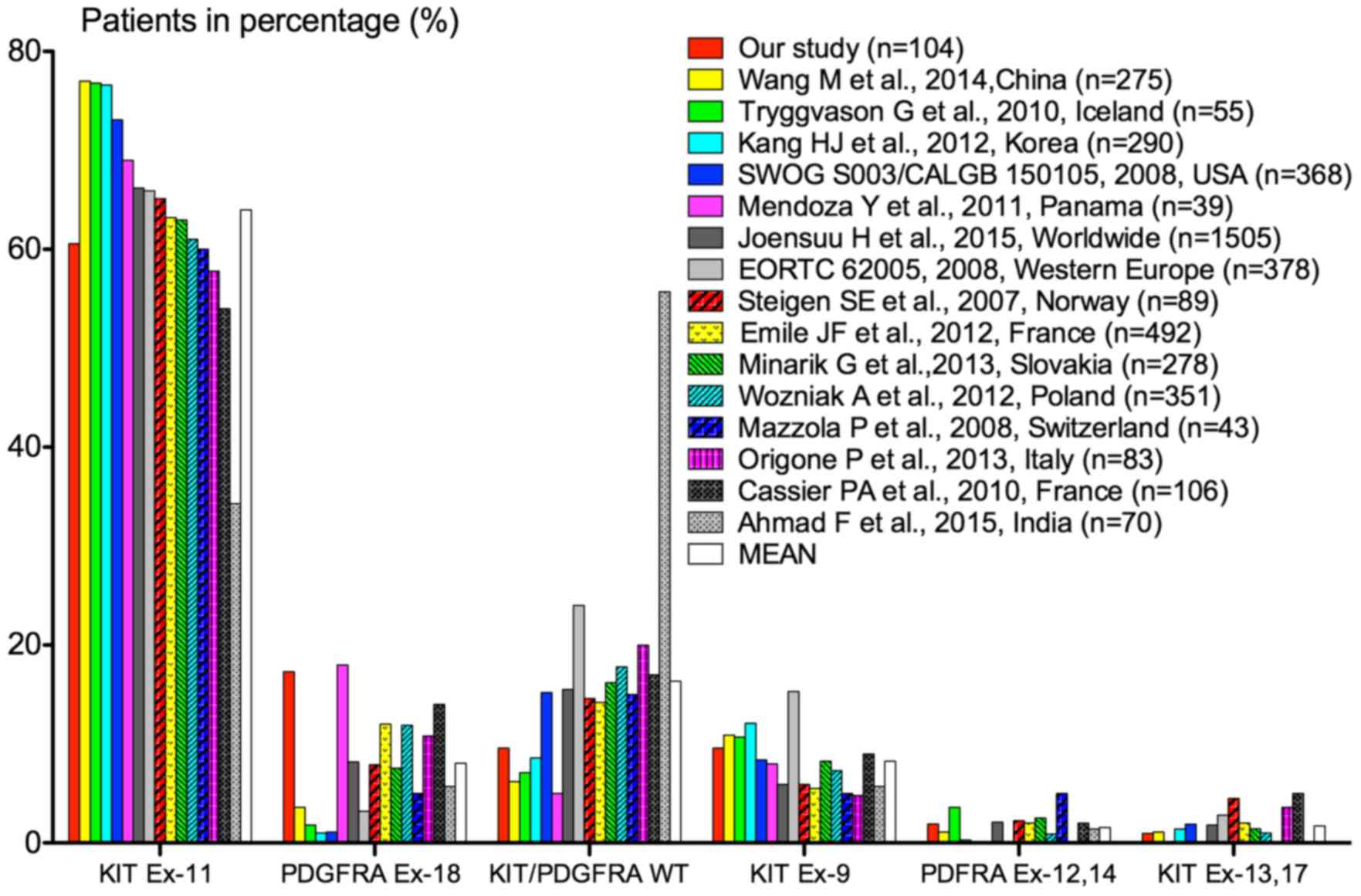
clinical trials (Fig. 3) (4,21,37,38,42–52).
It appeared that the distribution of mutations greatly varied
according to the population studied. In comparison with other
studies, more PDGFRA exon 18 mutations and less
KIT/PDGFRA wild-type GISTs were found in the present
study. For KIT exons 9, 11, 13 and 17 and for PDGFRA
exon 12, our results were however in the same range.
In addition, all KIT/PDGFRA wild-type
tumors (n=10) were tested for the presence of BRAF codon 600
and KRAS codon 12 and 13 mutations. No mutation was
detected. Of note, one patient with wild-type GIST has been
diagnosed with type I neurofibromatosis.
Association of tumor genotype with
clinicopathological characteristics
Detailed distribution of patients according to
mutations and clinicopathological characteristics is shown in
Table IV.
| Table IV.Distribution of patients according to
mutations and clinicopathological characteristics. |
Table IV.
Distribution of patients according to
mutations and clinicopathological characteristics.
|
| KIT-mutated
tumors |
|
|---|
|
|
|
|
|---|
|
| Exon 11 |
|
|
|---|
|
|
|
|
|
|---|
|
| All patients | All
KIT/PDGFRA-mutated tumors n(%) |
KIT/PDGFRA wild-type tumors
n(%) | All n(%) | Exon 9 n(%) | All n(%) | Deletion n(%) | Substitution
n(%) | Duplication
n(%) | Complex mutations
n(%) | Exon 13 n(%) | All PDGFRA-
mutated tumors n(%) |
|---|
| Gender |
|
Male | 60 (57.7) | 52 (55.3) | 8 (80) | 39 (52.7) | 6 (60) | 32 (50.8) | 12 (57.1) | 13 (46.4) | 3 (60) | 4 (44.4) | 1 (100) | 13 (65) |
|
Female | 44 (42.3) | 42 (44.7) | 2 (20) | 35 (47.3) | 4 (40) | 31 (49.2) | 9 (42.9) | 15 (53.6) | 2 (40) | 5 (55.6) | 0 (0) | 7 (35) |
| Age (years) |
|
Median | 66.37 | 66.4 | 65.8 | 66.6 | 66.6 | 66.3 | 63.6 | 69.5 | 64 | 63.9 | 63 | 68.85 |
|
Range | 29–92 | 29–92 | 27–80 | 39.8 | 39–88 | 29–88 | 41–84 | 29–88 | 39–82 | 56–76 | 44–80 | 46–92 |
| Primary tumor
site |
|
Stomach | 67 (64.4) | 61 (64.9) | 6 (60) | 44 (55.4) | 0 (0) | 41 (65.1) | 16 (76.2) | 16 (57.1) | 4 (80) | 5 (55.6) | 0 (0) | 20 (100) |
| Small
bowel | 32 (30.8) | 29 (30.9) | 3 (30) | 29 (39.2) | 10 (100) | 18 (28.6) | 3 (14.3) | 10 (35.7) | 1 (20) | 4 (44.4) | 1 (100) | 0 (0) |
|
Other | 5 (4.8) | 4 (4.3) | 1 (10) | 4 (5.4) | 0 (0) | 4 (6.3) | 2 (9.5) | 2 (7.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Synchronous
metastases |
|
Localized tumor | 93 (89.4) | 85 (90.4) | 8 (80) | 65 (87.8) | 8 (80) | 56 (88.9) | 19 (90.5) | 26 (92.9) | 3 (60) | 8 (88.9) | 1 (100) | 20 (100) |
|
Metastatic | 11 (10.6) | 9 (9.6) | 2 (20) | 9 (12.2) | 2 (20) | 7 (11.1) | 2 (9.5) | 2 (7.1) | 2 (40) | 1 (11.1) | 0 (0) | 0 (0) |
| Cell type |
| Spindle
cell | 58 (55.8) | 52 (55.3) | 6 (60) | 47 (63.5) | 7 (70) | 39 (61.9) | 14 (66.7) | 18 (64.3) | 3 (60) | 4 (44.4) | 1 (100) | 5 (25) |
|
Epithelioid | 13 (12.5) | 13 (13.8) | 0 (0) | 8 (10.8) | 1 (10) | 7 (11.1) | 3 (14.3) | 0 (0) | 1 (20) | 3 (33.3) | 0 (0) | 5 (25) |
|
Mixed | 18 (17.3) | 15 (16) | 3 (30) | 8 (10.8) | 1 (10) | 7 (11.1) | 0 (0) | 7 (25) | 0 (0) | 0 (0) | 0 (0) | 7 (35) |
| NA | 15 (14.4) | 14 (14.9) | 1 (10) | 11 (14.9) | 1 (10) | 10 (15.9) | 4 (19) | 3 (10.7) | 1 (20) | 2 (22.2) | 0 (0) | 3 (15) |
| Mitotic index |
|
≤5/50 | 68 (65.4) | 62 (66) | 6 (60) | 47 (63.5) | 4 (40) | 42 (66.7) | 10 (47.6) | 24 (85.7) | 3 (60) | 5 (55.6) | 1 (100) | 15 (75) |
|
>5/50 | 27 (26) | 24 (25.5) | 3 (30) | 22 (29.7) | 5 (50) | 17 (27) | 9 (42.9) | 3 (10.7) | 2 (40) | 3 (33.3) | 0 (0) | 2 (10) |
| NA | 9 (8.7) | 8 (8.5) | 1 (10) | 5 (6.8) | 1 (10) | 4 (6.3) | 2 (9.5) | 1 (3.6) | 0 (0) | 1 (11.1) | 0 (0) | 3 (15) |
| Tumor size
(cm) |
| ≤2 | 15 (14.4) | 13 (13.8) | 2 (20) | 10 (13.5) | 1 (10) | 9 (14.3) | 4 (19) | 4 (14.3) | 0 (0) | 1 (11.1) | 0 (0) | 3 (15) |
| >2
to ≤5 | 35 (33.7) | 31 (33) | 4 (40) | 27 (36.5) | 2 (20) | 24 (38.1) | 4 (19) | 14 (50) | 2 (40) | 4 (44.4) | 1 (100) | 4 (20) |
|
5–10 | 26 (25) | 26 (27.7) | 0 (0) | 17 (23) | 5 (50) | 12 (19) | 4 (19) | 5 (17.9) | 1 (20) | 2 (22.2) | 0 (0) | 9 (45) |
|
>10 | 18 (17.3) | 15 (16) | 3 (30) | 14 (18.9) | 2 (20) | 12 (19) | 6 (28.6) | 4 (14.3) | 1 (20) | 1 (11.1) | 0 (0) | 1 (5) |
| NA | 10 (9.6) | 9 (9.6) | 1 (10) | 6 (8.1) | 0 (0) | 6 (9.5) | 3 (14.3) | 1 (3.6) | 1 (20) | 1 (11.1) | 0 (0) | 3 (15) |
| Risk |
|
None | 19 (18.3) | 17 (18.1) | 2 (20) | 13 (17.6) | 1 (10) | 12 (19) | 4 (19) | 5 (17.9) | 0 (0) | 3 (33.3) | 0 (0) | 4 (20) |
| Very low | 21 (20.2) | 18 (19.1) | 3 (30) | 15 (20.3) | 0 (0) | 15 (23.8) | 3 (14.3) | 9 (32.1) | 2 (40) | 1 (11.1) | 0 (0) | 3 (15) |
|
Low | 16 (15.4) | 15 (16) | 1 (10) | 9 (12.2) | 2 (20) | 6 (9.5) | 0 (0) | 5 (17.9) | 0 (0) | 1 (11.1) | 1 (100) | 6 (30) |
|
Intermediate | 14 (13.5) | 13 (13.8) | 1 (10) | 12 (16.2) | 1 (10) | 11 (17.5) | 4 (19) | 5 (17.9) | 1 (20) | 1 (11.1) | 0 (0) | 1 (5) |
|
High | 23 (22.1) | 21 (22.3) | 2 (20) | 19 (25.7) | 5 (50) | 14 (22.2) | 7 (33.3) | 3 (10.7) | 2 (40) | 2 (22.2) | 0 (0) | 2 (10) |
|
n.a. | 11 (10.6) | 10 (10.6) | 1 (10) | 6 (8.1) | 1 (10) | 5 (7.9) | 3 (14.3) | 1 (3.6) | 0 (0) | 1 (11.1) | 0 (0) | 4 (20) |
| Other
malignancies |
|
Synchronous | 13 (12.5) | 11 (11.7) | 2 (20) | 6 (8.1) | 0 (0) | 5 (7.9) | 2 (9.5) | 3 (10.7) | 0 (0) | 0 (0) | 1 (100) | 5 (25) |
| Before
GIST | 2 (1.9) | 2 (2.1) | 0 (0) | 2 (2.7) | 1 (10) | 1 (1.6) | 0 (0) | 1 (3.6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| After
GIST | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
It can be noted that GISTs with PDGFRA exon
18 mutations (n=18) were associated with primary gastric
localization (18/18), tumors with KIT exon 9 mutations
(n=10) were exclusively localized in the small bowel (10/10) while
tumors with KIT exon 11 were respectively localized in the
stomach (41/63), small bowel (18/63) and other sites (4/63)
(p<0.001).
No other significant association was observed
between KIT and PDGFRA mutations and the
clinicopathological features of the GISTs. Furthermore, spindle
cell type and mitotic index >5/50 mm2 was less
frequent in tumors harboring PDGFRA exon 18 mutation than in
the whole series. Despite a tumor size greater than other GISTs,
the estimated risk of relapse was more frequently very low/low in
this subset of tumors (Table
IV).
Advanced GISTs were diagnosed in 11 cases in our
series and no association with a specific mutation was found.
Finally, 15 patients (14%) also presented another malignancy of
which 13 were synchronous. Of note, 25% of patients with
PDGFRA mutations had other malignancies.
Molecular pattern of KIT exon 11
substitutions
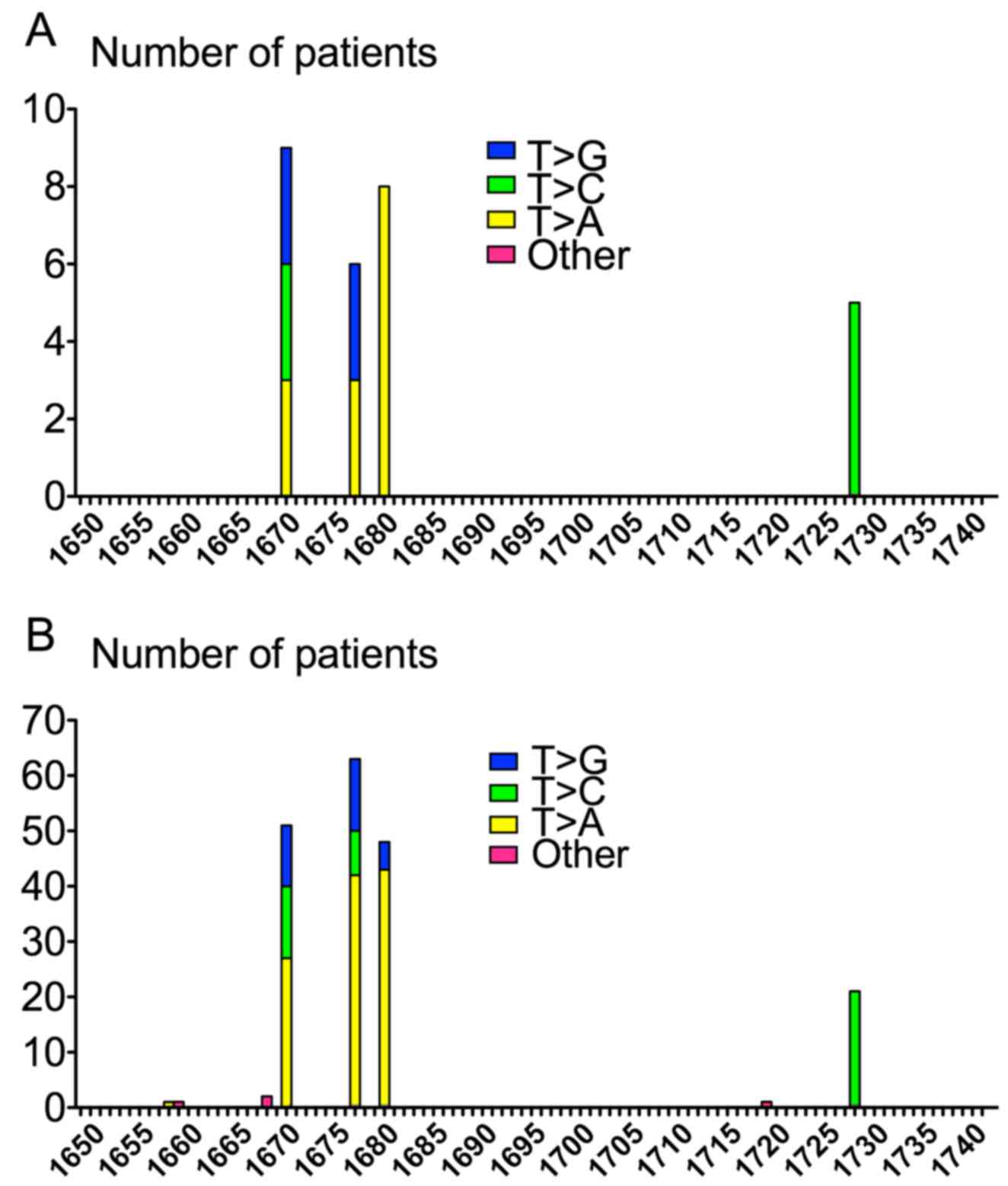
Single substitutions in KIT exon 11 occurred,
in decreased frequency, at codons 557 (n=9; p.Trp557Gly,
p.Trp557Arg), 560 (n=8; p.Val560Asp), 559 (n=6; p.Val559Gly,
p.Val559Asp) and 576 (n=5; p.Leu576Pro). Strikingly, all KIT
exon 11 substitutions (n=28) shared the same T>N molecular
pattern. These substitutions occurred at nucleotides 1669, 1676,
1679 and 1727. Half of these point mutations involved T>A
transversion, 8 T>C transition and 6 T>G transversion
(Table V and Fig. 4). No significant association was
found between KIT exon 11 substitution and
clinicopathological characteristics. However, patients with such
mutation tended to be older than other patients of the present
series (median age 69.5 vs. 66.37 in the whole cohort). Of note, in
comparison with all patients, no epithelioid tumor was observed in
patients with KIT exon 11 substitution, mitotic index was
≤5/50 mm2 in 86 vs. 65% and tumor size was ≤5 cm in 66%
in this subset of patients vs. 54% in all cases (Table IV).
| Table V.Detailed clinicopathological features
of GISTs with KIT exon 11 substitutions. |
Table V.
Detailed clinicopathological features
of GISTs with KIT exon 11 substitutions.
| No. | Age (years) | Gender | Metastases | Morphology | Tumor site | Mitotic index | Risk | Other
malignancies | Nucleotide
change | Amino acid
change |
|---|
| 1 | 66 | F | 0 | Spindle cell | Stomach | ≤5 | 1 | Prostate
cancer | 1669T>A | Trp557Arg |
| 2 | 72 | M | 0 | Mixed | Stomach | ≤5 | 1 | NA | 1669T>A | Trp557Arg |
| 3 | 85 | M | 0 | Mixed | Stomach | ≤5 | 1 | 0 | 1669T>A | Trp557Arg |
| 4 | 43 | F | Synchronous | Spindle cell | Small bowel | ≤5 | 2 | 0 | 1669T>C | Trp557Arg |
| 5 | 62 | F | 0 | Spindle cell | Stomach | ≤5 | 1 | 0 | 1669T>C | Trp557Arg |
| 6 | 85 | F | 0 | Mixed | Small bowel | >5 | 3 | 0 | 1669T>C | Trp557Arg |
| 7 | 63 | M | 0 | Spindle cell | Stomach | ≤5 | 2 | 0 | 1669T>G | Trp557Gly |
| 8 | 79 | F | 0 | NA | Small bowel | >5 | 3 | 0 | 1669T>G | Trp557Gly |
| 9 | 81 | F | 0 | Spindle cell | Stomach | >5 | 2 | 0 | 1669T>G | Trp557Gly |
| 10 | 70 | F | Synchronous | Mixed | Stomach | ≤5 | 2 | 0 | 1676T>A | Val559Asp |
| 11 | 76 | M | 0 | Spindle cell | Small bowel | ≤5 | 1 | Gastric
adenocarcinoma | 1676T>A | Val559Asp |
| 12 | 79 | F | 0 | Spindle cell | Stomach | ≤5 | 1 | NA | 1676T>A | Val559Asp |
| 13 | 29 | F | 0 | Spindle cell | Stomach | ≤5 | 1 | 0 | 1676T>G | Val559Gly |
| 14 | 77 | F | 0 | Spindle cell | Small bowel | ≤5 | 2 | 0 | 1676T>G | Val559Gly |
| 15 | 83 | M | 0 | Spindle cell | Small bowel | ≤5 | 1 | 0 | 1676T>G | Val559Gly |
| 16 | 49 | F | 0 | Spindle cell | Stomach | ≤5 | 1 | Ovarian
adenocarcinoma | 1679T>A | Val560Asp |
| 17 | 58 | F | 0 | Spindle cell | Stomach | ≤5 | 1 | 0 | 1679T>A | Val560Asp |
| 18 | 63 | M | 0 | Spindle cell | Stomach | ≤5 | 1 | 0 | 1679T>A | Val560Asp |
| 19 | 69 | M | 0 | Mixed | Stomach | ≤5 | 1 | 0 | 1679T>A | Val560Asp |
| 20 | 77 | M | 0 | NA | Small bowel | ≤5 | 1 | 0 | 1679T>A | Val560Asp |
| 21 | 78 | F | 0 | Mixed | Stomach | ≤5 | 1 | 0 | 1679T>A | Val560Asp |
| 22 | 79 | M | 0 | NA | Colon | n.a. |
| NA | 1679T>A | Val560Asp |
| 23 | 88 | M | 0 | Spindle cell | Small bowel | ≤5 | 1 | 0 | 1679T>A | Val560Asp |
| 24 | 60 | M | 0 | Mixed | Rectum | ≤5 | 1 | Rectal
adenocarcinoma | 1727T>C | Leu576Pro |
| 25 | 63 | M | 0 | Spindle cell | Small bowel | ≤5 | 3 | 0 | 1727T>C | Leu576Pro |
| 26 | 66 | M | 0 | Spindle cell | Small bowel | ≤5 | 1 | 0 | 1727T>C | Leu576Pro |
| 27 | 72 | F | 0 | Spindle cell | Stomach | ≤5 | 1 | 0 | 1727T>C | Leu576Pro |
| 28 | 74 | F | 0 | Spindle cell | Stomach | ≤5 | 1 | NA | 1727T>C | Leu576Pro |
Discussion
Here, we provide a prospective study of
clinicopathological and molecular characteristics of 104 GISTs from
a Northeastern French population.
All patients with GISTs diagnosed between August
2005 and October 2014 at the University Hospital of Besançon
benefited from a routine molecular diagnosis as recommended by the
French National Cancer Institute (INCa). Thus, our study reflects
the distribution of clinicopathological and molecular features of
GISTs in real life with the accuracy and the management of quality
from a clinical laboratory.
The detailed molecular characterization of GISTs has
become of great prognosis and therapeutic value in the past few
years.
Indeed, treatment with the tyrosine kinase inhibitor
imatinib led to significant improvement of survival of patients
with KIT and PDGFRA mutated GISTs. Imatinib has been
approved as the first line treatment of patients with advanced
GISTs and substantially increased survival of these patients (10–20
vs. 51–57 months median survival) (53–55).
Subsequently, it has been shown that the position of KIT or
PDGFRA mutations influences the response to imatinib. Thus,
GISTs with KIT exon 11 mutant genotype are
imatinib-responsive whereas mutations in PDGFRA exon 18
(mostly Asp842Val) are associated with resistance to imatinib.
GISTs with a mutation in KIT exon 9 (mostly Ala-Tyr502-503
duplications) are imatinib-responsive but doubling the dose of
imatinib (400 mg twice daily) increases the progression-free
survival significantly (56).
Treatment of KIT/PDGFRA wild-type tumors is not
currently standardized and the administration of imatinib in these
patients remains controversial.
Additionally, tumor genotype has been shown to have
an independent prognostic relevance in patients with GISTs.
KIT exon 9 duplications and KIT exon 11 deletions are
known to be associated with aggressive tumor behavior and poor
prognosis whereas patients with PDGFRA Asp842Val mutant
GISTs usually have a favorable outcome (57,58).
Recently, Joensuu et al have shown that patients with
PDGFRA mutations and those with KIT exon 11
duplication or deletion of one codon have favorable relapse-free
survival (RFS) with surgery alone (47). Thus, KIT and PDGFRA
mutation analysis provides important information to estimate the
risk of recurrence in patients with localized GISTs and deserve to
be investigated to select candidates for adjuvant therapy.
The distribution of somatic mutations in GISTs has
previously been characterized in large population-based studies and
varies widely from one region of the globe to another but the
reasons for these variations still remain unknown.
Thus, KIT and PDGFRA mutations are
found respectively in 70 and 10% of cases in the USA (59), 70.7 and 20% in France (4), 67.9 and 1% in China (60), and 72.4 and 6.5% in South Africa
(61). Notably, the variation of
the genotype mainly involves the proportion of
PDGFRA-mutated tumors. Such variations may be explained by
several factors. First, it may be the result of variable diagnosis
delays. PDGFRA-mutated tumors are known to evolve more
slowly than KIT-mutated tumors. Consequently, the series
that comprised a higher proportion of advanced GISTs had less
PDGFRA-mutated tumors. Secondly, the technical procedures
used to assess the mutational status of GISTs can influence the
proportion of mutations in these different series. The Sanger
sequencing probably allows a more extensive detection of rare
variants compared with targeted methods. Thus, it may be assumed
that the implementation of next-generation sequencing in clinical
laboratories will change the current molecular epidemiology of
GISTs. Finally, PDGFRA mutations may vary with the ethnic
origins of patients with GISTs as shown in non-small cell lung
cancer in which a higher proportion of somatic epidermal growth
factor receptor (EGFR) mutations has been observed in the
Asian population. In our series the distribution of KIT and
PDGFRA mutations was quite similar to those of the MolecGIST
study that reviewed tumor samples from 596 patients from all over
France during a 24-month period. Notably, we observed a higher
proportion of KIT exon 11 substitutions in the present study
compared with MolecGIST (44.4 vs. 34.1%). A focused analysis of
these substitutions has displayed a common molecular pattern
consisting in all cases of a T>N point mutation located at
codons 557, 559, 560 and 576. Analysis of the molecular pattern of
KIT exon 11 substitutions in the MolecGIST cohort showed the
same distribution with 97.9% mutations affecting a thymine at 4
different loci. Surprisingly, a recent Indian study of 70 GISTs
revealed a different distribution with only 40% thymine
substitutions among all KIT exon 11 point mutations
(49). Thus, we suggest that
environment and/or genetic background may affect the distribution
of point mutations in GISTs.
Environmental risks of cancer usually include
exposure to carcinogens. Characteristic mutations in KIT
exon 11 in GISTs may be mutational signatures that are linked to
specific mutagens. Despite an increasing number of studies, little
is known about the natural history of GISTs. Notably, the role of
non-genetic risk factors, such as exposure to carcinogens, is not
currently known.
Genetics risks include constitutional genomic
instability and DNA repair defects. Such alterations have already
been suggested to play a role in the oncogenesis of GISTs. Thus,
methylation of mutL homolog 1 and MGMT have been observed in
60 and 49% of GISTs respectively and single-nucleotide
polymorphisms (SNPs) in two other DNA repair genes, RAD23B
and ERCC2, were associated with KIT exon 11 mutations
(59,62).
Despite the advent of targeted therapies, the
prognosis of GISTs, especially in advanced stages, is still poor
and a better comprehension of genetic and environmental risk
factors may allow the development of preventive and/or screening
strategies for GISTs.
In conclusion, this study confirms existing data
and enriches the knowledge of the genotypes of GISTs which is
essential for therapeutic innovation. By describing 13 novel
mutations in KIT, our data contribute to widen the spectrum
of known mutations in GISTs and to confirm the most frequently
altered regions underlying GIST development. It also confirms that
KRAS exon 2 and BRAF V600 mutations are very scarce
since no mutation was found in the wild-type GISTs in our
series.
Finally, this study highlights the importance of
taking into consideration the genetic and environmental risk
factors favoring GIST development since the current scientific
knowledge on this topic is still poor.
Acknowledgements
We would like to thank Alice Brunier, Evelyne
Chezy, Laurence Madoz and Lise Rognon for the achievement of the
technical procedures used in this study. We also acknowledge the
Institut National du Cancer (INCa) for the financial support of the
molecular diagnosis of GISTs in France. Finally, we thank the
pathologists who participated in this study: Séverine Valmary
Degano, Isabelle Bedgedjian, Franck Vitte, Yannick Jeffredo and
Alain Petitjean.
Glossary
Abbreviations
Abbreviations:
|
AFIP
|
Armed Forces Institute of
Pathology
|
|
COSMIC
|
Catalogue of Somatic Mutations in
Cancer
|
|
EGFR
|
epidermal growth factor receptor
|
|
FFPE
|
formalin-fixed paraffin-embedded
|
|
GISTs
|
gastrointestinal stromal tumors
|
|
KRAS
|
Kirsten rat sarcoma
|
|
MLH1
|
mutL homolog 1
|
|
DNA
|
deoxyribonucleic acid
|
|
NIH
|
National Institutes of Health
|
|
PDGFRA
|
platelet-derived growth factor
receptor α
|
|
SDHB
|
succinate dehydrogenase complex,
subunit B
|
|
SNP
|
single-nucleotide polymorphism
|
References
|
1
|
Corless CL, Barnett CM and Heinrich MC:
Gastrointestinal stromal tumours: Origin and molecular oncology.
Nat Rev Cancer. 11:865–878. 2011.PubMed/NCBI
|
|
2
|
Joensuu H, Hohenberger P and Corless CL:
Gastrointestinal stromal tumour. Lancet. 382:973–983. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Monges G, Bisot-Locard S, Blay JY, Bouvier
AM, Urbieta M, Coindre JM and Scoazec JY: The estimated incidence
of gastrointestinal stromal tumors in France. Results of PROGIST
study conducted among pathologists. Bull Cancer. 97:E16–E22.
2010.PubMed/NCBI
|
|
4
|
Emile JF, Brahimi S, Coindre JM, Bringuier
PP, Monges G, Samb P, Doucet L, Hostein I, Landi B, Buisine MP, et
al: Frequencies of KIT and PDGFRA mutations in the MolecGIST
prospective population-based study differ from those of advanced
GISTs. Med Oncol. 29:1765–1772. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sarlomo-Rikala M, Kovatich AJ,
Barusevicius A and Miettinen M: CD117: A sensitive marker for
gastrointestinal stromal tumors that is more specific than CD34.
Mod Pathol. 11:728–734. 1998.PubMed/NCBI
|
|
6
|
Hirota S, Isozaki K, Moriyama Y, Hashimoto
K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M,
et al: Gain-of-function mutations of c-kit in human
gastrointestinal stromal tumors. Science. 279:577–580. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heinrich MC, Corless CL, Duensing A,
McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A,
Town A, et al: PDGFRA activating mutations in gastrointestinal
stromal tumors. Science. 299:708–710. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hirota S, Ohashi A, Nishida T, Isozaki K,
Kinoshita K, Shinomura Y and Kitamura Y: Gain-of-function mutations
of platelet-derived growth factor receptor α gene in
gastrointestinal stromal tumors. Gastroenterology. 125:660–667.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nannini M, Biasco G, Astolfi A and
Pantaleo MA: An overview on molecular biology of KIT/PDGFRA wild
type (WT) gastrointestinal stromal tumours (GIST). J Med Genet.
50:653–661. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Agaimy A, Terracciano LM, Dirnhofer S,
Tornillo L, Foerster A, Hartmann A and Bihl MP: V600E BRAF
mutations are alternative early molecular events in a subset of
KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin
Pathol. 62:613–616. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Agaram NP, Wong GC, Guo T, Maki RG, Singer
S, Dematteo RP, Besmer P and Antonescu CR: Novel V600E BRAF
mutations in imatinib-naive and imatinib-resistant gastrointestinal
stromal tumors. Genes Chromosomes Cancer. 47:853–859. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hostein I, Faur N, Primois C, Boury F,
Denard J, Emile JF, Bringuier PP, Scoazec JY and Coindre JM: BRAF
mutation status in gastrointestinal stromal tumors. Am J Clin
Pathol. 133:141–148. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miettinen M, Killian JK, Wang Z-F, Lasota
J, Lau C, Jones L, Walker R, Pineda M, Zhu YJ, Kim SY, et al:
Immunohistochemical loss of succinate dehydrogenase subunit A
(SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA
germline mutation. Am J Surg Pathol. 37:234–240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jones DH, Caracciolo JT, Hodul PJ,
Strosberg JR, Coppola D and Bui MM: Familial gastrointestinal
stromal tumor syndrome: Report of 2 cases with KIT exon 11
mutation. Cancer Control. 22:102–108. 2015.PubMed/NCBI
|
|
15
|
Janeway KA, Kim SY, Lodish M, Nosé V,
Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, et al: NIH
Pediatric and Wild-Type GIST Clinic: Defects in succinate
dehydrogenase in gastrointestinal stromal tumors lacking KIT and
PDGFRA mutations. Proc Natl Acad Sci USA. 108:314–318. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pantaleo MA, Astolfi A, Urbini M, Nannini
M, Paterini P, Indio V, Saponara M, Formica S, Ceccarelli C,
Casadio R, et al: GIST Study Group: Analysis of all subunits, SDHA,
SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in
KIT/PDGFRA wild-type GIST. Eur J Hum Genet. 22:32–39. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Joensuu H, Vehtari A, Riihimäki J, Nishida
T, Steigen SE, Brabec P, Plank L, Nilsson B, Cirilli C, Braconi C,
et al: Risk of recurrence of gastrointestinal stromal tumour after
surgery: An analysis of pooled population-based cohorts. Lancet
Oncol. 13:265–274. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bischof DA, Kim Y, Dodson R, Jimenez MC,
Behman R, Cocieru A, Fisher SB, Groeschl RT, MH III Squires,
Maithel SK, et al: Conditional disease-free survival after surgical
resection of gastrointestinal stromal tumors: A multi-institutional
analysis of 502 patients. JAMA Surg. 150:299–306. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fletcher CDM, Berman JJ, Corless C,
Gorstein F, Lasota J, Longley BJ, Miettinen M, O'Leary TJ, Remotti
H, Rubin BP, et al: Diagnosis of gastrointestinal stromal tumors: A
consensus approach. Hum Pathol. 33:459–465. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miettinen M and Lasota J: Gastrointestinal
stromal tumors: Pathology and prognosis at different sites. Semin
Diagn Pathol. 23:70–83. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wozniak A, Rutkowski P, Piskorz A,
Ciwoniuk M, Osuch C, Bylina E, Sygut J, Chosia M, Rys J, Urbanczyk
K, et al: Polish Clinical GIST Registry: Prognostic value of
KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST):
Polish Clinical GIST Registry experience. Ann Oncol. 23:353–360.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mol CD, Dougan DR, Schneider TR, Skene RJ,
Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC and Wilson KP:
Structural basis for the autoinhibition and STI-571 inhibition of
c-Kit tyrosine kinase. J Biol Chem. 279:31655–31663. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tuveson DA, Willis NA, Jacks T, Griffin
JD, Singer S, Fletcher CD, Fletcher JA and Demetri GD: STI571
inactivation of the gastrointestinal stromal tumor c-KIT
oncoprotein: Biological and clinical implications. Oncogene.
20:5054–5058. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Heinrich MC, Griffith DJ, Druker BJ, Wait
CL, Ott KA and Zigler AJ: Inhibition of c-kit receptor tyrosine
kinase activity by STI 571, a selective tyrosine kinase inhibitor.
Blood. 96:925–932. 2000.PubMed/NCBI
|
|
25
|
Demetri GD, von Mehren M, Blanke CD, van
den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA,
Singer S, Janicek M, et al: Efficacy and safety of imatinib
mesylate in advanced gastrointestinal stromal tumors. N Engl J Med.
347:472–480. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Heinrich MC, Corless CL, Demetri GD,
Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, van den
Abbeele AD, Druker BJ, et al: Kinase mutations and imatinib
response in patients with metastatic gastrointestinal stromal
tumor. J Clin Oncol. 21:4342–4349. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Heinrich MC, Maki RG, Corless CL,
Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou WB,
Fletcher JA, et al: Primary and secondary kinase genotypes
correlate with the biological and clinical activity of sunitinib in
imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol.
26:5352–5359. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen LL, Trent JC, Wu EF, Fuller GN,
Ramdas L, Zhang W, Raymond AK, Prieto VG, Oyedeji CO, Hunt KK, et
al: A missense mutation in KIT kinase domain 1 correlates with
imatinib resistance in gastrointestinal stromal tumors. Cancer Res.
64:5913–5919. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Heinrich MC, Corless CL, Blanke CD,
Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von Mehren M,
Fletcher CD, Sandau K, et al: Molecular correlates of imatinib
resistance in gastrointestinal stromal tumors. J Clin Oncol.
24:4764–4774. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wardelmann E, Thomas N, Merkelbach-Bruse
S, Pauls K, Speidel N, Büttner R, Bihl H, Leutner CC, Heinicke T
and Hohenberger P: Acquired resistance to imatinib in
gastrointestinal stromal tumours caused by multiple KIT mutations.
Lancet Oncol. 6:249–251. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liegl B, Kepten I, Le C, Zhu M, Demetri
GD, Heinrich MC, Fletcher CD, Corless CL and Fletcher JA:
Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J
Pathol. 216:64–74. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nishida T, Kanda T, Nishitani A, Takahashi
T, Nakajima K, Ishikawa T and Hirota S: Secondary mutations in the
kinase domain of the KIT gene are predominant in imatinib-resistant
gastrointestinal stromal tumor. Cancer Sci. 99:799–804. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Demetri GD, van Oosterom AT, Garrett CR,
Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich
MC, Morgan JA, et al: Efficacy and safety of sunitinib in patients
with advanced gastrointestinal stromal tumour after failure of
imatinib: A randomised controlled trial. Lancet. 368:1329–1338.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
George S, Wang Q, Heinrich MC, Corless CL,
Zhu M, Butrynski JE, Morgan JA, Wagner AJ, Choy E, Tap WD, et al:
Efficacy and safety of regorafenib in patients with metastatic
and/or unresectable GI stromal tumor after failure of imatinib and
sunitinib: A multicenter phase II trial. J Clin Oncol.
30:2401–2407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guo T, Agaram NP, Wong GC, Hom G, D'Adamo
D, Maki RG, Schwartz GK, Veach D, Clarkson BD, Singer S, et al:
Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper
mutation in gastrointestinal stromal tumor. Clin Cancer Res.
13:4874–4881. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cullinane C, Natoli A, Hui Y, Conus N,
Jackson S, Brüggen J, Manley PW and McArthur GA: Preclinical
evaluation of nilotinib efficacy in an imatinib-resistant
KIT-driven tumor model. Mol Cancer Ther. 9:1461–1468. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Steigen SE, Eide TJ, Wasag B, Lasota J and
Miettinen M: Mutations in gastrointestinal stromal tumors - a
population-based study from Northern Norway. APMIS. 115:289–298.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mazzola P, Spitale A, Banfi S,
Mazzucchelli L, Frattini M and Bordoni A: Epidemiology and
molecular biology of gastrointestinal stromal tumors (GISTs): A
population-based study in the South of Switzerland, 1999–2005.
Histol Histopathol. 23:1379–1386. 2008.PubMed/NCBI
|
|
39
|
Nowak F, Soria JC and Calvo F: Tumour
molecular profiling for deciding therapy-the French initiative. Nat
Rev Clin Oncol. 9:479–486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ye J, Coulouris G, Zaretskaya I,
Cutcutache I, Rozen S and Madden TL: Primer-BLAST: A tool to design
target-specific primers for polymerase chain reaction. BMC
Bioinformatics. 13:1342012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Magnin S, Viel E, Baraquin A,
Valmary-Degano S, Kantelip B, Pretet JL, Mougin C, Bigand M,
Girardo B, Borg C, et al: A multiplex SNaPshot assay as a rapid
method for detecting KRAS and BRAF mutations in advanced colorectal
cancers. J Mol Diagn. 13:485–492. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cassier PA, Ducimetière F, Lurkin A,
Ranchère-Vince D, Scoazec JY, Bringuier PP, Decouvelaere AV, Méeus
P, Cellier D, Blay JY, et al: A prospective epidemiological study
of new incident GISTs during two consecutive years in Rhône A lpes
region: Incidence and molecular distribution of GIST in a European
region. Br J Cancer. 103:165–170. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mendoza Y, Singh C, Mewa J Castillo,
Fonseca E, Smith R and Pascale JM: Beginning of personalized
medicine in Panama: Molecular and pathological characteristics of
gastrointestinal stromal tumors from archival paraffin-embedded
tissue. Oncol Lett. 2:941–947. 2011.PubMed/NCBI
|
|
44
|
Heinrich MC, Owzar K, Corless CL, Hollis
D, Borden EC, Fletcher CD, Ryan CW, von Mehren M, Blanke CD, Rankin
C, et al: Correlation of kinase genotype and clinical outcome in
the North American Intergroup Phase III Trial of imatinib mesylate
for treatment of advanced gastrointestinal stromal tumor: CALGB
150105 Study by Cancer and Leukemia Group B and Southwest Oncology
Group. J Clin Oncol. 26:5360–5367. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sciot R, Debiec-Rychter M, Daugaard S,
Fisher C, Collin F, van Glabbeke M, Verweij J, Blay JY; Hogendoorn
PCEORTC Soft Tissue and Bone Sarcoma Group, ; et al: Australasian
Trials Group: Distribution and prognostic value of histopathologic
data and immunohistochemical markers in gastrointestinal stromal
tumours (GISTs): An analysis of the EORTC phase III trial of
treatment of metastatic GISTs with imatinib mesylate. Eur J Cancer.
44:1855–1860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kang HJ, Ryu MH, Kim KM, Park YS, Choi J,
Ryoo BY, Kim WH, Im SA, Bang YJ, Park SH, et al: Imatinib efficacy
by tumor genotype in Korean patients with advanced gastrointestinal
stromal tumors (GIST): The Korean GIST Study Group (KGSG) study.
Acta Oncol. 51:528–536. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Joensuu H, Rutkowski P, Nishida T, Steigen
SE, Brabec P, Plank L, Nilsson B, Braconi C, Bordoni A, Magnusson
MK, et al: KIT and PDGFRA mutations and the risk of GI stromal
tumor recurrence. J Clin Oncol. 33:634–642. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Origone P, Gargiulo S, Mastracci L,
Ballestrero A, Battistuzzi L, Casella C, Comandini D, Cusano R, Dei
Tos AP, Fiocca R, et al: Liguria GIST Unit: Molecular
characterization of an Italian series of sporadic GISTs. Gastric
Cancer. 16:596–601. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ahmad F, Lad P, Bhatia S and Das BR:
Molecular spectrum of c-KIT and PDGFRA gene mutations in gastro
intestinal stromal tumor: Determination of frequency, distribution
pattern and identification of novel mutations in Indian patients.
Med Oncol. 32:4242015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang M, Xu J, Zhao W, Tu L, Qiu W, Wang C,
Shen Y, Liu Q and Cao H: Prognostic value of mutational
characteristics in gastrointestinal stromal tumors: A single-center
experience in 275 cases. Med Oncol. 31:8192014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Minárik G, Plank L, Lasabová Z, Szemes T,
Burjanivová T, Szépe P, Buzalková V, Porubský D and Sufliarsky J:
Spectrum of mutations in gastrointestinal stromal tumor patients -
a population-based study from Slovakia. APMIS. 121:539–548. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tryggvason G, Hilmarsdottir B, Gunnarsson
GH, Jónsson JJ, Jónasson JG and Magnússon MK: Tyrosine kinase
mutations in gastrointestinal stromal tumors in a nation-wide study
in Iceland. APMIS. 118:648–656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Blanke CD, Rankin C, Demetri GD, Ryan CW,
von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki
RG, et al: Phase III randomized, intergroup trial assessing
imatinib mesylate at two dose levels in patients with unresectable
or metastatic gastrointestinal stromal tumors expressing the kit
receptor tyrosine kinase: S0033. J Clin Oncol. 26:626–632. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Blanke CD, Demetri GD, von Mehren M,
Heinrich MC, Eisenberg B, Fletcher JA, Corless CL, Fletcher CD,
Roberts PJ, Heinz D, et al: Long-term results from a randomized
phase II trial of standard- versus higher-dose imatinib mesylate
for patients with unresectable or metastatic gastrointestinal
stromal tumors expressing KIT. J Clin Oncol. 26:620–625. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Joensuu H, Fletcher C, Dimitrijevic S,
Silberman S, Roberts P and Demetri G: Management of malignant
gastrointestinal stromal tumours. Lancet Oncol. 3:655–664. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gastrointestinal Stromal Tumor
Meta-Analysis Group (MetaGIST), . Comparison of two doses of
imatinib for the treatment of unresectable or metastatic
gastrointestinal stromal tumors: A meta-analysis of 1,640 patients.
J Clin Oncol. 28:1247–1253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wardelmann E, Losen I, Hans V, Neidt I,
Speidel N, Bierhoff E, Heinicke T, Pietsch T, Büttner R and
Merkelbach-Bruse S: Deletion of Trp-557 and Lys-558 in the
juxtamembrane domain of the c-kit protooncogene is associated with
metastatic behavior of gastrointestinal stromal tumors. Int J
Cancer. 106:887–895. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Andersson J, Bümming P, Meis-Kindblom JM,
Sihto H, Nupponen N, Joensuu H, Odén A, Gustavsson B, Kindblom LG
and Nilsson B: Gastrointestinal stromal tumors with KIT exon 11
deletions are associated with poor prognosis. Gastroenterology.
130:1573–1581. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
O'Brien KM, Orlow I, Antonescu CR, Ballman
K, McCall L, DeMatteo R and Engel LS: Gastrointestinal stromal
tumors, somatic mutations and candidate genetic risk variants. PLoS
One. 8:e621192013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
He HY, Fang WG, Zhong HH, Li Y, Zheng J,
Du J, Heng WJ and Wu BQ: Status and clinical implication of c-kit
and PDGFRA mutations in 165 cases of gastrointestinal stromal tumor
(GIST). Zhonghua Bing Li Xue Za Zhi. 35:262–266. 2006.(In Chinese).
PubMed/NCBI
|
|
61
|
Baker G, Babb C, Schnugh D, Nayler S, Louw
M, Goedhals J, Bringuier PP, Blay JY and Willem P: Molecular
characterisation of gastrointestinal stromal tumours in a South
African population. Oncol Lett. 5:155–160. 2013.PubMed/NCBI
|
|
62
|
Saito K, Sakurai S, Sano T, Sakamoto K,
Asao T, Hosoya Y, Nakajima T and Kuwano H: Aberrant methylation
status of known methylation-sensitive CpG islands in
gastrointestinal stromal tumors without any correlation to the
state of c-kit and PDGFRA gene mutations and their malignancy.
Cancer Sci. 99:253–259. 2008. View Article : Google Scholar : PubMed/NCBI
|