Introduction
One of the most distinguishing characteristics
between normal and tumorigenic cells is altered glucose metabolism,
which is listed as one of the ten hallmarks of cancer (1). Under aerobic conditions, normal
differentiated cells extract energy from glucose chiefly through
oxidative phosphorylation, but tumor cells metabolize more glucose
to lactate. This phenomenon is termed the Warburg effect (aerobic
glycolysis) (2,3). The preference to utilize glycolysis
offers several advantages to cancer cells including adaptation in
hypoxic and acidic environments (lactate production), which help
cancer cells in invasion and apoptosis resistance (4–7).
Additionally, increased glycolysis in cancer tissues allows the
diversion of glycolytic intermediates into various biosynthetic
pathways that can synthesize macromolecules and organelles required
for the assembly of new cancer cells, which is important for tumor
cell proliferation (8,9). Until now, few studies on prostate
cancer energy metabolism have been reported.
The high rate of glycolysis commonly observed in
cancers is presumably ascribed to upregulation of key enzymes in
glycolysis including hexokinase 2 (HK2), 5
glyceraldehyde-3-phosphate dehydrogenase (GAPDH),
6-phosphofructo-1-kinase (PFK1) (10,11).
HK2, coding for the first rate-limiting enzyme of glycolysis, is
found to be overexpressed in some malignant tumors. As reported,
HK2 could be bound to the outer mitochondrial membrane (12,13).
At this location, it helps couple ATP formation in mitochondria to
the phosphorylation of glucose, thus conferring cancer cells with a
highly glycolytic phenotype and ample biosynthetic precursor
(14). In this study, we firstly
reported that HK2 was elevated in prostate cancer tissues compared
to prostatic hyperplasia tissues. Moreover, its expression
significantly increased in castration-resistant prostate cancer
(CRPC) compared to androgen-dependent prostate cancer (ADPC).
Further study revealed that HK2 regulated genes primarily
associated with glycometabolism and cell cycle progression in
prostate cancer. In addition, the results showed that HK2 was
directly regulated by EZH2/miR-181b axis at post-transcriptional
level. EZH2/miR-181b axis promoted aerobic glycolysis by
upregulating HK2 in prostate cancer.
Materials and methods
Cell culture and reagents
The human PCa cell line PC-3 was purchased from
KeyGene Biotech (Nanjing, China). Cells were cultured in RPMI-1640
supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and
100 µg/ml streptomycin at 37°C in a humidified atmosphere with 5%
CO2. Transfections of sh-EZH2, miR-181 mimics,
As-miR-181b, and negative control (NC) (GenePharma; Shanghai,
China) were performed with Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA).
Data mining and bioinformation
analysis
The GSE35988 and GSE21032 datasets were downloaded
from GEO database (http://www.ncbi.nlm.nih.gov/geo) and re-analyzed with
R (15,16). Pearson's correlation was used to
analyze the relationship between EZH2 and all identified genes with
Matlab software (P<0.05). GSEA (http://www.broadinstitute.org/gsea/) analysis was used
to identify pathway gene sets that are correlated with the HK2
expression profile. The gene sets were derived from the Molecular
Signatures Database (http://www.broadinstitute.org/gsea/msigdb/index.jsp).
Oligonucleotides and cell
transfection
The EZH2-shRNA and HK2-shRNA oligonucleotide were
cloned into vectors U6/GPF/Neo by GenePharma. A control shRNA
unrelated to any human sequence was used as the negative control
(NC). Oligonucleotides were chemically synthesized by GenePharma
according to the sequences of hsa-miR-181b mimic (miR-181b),
5′-AACAUUCAUUGCUGUCGGUGGGU-3′; hsa-miR-181b antisense
oligonucleotide (As-miR-181b), 5′-ACCCACCGACAGCAATGAATGTT-3′;
scrambled miRNA mimic, 5′-UUCUCCGAACGUGUCACGUTT-3′; scrambled
microRNA inhibitor, 5′-CAGUACUUUUGUGUAGUACAA-3′. Cells at 60–70%
confluence were transfected using Lipofectamine 2000 reagent
(Invitrogen). The oligonucleotides and plasmids formed transfection
complexes with Lipofectamine 2000 reagent. The transfection
complexes were added to PCa cells at different concentrations and
then incubated for 6–8 h before changing the medium. The lentiviral
vector containing EZH2-siRNA and NC sequence was generated by
GeneChem Inc.
Colony formation assay
Cells were plated onto dishes in RPMI-1640 culture.
After 48 h of EZH2-shRNA processing, the cells were washed thrice
with phosphate-buffered saline (PBS), and fresh broth was supplied.
After two weeks, the cells were fixed in 5 ml of 4%
paraformaldehyde for 15 min. Giemsa staining was performed for 20
min, and the cells were washed thrice with PBS. The clone number
was counted under a microscope.
Glycolysis stress test
The extracellular acidification rate (ECAR) was
measured using the Seahorse XF96 Analyzer Glycolysis which
calculates the net production and extrusion of protons into the
extracellular medium. As glycolysis occurs, the resulting
acidification of the medium surrounding the cells is measured
directly by the XF analyzer and reported as the ECAR. Initially,
cells are incubated in glycolysis stress test medium without
glucose. The ECAR refers to non-glycolytic acidification, which
includes CO2 evolution followed by its hydration to
carbonic acid and bicarbonate, as well as proton extrusion. The
first injection is a saturating concentration of glucose. Glucose
is taken up by the cells and catabolized to lactate, producing ATP
and protons, with a corresponding rapid increase in ECAR. This
glucose-induced response is reported as the rate of glycolysis
under basal conditions. The second injection is oligomycin. It
inhibits mitochondrial ATP production and thus shifts the energy
production to glycolysis, with the increase in ECAR revealing the
maximum glycolytic capacity. The final injection is 2-DG, a glucose
analog, which inhibits glycolysis through competitively binding to
glucose hexokinase. The resulting decrease in ECAR further confirms
that the ECAR produced in the experiment is due to glycolysis.
Quantitative RT-PCR
RNA was extracted from cells or tissues using TRIzol
(Invitrogen). miR-181b (qRT-PCR) reactions were performed using
Fermentas reverse transcription reagents and SYBR Green PCR Master
Mix (GenePharma) according to manufacturer's protocols. U6 was used
for normalisation. Relative gene expression was calculated by the
2−∆∆Ct method.
Western blot analysis
Western blot analysis was performed as previously
described (17). Immunoblot was
performed using appropriate primary antibodies: EZH2 (1:1000; Cell
Signaling Technology, Inc., Danvers, MA, USA), HK2 (1:1000; Abcam,
Cambridge, UK), and β-actin (1:1000; Santa Cruz Biotechnology;
Santa Cruz, CA, USA).
Luciferase reporter assay
An HK2 3′UTR-Luc reporter was created by ligating
the HK2 3′UTR PCR product into the XbaI site of the pGL3
control vector (Invitrogen). A mutant reporter was generated from
pGL3-WT-HK2 3′UTR-Luc by deleting the binding site for miR-181b
‘UGAAUGU’. A 1.9 kb fragment upstream of human pri-miR-181b
stem-loop was constructed into pGL3-basic plasmids (Invitrogen).
Luciferase activity was measured using the Dual-Luciferase Reporter
Assay System (Promega, Madison, WI, USA).
Xenograft tumor assay
BALB/c nude mice at 4 weeks of age were purchased
from the Animal Center of the Cancer Institute at the Chinese
Academy of Medical Science. All mice were divided randomly into
negative control group and EZH2 inhibition group. The guidelines
for animal welfare were approved by the Ethics Committee on Animal
Research of Southeast University. To test the effect of EZH2 on
prostate cancer growth and downstream pathway in vivo,
4×106 PC-3 cells infected with EZH2-siRNA lentivirus
were implanted into subcutaneous tissue of each nude mouse in EZH2
inhibition group. The tumor volumes were measured every 7 days by
vernier calipers.
Immunohistochemical staining (IHC) and
in situ hybridization (ISH)
IHC and ISH were performed as previously described
(18). IHC was performed using
appropriate primary antibodies according to the manufacturer's
instructions. Tissue sections were stained with
3′3-diaminobenzidine solution (DAB). ISH was performed using
antisense locked nucleic acid (LNA)-modified probes based on the
sequence of hsa-miR-181b (Boster, Wuhan, China). The hybridized
probes were also detected by incubating with DAB.
Statistical analysis
Data were obtained from at least three independent
experiments and presented as mean ± SD. The significance of
differences was calculated using one-way ANOVA for three-group
comparisons and t-tests for two-group comparisons with SPSS 13.0
software package. Pearson's correlation analysis was performed
using Matlab software. P<0.05 was considered to indicate a
statistically significant difference.
Results
EZH2 expression and function in
PCa
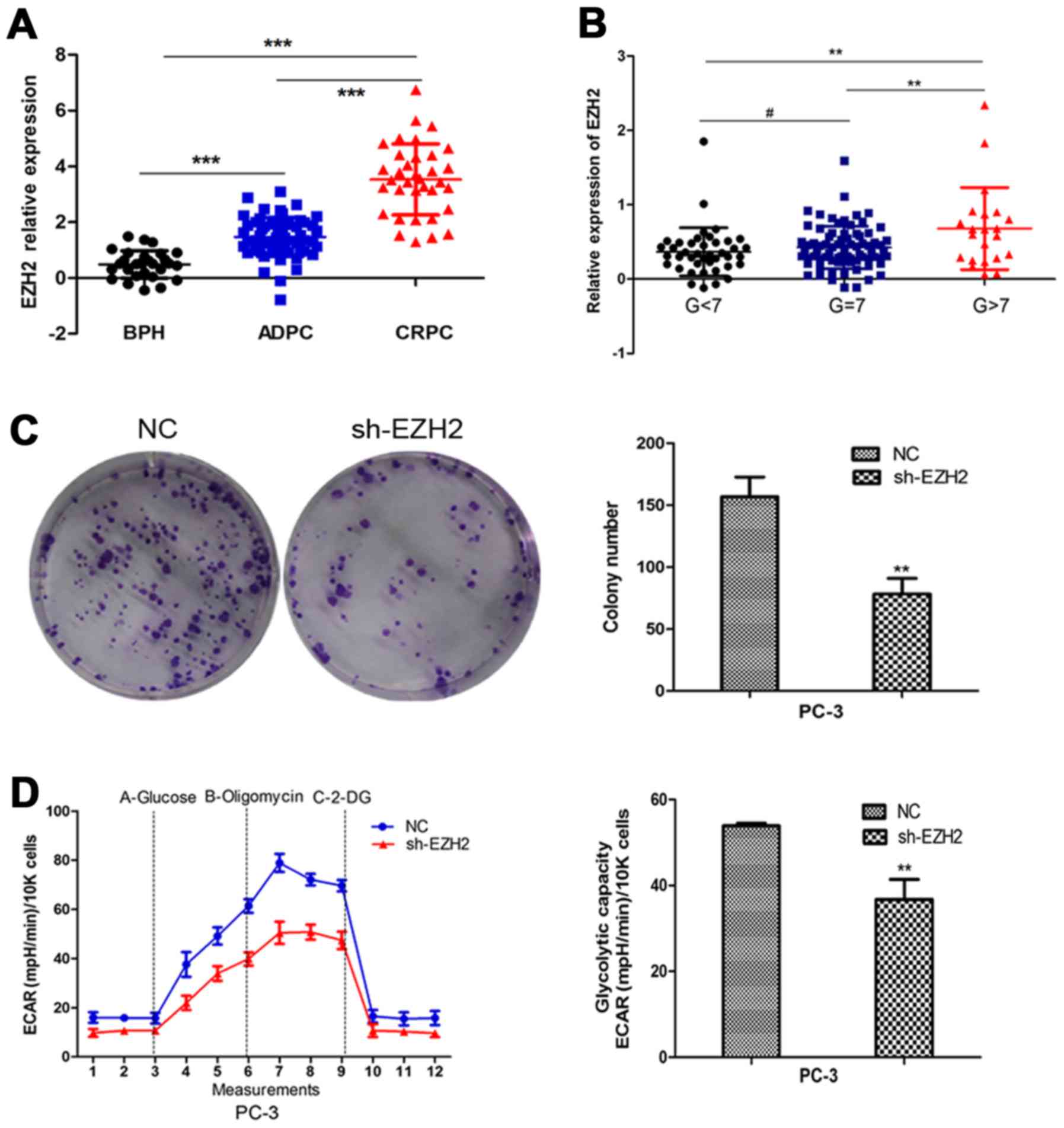
Firstly, we employed the GEO database to analyze the
expression of EZH2 in benign prostatic hyperplasia tissues and
prostate cancer tissues in different stages and different
pathological levels. The results indicated that EZH2 expression was
elevated in prostate cancer tissues compared to prostatic
hyperplasia tissues. Further, EZH2 expression significantly
increased in CRPC compared to ADPC (Fig. 1A). Besides, EZH2 expression was
higher in Gleason >7 prostate cancer tissues compared to Gleason
≤7 ones (Fig. 1B). To further
explore the effect of EZH2 on prostate cancer cells, EZH2 was
downregulated by sh-EZH2. Cell tablet assays revealed that EZH2
inhibition can significantly inhibit the number of cell colonies
(Fig. 1C). To assess the function
of EZH2 in cellular energy metabolism, we performed a glycolysis
stress test. Inhibition of EZH2 weakened the glycolytic abilities
of prostate cancer cells compared with negative control (Fig. 1D).
EZH2 correlates with
glycometabolism-related genes in PCa
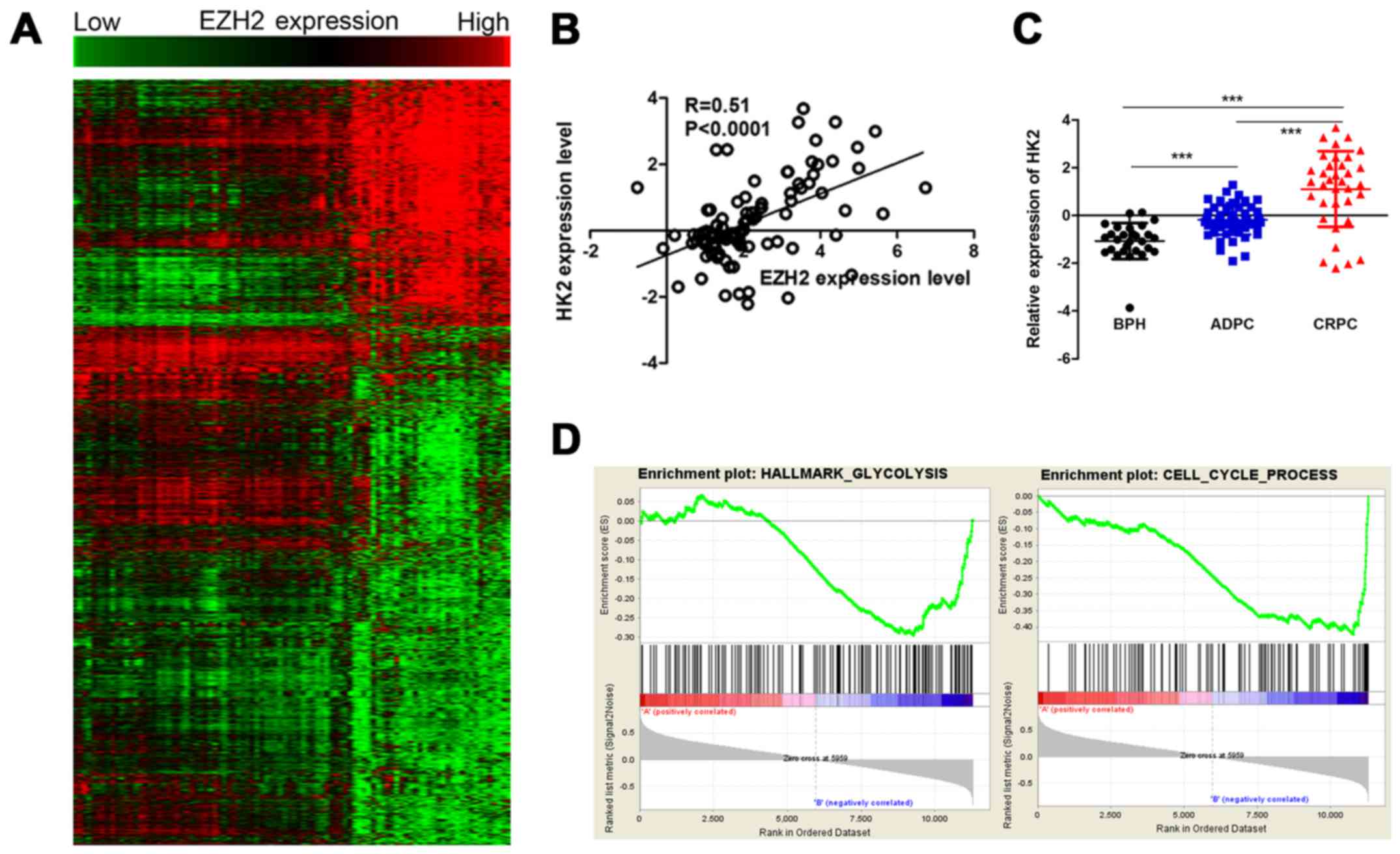
To evaluate the potential mechanism in aberrant
expression of EZH2 and cellular metabolism and growth, Pearson's
correlations were used to analysis the relationship between EZH2
and all identified genes. The EZH2 specific gene signature included
925 gene positively correlated with EZH2 expression, and 1,952 gene
negatively correlated with EZH2 expression (Fig. 2A). Among them, a set of
glycometabolism-related genes were positively correlated with EZH2
expression, including HK2, solute carrier family 2 member 1
(SLC2A1) and ribosomal protein S6 kinase B1 (RPS6KB1). HK2 was a
metabolic enzyme that executes the first step of aerobic glycolysis
(Fig. 2B). It was elevated in
prostate cancer tissues compared to prostatic hyperplasia tissues.
Moreover, its expression significantly increased in CRPC compared
to ADPC (Fig. 2C). Therefore, it
was chosen for further study. GSEA was used to evaluate the
pathways that were differentially expressed between patients with
high levels of HK2 expression and those with low levels of HK2
expression. The result revealed that genes correlated with HK2 were
primarily associated with glycometabolism and cell cycle
progression (Fig. 2D). Glycolysis
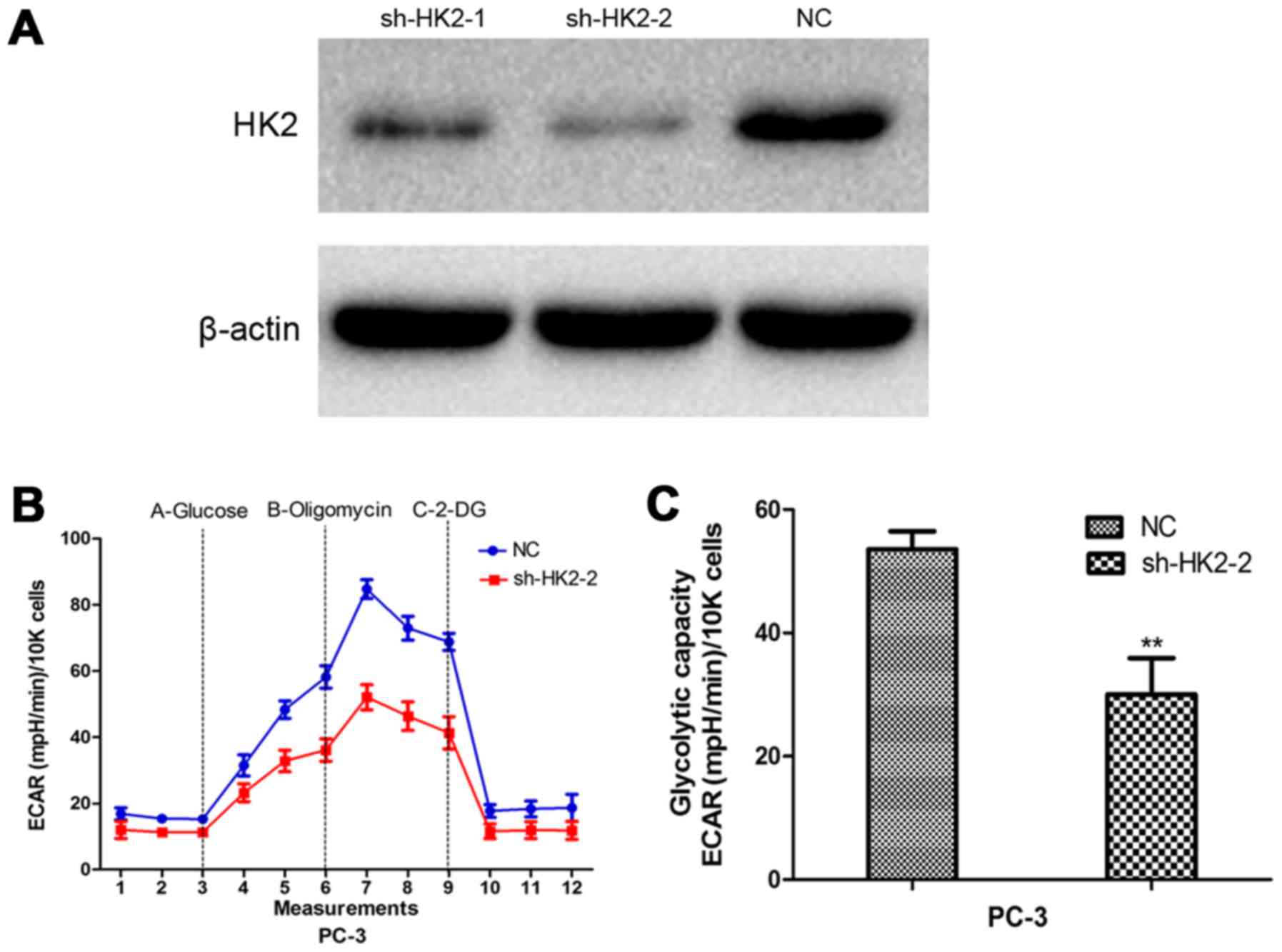
stress tests demonstrated sh-HK2 significantly inhibited the
glycolytic ability of prostate cancer cells (Fig. 3).
miR-181b is an important mediator
between EZH2 and HK2
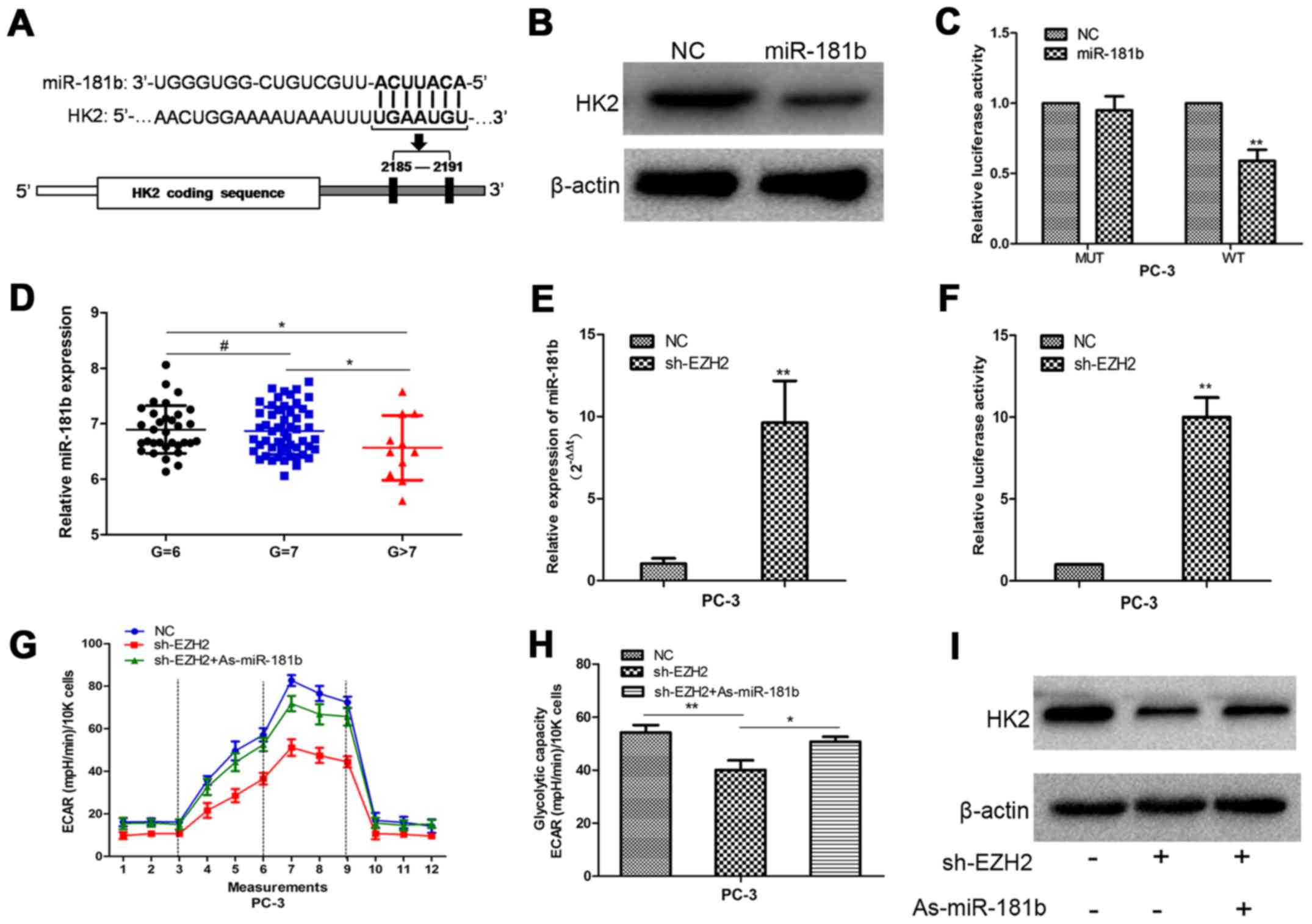
To detect the potential mechanism by which EZH2
regulates HK2, we performed bioinformatic analysis to identify HK2
as the potential target of miR-181b (Fig. 4A). Western blot analysis showed that
HK2 expression was decreased in prostate cancer cells with
upregulation of miR-181b (Fig. 4B).
To determine the direct interaction between miR-181b and its
binding site within HK2 mRNA, we created constructs containing
wild-type (pGL3-WT-HK2–3′UTR) and mutant (pGL3-MUT-HK2-3′UTR) HK2
3′UTRs. Luciferase reporter assays showed that over-expression of
miR-181b led to a marked decrease in luciferase activity of the
pGL3-WT-HK2-3′UTR plasmid in PC-3 cells without significant change
in luciferase activity of the pGL3-MUT- HK2-3′UTR plasmid (Fig. 4C). Our data provided strong evidence
that miR-181b directly regulated HK2 expression by binding to the
3′UTR of HK2 mRNA in prostate cancer cells.
Next, we found that the expression of the miR-181b
in the high pathological level group was lower than that in the low
pathological level group in prostate cancer database (Fig. 4D). Then, PC-3 cells were treated
with sh-EZH2. miR-181b expression was determined in scrambled and
sh-EZH2-treated cells by qRT-PCR. Compared with the scrambled
control cells, the sh-EZH2-treated cells showed higher miR-181b
expression (Fig. 4E). A 1.9 kb
fragment upstream of human pri-miR-181b stem-loop was constructed
and inserted into the luciferase reporter plasmid pGL3. The plasmid
was co-transfected with shEZH2 or NC into PC-3 cells. The depletion
of EZH2 increased the activity of luciferase construction (Fig. 4F). These results indicated EZH2
regulated miR-181b expression at transcriptional level in PCa
cells. Furthermore, inhibition of miR-181b in EZH2-depleted cells
largely abrogated the effect of sh-EZH2 on cellular glucose
metabolism and the decreased HK2 expression in prostate cells,
suggesting that EZH2 regulates prostate cancer energy metabolism at
least partially in a miR-181b-dependent manner (Fig. 4G-I).
EZH2 inhibition suppresses tumor
growth and modulates miR-181b/HK2 axis in vivo
To further investigate the role of EZH2/miR-181b/HK2
signaling in tumor growth of prostate cancer in vivo, we
extended our investigation by a nude mouse prostate cancer
xenograft model. PC-3 cells were infected with
EZH2-siRNA-lentivirus. Inhibition of EZH2 repressed prostate cancer
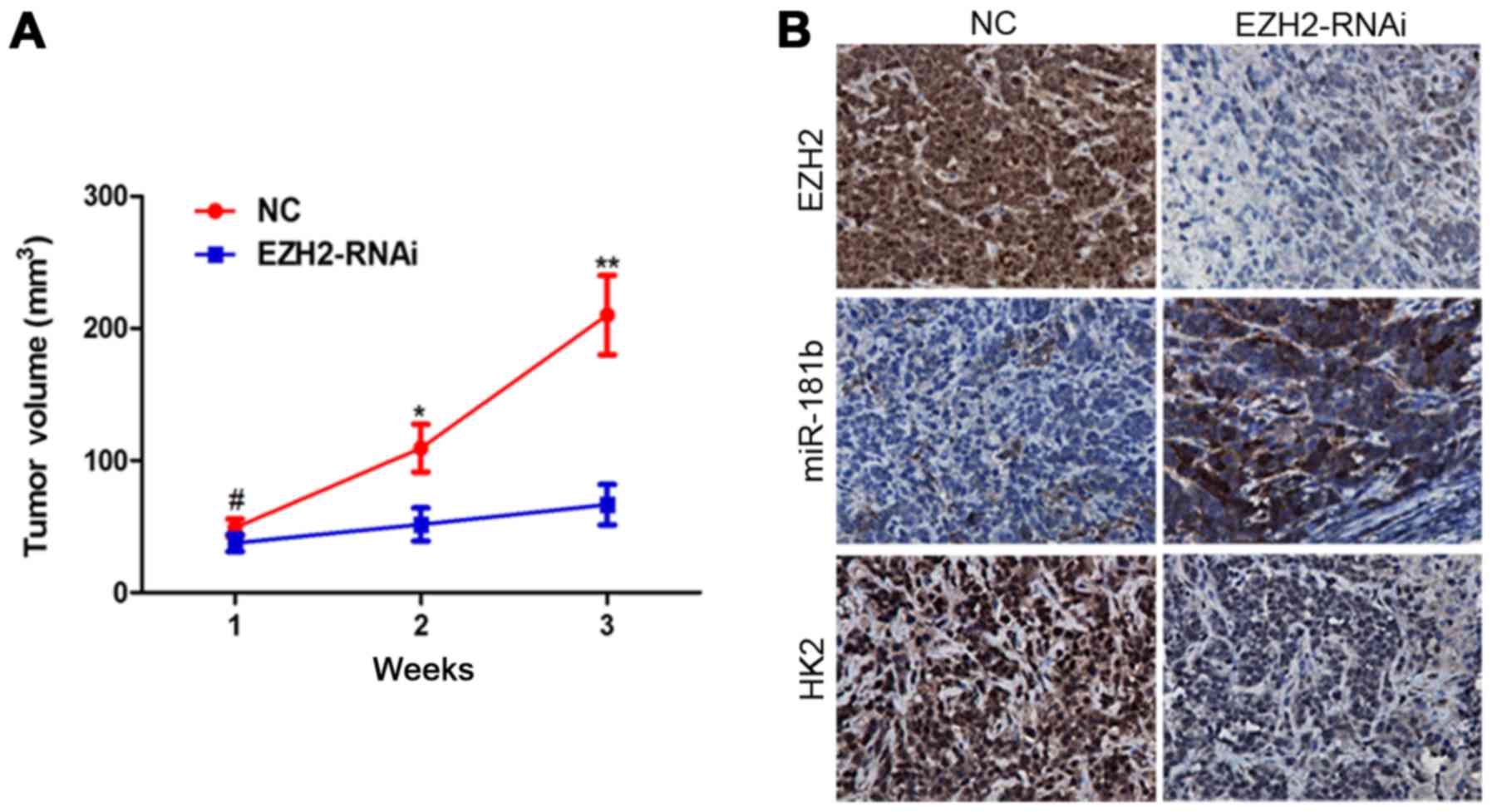
xenograft growth in vivo (Fig.
5A). ISH and IHC analysis revealed that miR-181b and HK2
expression were markedly changed following EZH2 inhibition
(Fig. 5B).
Discussion
Prostate cancer is a common malignant tumor in male
genito-urinary system. Its incidence has obvious geographical and
ethnic differences. In the United States, prostate cancer is the
most commonly diagnosed cancer and the second leading cause of
cancer mortalities in men (19). In
Europe, approximately 75,800 patients were estimated to die from
prostate cancer in 2016 (20). Its
mortality ranks the third in all male malignant tumors. It has
become a major health concern in the older male population in the
world. At present, radical prostatectomy and external radiation
therapy are main methods of treatment for localized prostate
cancer. Additionally, hormonal therapy is also an important
treatment method for some complicated cases such as patients with
distant metastasis or recurrence after treatment. More than 80% of
the patients can alleviate disease through androgen deprivation
therapy. However, after 14–30 months, almost all patients with
lesions will be gradually transformed to CRPC, which leads to very
poor prognosis. Although a variety of drugs are used for CRPC
patients, including docetaxel, abiraterone, and enzalutamide, their
curative effects are still limited (21). CRPC has become the main cause of
death in patients with advanced prostate cancer. It is necessary to
further study tumor progression mechanism and novel molecular
targets.
EZH2 is the catalytic member of the polycomb
repressive complex 2 (PRC2). It comprises a SET domain which is
recognized as the signature of methyltransferases as it provides
the active site for the covalent methylation reaction, resulting in
trimethylation (me3) of histone 3 (H3) at lysine 27 (K27), as well
as at lysine 9 (K9) albeit to a much lesser extent. EZH2 alone
exhibits no intrinsic enzymatic function, in order to be
catalytically active it must interact with at least two proteins,
embryonic ectoderm development (EED) and suppressor of zeste 12
(SUZ12) (22–24).
Studies have found that EZH2 is dysregulated in
breast cancer, bladder cancer, gastric cancer, lung cancer, and
liver cancer, which indicating poor prognosis (25,26).
Also there is higher expression in metastatic prostate cancer
tissues than in localized ones (27,28).
It was reported that EZH2 promotes the invasive ability of tumor
cells through catalyzing H3K27me3 and then inhibiting the
expression of ADRB2 and CDH1 (29,30).
However, the expression of EZH2 is not clear in prostate cancer of
different stages and different pathological levels. In addition, we
do not know the potential biological function of EZH2 and its
relationship with non-coding RNA signaling pathways. In this study,
we further evaluated the EZH2 biological behavior in prostate
cancer progression and its potential molecular mechanisms by a
series of methods including clinical sample analysis,
bioinformatics, microarray, cell function experiments, molecular
biology experiments and animal experiments in vivo. The
result indicated that EZH2 expression was elevated in prostate
cancer tissues compared to normal prostate tissues.
Further, EZH2 expression significantly increased in
CRPC compared to ADPC. Its expression was significantly elevated in
Gleason >7 prostate cancer tissues compared to Gleason ≤7 ones.
EZH2 depletion inhibited cellular colony formation and aerobic
glycolysis process in vitro. Additionally, EZH2 inhibition
significantly slowed tumor growth rate in nude mouse tumor
xenograft experiments. Next, we used gene expression profile
microarray to analyze the metabolism-related genes which were
correlated with EZH2. The results showed that the expression of HK2
was positively correlated with EZH2. Real-time PCR and luciferase
reporter assays showed that EZH2 inversely modulated miR-181b at
transcriptional level. Further study indicated that HK2 was a
direct target of miR-181b. Moreover, decreased miR-181b expression
largely abrogated the effect of sh-EZH2 on HK2 expression and
HK2-induced glucose metabolism process. The results indicate that
miR-181b serves as important mediator between EZH2 and its
downstream pathway.
Above all, we discover that EZH2 was able to
regulate glucose metabolism in prostate cancer, acquired the EZH2
related gene profile and construct EZH2/miRNA axis and its
downstream pathway in prostate cancer. These accomplishments
establish a foundation for further research on miRNA-mediated
EZH2-related molecule network. Next we are aiming to reveal the
network model, providing novel targets and contributing to the
optimizing treatment strategies for molecular targeted therapy in
prostate cancer.
Acknowledgements
This study was supported by National Natural Science
Foundation of China (81370849, 81300472, and 81572517), and Anhui
Natural Science Foundation (1608085MH166 and 1708085QH202).
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heiden MG Vander, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kroemer G and Pouyssegur J: Tumor cell
metabolism: Cancer's Achilles' heel. Cancer Cell. 13:472–482. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Heiden MG Vander: Targeting cancer
metabolism: A therapeutic window opens. Nat Rev Drug Discov.
10:671–684. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singleterry J, Sreedhar A and Zhao Y:
Components of cancer metabolism and therapeutic interventions.
Mitochondrion. 17:50–55. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Swietach P, Vaughan-Jones RD and Harris
AL: Regulation of tumor pH and the role of carbonic anhydrase 9.
Cancer Metastasis Rev. 26:299–310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tao T, Li G, Dong Q, Liu D, Liu C, Han D,
Huang Y, Chen S, Xu B and Chen M: Loss of SNAIL inhibits cellular
growth and metabolism through the miR-128-mediated
RPS6KB1/HIF-1α/PKM2 signaling pathway in prostate cancer cells.
Tumour Biol. 35:8543–8550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warburg O: On respiratory impairment in
cancer cells. Science. 124:269–270. 1956.PubMed/NCBI
|
|
10
|
Yeung SJ, Pan J and Lee MH: Roles of p53,
MYC and HIF-1 in regulating glycolysis - the seventh hallmark of
cancer. Cell Mol Life Sci. 65:3981–3999. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hsu PP and Sabatini DM: Cancer cell
metabolism: Warburg and beyond. Cell. 134:703–707. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mathupala SP, Ko YH and Pedersen PL:
Hexokinase-2 bound to mitochondria: Cancer's stygian link to the
‘Warburg Effect’ and a pivotal target for effective therapy. Semin
Cancer Biol. 19:17–24. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pedersen PL, Mathupala S, Rempel A,
Geschwind JF and Ko YH: Mitochondrial bound type II hexokinase: A
key player in the growth and survival of many cancers and an ideal
prospect for therapeutic intervention. Biochim Biophys Acta.
1555:14–20. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wolf A, Agnihotri S, Micallef J, Mukherjee
J, Sabha N, Cairns R, Hawkins C and Guha A: Hexokinase 2 is a key
mediator of aerobic glycolysis and promotes tumor growth in human
glioblastoma multiforme. J Exp Med. 208:313–326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Taylor BS, Schultz N, Hieronymus H,
Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva
B, et al: Integrative genomic profiling of human prostate cancer.
Cancer Cell. 18:11–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Grasso CS, Wu YM, Robinson DR, Cao X,
Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC,
et al: The mutational landscape of lethal castration-resistant
prostate cancer. Nature. 487:239–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang C, Zhang J, Hao J, Shi Z, Wang Y,
Han L, Yu S, You Y, Jiang T, Wang J, et al: High level of
miR-221/222 confers increased cell invasion and poor prognosis in
glioma. J Transl Med. 10:1192012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang XF, Shi ZM, Wang XR, Cao L, Wang YY,
Zhang JX, Yin Y, Luo H, Kang CS, Liu N, et al: MiR-181d acts as a
tumor suppressor in glioma by targeting K-ras and Bcl-2. J Cancer
Res Clin Oncol. 138:573–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Malvezzi M, Carioli G, Bertuccio P, Rosso
T, Boffetta P, Levi F, La Vecchia C and Negri E: European cancer
mortality predictions for the year 2016 with focus on leukaemias.
Ann Oncol. 27:725–731. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tao T, Liu D, Liu C, Xu B, Chen S, Yin Y,
Ang L, Huang Y, Zhang X and Chen M: Autoregulatory feedback loop of
EZH2/miR-200c/E2F3 as a driving force for prostate cancer
development. Biochim Biophys Acta. 1839:858–865. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang YA and Yu J: EZH2, an epigenetic
driver of prostate cancer. Protein Cell. 4:331–341. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim J and Yu J: Interrogating genomic and
epigenomic data to understand prostate cancer. Biochim Biophys
Acta. 1825:186–196. 2012.PubMed/NCBI
|
|
24
|
Varambally S, Cao Q, Mani RS, Shankar S,
Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, et al:
Genomic loss of microRNA-101 leads to overexpression of histone
methyltransferase EZH2 in cancer. Science. 322:1695–1699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sauvageau M and Sauvageau G: Polycomb
group proteins: Multi-faceted regulators of somatic stem cells and
cancer. Cell Stem Cell. 7:299–313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kleer CG, Cao Q, Varambally S, Shen R, Ota
I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, et al: EZH2
is a marker of aggressive breast cancer and promotes neoplastic
transformation of breast epithelial cells. Proc Natl Acad Sci USA.
100:11606–11611. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Berezovska OP, Glinskii AB, Yang Z, Li XM,
Hoffman RM and Glinsky GV: Essential role for activation of the
Polycomb group (PcG) protein chromatin silencing pathway in
metastatic prostate cancer. Cell Cycle. 5:1886–1901. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hoffmann MJ, Engers R, Florl AR, Otte AP,
Muller M and Schulz WA: Expression changes in EZH2, but not in
BMI-1, SIRT1, DNMT1 or DNMT3B are associated with DNA methylation
changes in prostate cancer. Cancer Biol Ther. 6:1403–1412. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani
RS, Tomlins SA, Mehra R, Laxman B, Cao X, Yu J, et al: Repression
of E-cadherin by the polycomb group protein EZH2 in cancer.
Oncogene. 27:7274–7284. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu J, Cao Q, Mehra R, Laxman B, Yu J,
Tomlins SA, Creighton CJ, Dhanasekaran SM, Shen R, Chen G, et al:
Integrative genomics analysis reveals silencing of beta-adrenergic
signaling by polycomb in prostate cancer. Cancer Cell. 12:419–431.
2007. View Article : Google Scholar : PubMed/NCBI
|